
PARP Inhibitors in Glioma: A Review of Therapeutic Opportunities



Abstract
:Simple Summary
Abstract
1. Introduction
2. Targeting DNA Damage Repair
- Temozolomide produces O6-methylguannine adducts which are incorrectly paired with thymine instead of cytosine during DNA synthesis. O6-methylguanine-DNA-methyltransferase (MGMT) is a DNA repair enzyme which removes the abnormal methyl groups to neutralize this DNA damage. MGMT promoter methylation reduces the expression of MGMT, which in turn improves the efficacy of temozolomide. Conversely, unmethylated tumors are associated with temozolomide resistance.
- Mismatch repair systems facilitate the abnormal pairing of O6-methylguannine adducts with thymine. They need to be intact for temozolomide-induced cytotoxicity. Conversely, mismatch repair deficiency leads to temozolomide resistance.
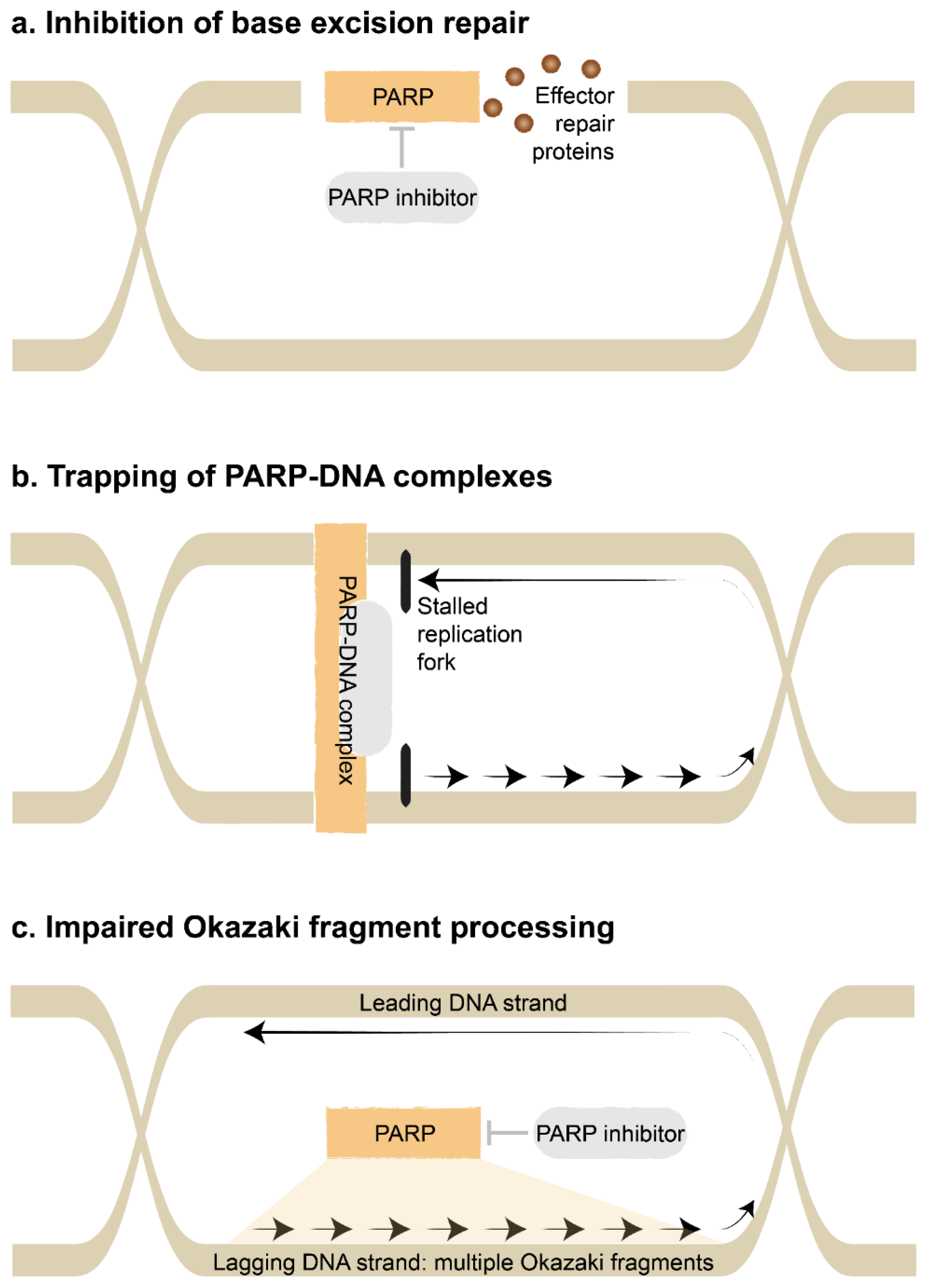
- Inhibition of base excision repair: Single-strand DNA damage is repaired by mismatch repair, nucleotide excision repair or base excision repair, whereas double-strand DNA damage is repaired by homologous recombination or an error-prone system of non-homologous end-joining [7]. The class of PARP inhibitors canonically interferes with the base excision repair pathway [8]. Ordinarily, PARP enzymes bind to sites of DNA damage, catalyze the attachment of polymers of ADP-Ribose in a process called PARylation, then recruit and regulate effector repair proteins. The catalytic activity of the PARP enzymes are blocked by PARP inhibitors. The base excision repair pathway is essential to repair damaged bases caused by ionizing radiation and alkylating agents such as temozolomide.
- Trapping of PARP-DNA complexes: PARP inhibitors also lead to the trapping of PARP1 at sites of DNA damage. These persistent PARP-DNA complexes interfere with DNA replication. They promote collapse of the replication fork, which is the region where the DNA helix is unwound and used as a template for DNA synthesis, in turn leading to double-strand DNA breaks [8]. Alternatively, the replication fork may reverse direction when encountering PARP-DNA complexes, then accelerate in an unrestrained manner to cause double-strand DNA breaks [9]. New generation PARP inhibitors such as talazoparib efficiently trap PARP-DNA complexes and have potent cytotoxicity [10]. This appears to be independent of blocking the catalytic activity of the PARP enzymes.
- Impaired Okazaki fragment processing: Finally, PARP inhibitors may interfere with the processing of Okazaki fragments [11]. During replication, the leading DNA strand (forward direction) is replicated in a continuous manner. However, the lagging DNA strand (reverse direction) is replicated in an interrupted manner as Okazaki fragments, since DNA polymerase can only work in a forward direction. These Okazaki fragments are later connected together by DNA ligase. Ordinarily, PARP1 acts as a key sensor for these unbound Okazaki fragments. Consequently, disruption by PARP inhibitors causes gaps to form within the lagging DNA strand. This cytotoxicity appears to be independent of the previous mechanisms.
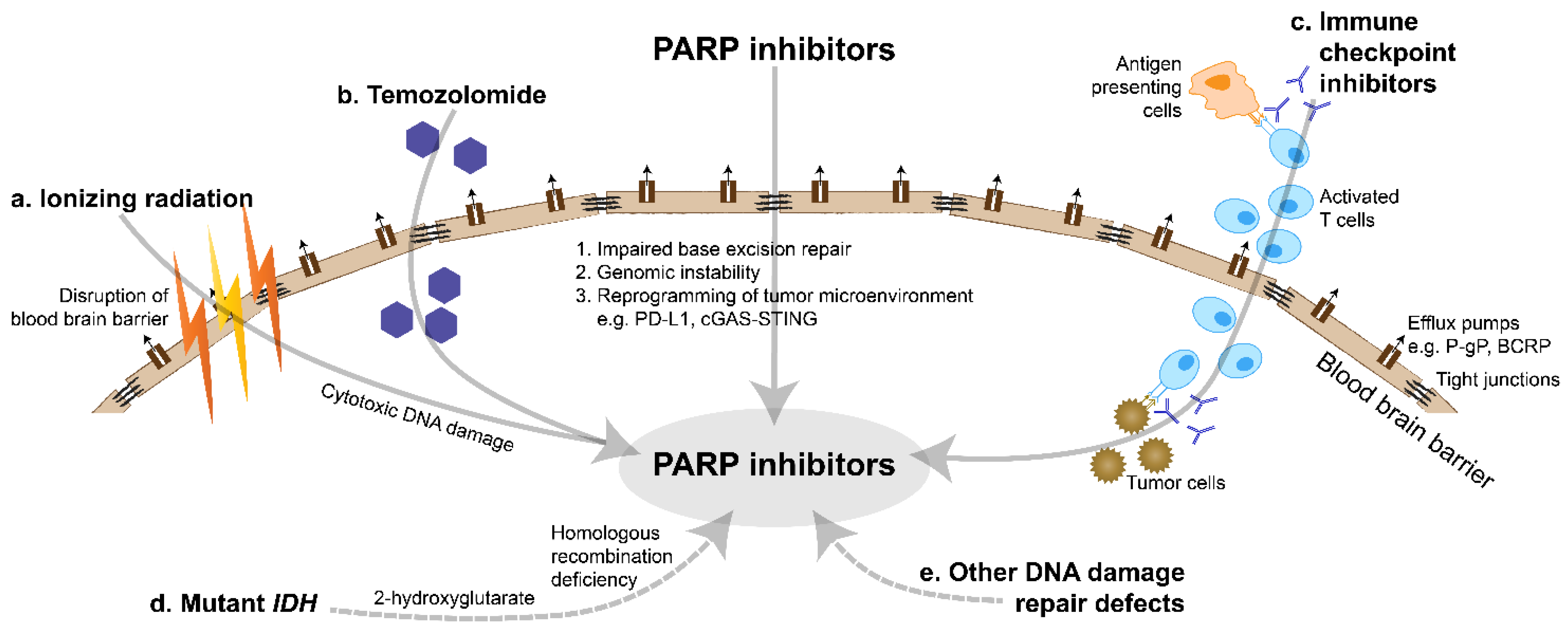
3. Blood Brain Barrier Uptake
4. PARP Inhibition with Radiotherapy or Tumor Treating Fields
- The base excision repair pathway is essential to repair damaged bases caused by ionizing radiation. The pathway is blocked by PARP inhibitors, which result in sensitizer enhancement ratios of 1.2–1.7 in glioma cells [47]. This radiosensitizing effect appears to increase in a replication-dependent manner, due to greater DNA turnover and damage with higher replication.
- Radiotherapy can effectively disrupt the blood brain barrier, augmented by modern localization techniques [53]. This can enhance the delivery of PARP inhibitors into tumor tissues.
5. PARP Inhibition with Chemotherapy or Antiangiogenics
6. PARP Inhibition with Immunotherapy
- By targeting the DNA damage repair pathways, PARP inhibitors cause an accumulation of DNA damage and genomic instability. This is expected to increase the tumor mutational burden and immunogenicity [72]. The efficacy of immune checkpoint inhibitors in highly mutated tumors has been well established [73].
- PD-L1 is a co-inhibitory ligand of the immune system which may be a predictive biomarker of response to immune checkpoint inhibitors [74]. PARP inhibitors eventually lead to double-strand DNA breaks within cancer cells, which in turn induce inflammatory pathways to upregulate PD-L1 expression. This includes activation of the cGAS-STING pathway, activation of the ATM-ATR-CHK1 pathway and inhibition of GSK-3β [72,75,76,77].
- Gliomas are characterized by a highly immunosuppressive environment dominated by glioma-associated macrophages which have limited innate activity [78]. PARP inhibitors contribute to reprogramming of the tumor microenvironment and may promote a favorable Th1-mediated immune response [79]. In particular, PARP inhibitor-mediated activation of the cGAS-STING pathway increases the number of tumor-infiltrating lymphocytes [72]. PARP inhibitors also appear to have other pleiotropic effects on the tumor microenvironment, including production of cytokines which regulate natural killer cells and angiogenesis [72,77,79].
7. PARP Inhibition in IDH-Mutant Glioma
8. Future Directions
Author Contributions
Funding
Conflicts of Interest
References
- Louis, D.N.; Perry, A.; Wesseling, P.; Brat, D.J.; Cree, I.A.; Figarella-Branger, D.; Hawkins, C.; Ng, H.K.; Pfister, S.M.; Reifenberger, G.; et al. The 2021 WHO Classification of Tumors of the Central Nervous System: A summary. Neuro-Oncology 2021, 23, 1231–1251. [Google Scholar] [CrossRef] [PubMed]
- Sim, H.-W.; Nowak, A.K.; Lwin, Z.; Khasraw, M. Management of glioblastoma: An Australian perspective. Chin. Clin. Oncol. 2021, 10, 42. [Google Scholar] [CrossRef] [PubMed]
- Tan, A.C.; Ashley, D.M.; López, G.Y.; Malinzak, M.; Friedman, H.S.; Khasraw, M. Management of glioblastoma: State of the art and future directions. CA Cancer J. Clin. 2020, 70, 299–312. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.; Miner, A.; Hennis, L.; Mittal, S. Mechanisms of temozolomide resistance in glioblastoma—A comprehensive review. Cancer Drug Resist. 2020, 4, 17–43. [Google Scholar] [CrossRef]
- Furgason, J.M.; Bahassi, E.M. Targeting DNA repair mechanisms in cancer. Pharmacol. Ther. 2013, 137, 298–308. [Google Scholar] [CrossRef]
- Faraoni, I.; Graziani, G. Role of BRCA Mutations in Cancer Treatment with Poly(ADP-Ribose) Polymerase (PARP) Inhibitors. Cancers 2018, 10, 487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fleck, O.; Nielsen, O. DNA repair. J. Cell Sci. 2004, 117, 515–517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farmer, H.; McCabe, N.; Lord, C.J.; Tutt, A.N.J.; Johnson, D.A.; Richardson, T.B.; Santarosa, M.; Dillon, K.J.; Hickson, I.; Knights, C.; et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005, 434, 917–921. [Google Scholar] [CrossRef]
- Maya-Mendoza, A.; Moudry, P.; Merchut-Maya, J.M.; Lee, M.; Strauss, R.; Bartek, J. High speed of fork progression induces DNA replication stress and genomic instability. Nature 2018, 559, 279–284. [Google Scholar] [CrossRef]
- Min, A.; Im, S.-A. PARP Inhibitors as Therapeutics: Beyond Modulation of PARylation. Cancers 2020, 12, 394. [Google Scholar] [CrossRef] [Green Version]
- Cong, K.; Peng, M.; Kousholt, A.N.; Lee, W.T.C.; Lee, S.; Nayak, S.; Krais, J.; VanderVere-Carozza, P.S.; Pawelczak, K.S.; Calvo, J.; et al. Replication gaps are a key determinant of PARP inhibitor synthetic lethality with BRCA deficiency. Mol. Cell 2021, 81, 3128–3144.e7. [Google Scholar] [CrossRef] [PubMed]
- Fong, P.C.; Boss, D.S.; Yap, T.A.; Tutt, A.; Wu, P.; Mergui-Roelvink, M.; Mortimer, P.; Swaisland, H.; Lau, A.; O’Connor, M.J.; et al. Inhibition of Poly(ADP-Ribose) Polymerase in Tumors fromBRCAMutation Carriers. N. Engl. J. Med. 2009, 361, 123–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moore, K.; Colombo, N.; Scambia, G.; Kim, B.-G.; Oaknin, A.; Friedlander, M.; Lisyanskaya, A.; Floquet, A.; Leary, A.; Sonke, G.S.; et al. Maintenance Olaparib in Patients with Newly Diagnosed Advanced Ovarian Cancer. N. Engl. J. Med. 2018, 379, 2495–2505. [Google Scholar] [CrossRef]
- Banerjee, S.; Moore, K.N.; Colombo, N.; Scambia, G.; Kim, B.-G.; Oaknin, A.; Friedlander, M.; Lisyanskaya, A.; Floquet, A.; Leary, A.; et al. Maintenance olaparib for patients with newly diagnosed advanced ovarian cancer and a BRCA mutation (SOLO1/GOG 3004): 5-year follow-up of a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2021, 22, 1721–1731. [Google Scholar] [CrossRef]
- Ray-Coquard, I.; Pautier, P.; Pignata, S.; Pérol, D.; González-Martín, A.; Berger, R.; Fujiwara, K.; Vergote, I.; Colombo, N.; Mäenpää, J.; et al. Olaparib plus Bevacizumab as First-Line Maintenance in Ovarian Cancer. N. Engl. J. Med. 2019, 381, 2416–2428. [Google Scholar] [CrossRef]
- Pujade-Lauraine, E.; Ledermann, J.A.; Selle, F.; Gebski, V.; Penson, R.T.; Oza, A.M.; Korach, J.; Huzarski, T.; Poveda, A.; Pignata, S.; et al. Olaparib tablets as maintenance therapy in patients with platinum-sensitive, relapsed ovarian cancer and a BRCA1/2 mutation (SOLO2/ENGOT-Ov21): A double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Oncol. 2017, 18, 1274–1284. [Google Scholar] [CrossRef] [Green Version]
- Poveda, A.; Floquet, A.; Ledermann, J.A.; Asher, R.; Penson, R.T.; Oza, A.M.; Korach, J.; Huzarski, T.; Pignata, S.; Friedlander, M.; et al. Olaparib tablets as maintenance therapy in patients with platinum-sensitive relapsed ovarian cancer and a BRCA1/2 mutation (SOLO2/ENGOT-Ov21): A final analysis of a double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Oncol. 2021, 22, 620–631. [Google Scholar] [CrossRef]
- Ledermann, J.; Harter, P.; Gourley, C.; Friedlander, M.; Vergote, I.; Rustin, G.; Scott, C.; Meier, W.; Shapira-Frommer, R.; Safra, T.; et al. Olaparib Maintenance Therapy in Platinum-Sensitive Relapsed Ovarian Cancer. N. Engl. J. Med. 2012, 366, 1382–1392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedlander, M.; Matulonis, U.; Gourley, C.; Du Bois, A.; Vergote, I.; Rustin, G.; Scott, C.; Meier, W.; Shapira-Frommer, R.; Safra, T.; et al. Long-term efficacy, tolerability and overall survival in patients with platinum-sensitive, recurrent high-grade serous ovarian cancer treated with maintenance olaparib capsules following response to chemotherapy. Br. J. Cancer 2018, 119, 1075–1085. [Google Scholar] [CrossRef] [Green Version]
- Matulonis, U.A.; Penson, R.T.; Domchek, S.M.; Kaufman, B.; Shapira-Frommer, R.; Audeh, M.W.; Kaye, S.; Molife, L.R.; Gelmon, K.A.; Robertson, J.D.; et al. Olaparib monotherapy in patients with advanced relapsed ovarian cancer and a germline BRCA1/2 mutation: A multistudy analysis of response rates and safety. Ann. Oncol. 2016, 27, 1013–1019. [Google Scholar] [CrossRef] [PubMed]
- Robson, M.; Im, S.-A.; Senkus, E.; Xu, B.; Domchek, S.M.; Masuda, N.; Delaloge, S.; Li, W.; Tung, N.; Armstrong, A.; et al. Olaparib for Metastatic Breast Cancer in Patients with a Germline BRCA Mutation. N. Engl. J. Med. 2017, 377, 523–533. [Google Scholar] [CrossRef] [PubMed]
- Golan, T.; Hammel, P.; Reni, M.; Van Cutsem, E.; Macarulla, T.; Hall, M.J.; Park, J.-O.; Hochhauser, D.; Arnold, D.; Oh, D.-Y.; et al. Maintenance Olaparib for Germline BRCA-Mutated Metastatic Pancreatic Cancer. N. Engl. J. Med. 2019, 381, 317–327. [Google Scholar] [CrossRef] [PubMed]
- De Bono, J.; Mateo, J.; Fizazi, K.; Saad, F.; Shore, N.; Sandhu, S.; Chi, K.N.; Sartor, O.; Agarwal, N.; Olmos, D.; et al. Olaparib for Metastatic Castration-Resistant Prostate Cancer. N. Engl. J. Med. 2020, 382, 2091–2102. [Google Scholar] [CrossRef]
- Coleman, R.L.; Oza, A.M.; Lorusso, D.; Aghajanian, C.; Oaknin, A.; Dean, A.; Colombo, N.; Weberpals, J.I.; Clamp, A.; Scambia, G.; et al. Rucaparib maintenance treatment for recurrent ovarian carcinoma after response to platinum therapy (ARIEL3): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2017, 390, 1949–1961. [Google Scholar] [CrossRef] [Green Version]
- Ledermann, J.A.; Oza, A.M.; Lorusso, D.; Aghajanian, C.; Oaknin, A.; Dean, A.; Colombo, N.; Weberpals, J.I.; Clamp, A.R.; Scambia, G.; et al. Rucaparib for patients with platinum-sensitive, recurrent ovarian carcinoma (ARIEL3): Post-progression outcomes and updated safety results from a randomised, placebo-controlled, phase 3 trial. Lancet Oncol. 2020, 21, 710–722. [Google Scholar] [CrossRef]
- Kristeleit, R.; Shapiro, G.I.; Burris, H.A.; Oza, A.M.; Lorusso, P.; Patel, M.R.; Domchek, S.M.; Balmaña, J.; Drew, Y.; Chen, L.-M.; et al. A Phase I–II Study of the Oral PARP Inhibitor Rucaparib in Patients with Germline BRCA1/2-Mutated Ovarian Carcinoma or Other Solid Tumors. Clin. Cancer Res. 2017, 23, 4095–4106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swisher, E.M.; Lin, K.K.; Oza, A.M.; Scott, C.L.; Giordano, H.; Sun, J.; Konecny, G.E.; Coleman, R.L.; Tinker, A.V.; O’Malley, D.M.; et al. Rucaparib in relapsed, platinum-sensitive high-grade ovarian carcinoma (ARIEL2 Part 1): An international, multicentre, open-label, phase 2 trial. Lancet Oncol. 2017, 18, 75–87. [Google Scholar] [CrossRef] [Green Version]
- Abida, W.; Patnaik, A.; Campbell, D.; Shapiro, J.; Bryce, A.H.; McDermott, R.; Sautois, B.; Vogelzang, N.J.; Bambury, R.M.; Voog, E.; et al. Rucaparib in Men with Metastatic Castration-Resistant Prostate Cancer Harboring a BRCA1 or BRCA2 Gene Alteration. J. Clin. Oncol. 2020, 38, 3763–3772. [Google Scholar] [CrossRef] [PubMed]
- Reiss, K.A.; Mick, R.; O’Hara, M.H.; Teitelbaum, U.; Karasic, T.B.; Schneider, C.; Cowden, S.; Southwell, T.; Romeo, J.; Izgur, N.; et al. Phase II Study of Maintenance Rucaparib in Patients with Platinum-Sensitive Advanced Pancreatic Cancer and a Pathogenic Germline or Somatic Variant in BRCA1, BRCA2, or PALB2. J. Clin. Oncol. 2021, 39, 2497–2505. [Google Scholar] [CrossRef]
- González-Martín, A.; Pothuri, B.; Vergote, I.; DePont Christensen, R.; Graybill, W.; Mirza, M.R.; McCormick, C.; Lorusso, D.; Hoskins, P.; Freyer, G.; et al. Niraparib in Patients with Newly Diagnosed Advanced Ovarian Cancer. N. Engl. J. Med. 2019, 381, 2391–2402. [Google Scholar] [CrossRef] [Green Version]
- Mirza, M.R.; Monk, B.J.; Herrstedt, J.; Oza, A.M.; Mahner, S.; Redondo, A.; Fabbro, M.; Ledermann, J.A.; Lorusso, D.; Vergote, I.; et al. Niraparib Maintenance Therapy in Platinum-Sensitive, Recurrent Ovarian Cancer. N. Engl. J. Med. 2016, 375, 2154–2164. [Google Scholar] [CrossRef] [PubMed]
- Mirza, M.R.; Benigno, B.; Dørum, A.; Mahner, S.; Bessette, P.; Barceló, I.B.; Berton-Rigaud, D.; Ledermann, J.; Rimel, B.; Herrstedt, J.; et al. Long-term safety in patients with recurrent ovarian cancer treated with niraparib versus placebo: Results from the phase III ENGOT-OV16/NOVA trial. Gynecol. Oncol. 2020, 159, 442–448. [Google Scholar] [CrossRef]
- Moore, K.N.; Secord, A.A.; Geller, M.A.; Miller, D.S.; Cloven, N.; Fleming, G.F.; Wahner Hendrickson, A.E.; Azodi, M.; DiSilvestro, P.; Oza, A.M.; et al. Niraparib monotherapy for late-line treatment of ovarian cancer (QUADRA): A multicentre, open-label, single-arm, phase 2 trial. Lancet Oncol. 2019, 20, 636–648. [Google Scholar] [CrossRef]
- Litton, J.K.; Rugo, H.S.; Ettl, J.; Hurvitz, S.A.; Gonçalves, A.; Lee, K.-H.; Fehrenbacher, L.; Yerushalmi, R.; Mina, L.A.; Martin, M.; et al. Talazoparib in Patients with Advanced Breast Cancer and a Germline BRCA Mutation. N. Engl. J. Med. 2018, 379, 753–763. [Google Scholar] [CrossRef] [PubMed]
- Heeke, A.L.; Pishvaian, M.J.; Lynce, F.; Xiu, J.; Brody, J.R.; Chen, W.-J.; Baker, T.M.; Marshall, J.L.; Isaacs, C. Prevalence of Homologous Recombination–Related Gene Mutations Across Multiple Cancer Types. JCO Precis. Oncol. 2018, 2018, 1–13. [Google Scholar] [CrossRef]
- Sulkowski, P.L.; Corso, C.D.; Robinson, N.D.; Scanlon, S.E.; Purshouse, K.R.; Bai, H.; Liu, Y.; Sundaram, R.K.; Hegan, D.C.; Fons, N.R.; et al. 2-Hydroxyglutarate produced by neomorphic IDH mutations suppresses homologous recombination and induces PARP inhibitor sensitivity. Sci. Transl. Med. 2017, 9, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pilié, P.G.; Gay, C.M.; Byers, L.A.; O’Connor, M.J.; Yap, T.A. PARP Inhibitors: Extending Benefit Beyond BRCA-Mutant Cancers. Clin. Cancer Res. 2019, 25, 3759–3771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abbott, N.J.; Patabendige, A.A.; Dolman, D.E.; Yusof, S.R.; Begley, D.J. Structure and function of the blood-brain barrier. Neurobiol. Dis. 2010, 37, 13–25. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, S.; Mittapalli, R.K.; Zellmer, D.M.; Gallardo, J.L.; Donelson, R.; Seiler, C.; Decker, S.A.; Santacruz, K.S.; Pokorny, J.L.; Sarkaria, J.N.; et al. Active Efflux of Dasatinib from the Brain Limits Efficacy against Murine Glioblastoma: Broad Implications for the Clinical Use of Molecularly Targeted Agents. Mol. Cancer Ther. 2012, 11, 2183–2192. [Google Scholar] [CrossRef] [Green Version]
- Hanna, C.; Kurian, K.M.; Williams, K.; Watts, C.; Jackson, A.; Carruthers, R.; Strathdee, K.; Cruickshank, G.; Dunn, L.; Erridge, S.; et al. Pharmacokinetics, safety, and tolerability of olaparib and temozolomide for recurrent glioblastoma: Results of the phase I OPARATIC trial. Neuro-Oncology 2020, 22, 1840–1850. [Google Scholar] [CrossRef]
- Sun, K.; Mikule, K.; Wang, Z.; Poon, G.; Vaidyanathan, A.; Smith, G.; Zhang, Z.-Y.; Hanke, J.; Ramaswamy, S.; Wang, J. A comparative pharmacokinetic study of PARP inhibitors demonstrates favorable properties for niraparib efficacy in preclinical tumor models. Oncotarget 2018, 9, 37080–37096. [Google Scholar] [CrossRef] [PubMed]
- Durmus, S.; Sparidans, R.W.; Van Esch, A.; Wagenaar, E.; Beijnen, J.H.; Schinkel, A.H. Breast Cancer Resistance Protein (BCRP/ABCG2) and P-glycoprotein (P-GP/ABCB1) Restrict Oral Availability and Brain Accumulation of the PARP Inhibitor Rucaparib (AG-014699). Pharm. Res. 2014, 32, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Parrish, K.E.; Cen, L.; Murray, J.W.; Calligaris, D.; Kizilbash, S.; Mittapalli, R.K.; Carlson, B.L.; Schroeder, M.A.; Sludden, J.; Boddy, A.; et al. Efficacy of PARP Inhibitor Rucaparib in Orthotopic Glioblastoma Xenografts Is Limited by Ineffective Drug Penetration into the Central Nervous System. Mol. Cancer Ther. 2015, 14, 2735–2743. [Google Scholar] [CrossRef] [Green Version]
- Kizilbash, S.; Gupta, S.K.; Chang, K.; Kawashima, R.; Parrish, K.E.; Carlson, B.L.; Bakken, K.K.; Mladek, A.C.; Schroeder, M.A.; Decker, P.A.; et al. Restricted Delivery of Talazoparib across the Blood–Brain Barrier Limits the Sensitizing Effects of PARP Inhibition on Temozolomide Therapy in Glioblastoma. Mol. Cancer Ther. 2017, 16, 2735–2746. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Delzer, J.; Voorman, R.; De Morais, S.M.; Lao, Y. Disposition and Drug-Drug Interaction Potential of Veliparib (ABT-888), a Novel and Potent Inhibitor of Poly(ADP-Ribose) Polymerase. Drug Metab. Dispos. 2011, 39, 1161–1169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiong, Y.; Guo, Y.; Liu, Y.; Wang, H.; Gong, W.; Liu, Y.; Wang, X.; Gao, Y.; Yu, F.; Su, D.; et al. Pamiparib is a potent and selective PARP inhibitor with unique potential for the treatment of brain tumor. Neoplasia 2020, 22, 431–440. [Google Scholar] [CrossRef] [PubMed]
- Dungey, F.A.; Löser, D.A.; Chalmers, A.J. Replication-Dependent Radiosensitization of Human Glioma Cells by Inhibition of Poly(ADP-Ribose) Polymerase: Mechanisms and Therapeutic Potential. Int. J. Radiat. Oncol. Biol. Phys. 2008, 72, 1188–1197. [Google Scholar] [CrossRef]
- Bao, S.; Wu, Q.; McLendon, R.E.; Hao, Y.; Shi, Q.; Hjelmeland, A.B.; Dewhirst, M.W.; Bigner, D.D.; Rich, J.N. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 2006, 444, 756–760. [Google Scholar] [CrossRef]
- Carruthers, R.D.; Ahmed, S.U.; Ramachandran, S.; Strathdee, K.; Kurian, K.M.; Hedley, A.; Gomez-Roman, N.; Kalna, G.; Neilson, M.P.; Gilmour, L.; et al. Replication Stress Drives Constitutive Activation of the DNA Damage Response and Radioresistance in Glioblastoma Stem-like Cells. Cancer Res. 2018, 78, 5060–5071. [Google Scholar] [CrossRef] [Green Version]
- Ali, M.; Kamjoo, M.; Thomas, H.D.; Kyle, S.; Pavlovska, I.; Babur, M.; Telfer, B.; Curtin, N.J.; Williams, K.J. The Clinically Active PARP Inhibitor AG014699 Ameliorates Cardiotoxicity but Does Not Enhance the Efficacy of Doxorubicin, despite Improving Tumor Perfusion and Radiation Response in Mice. Mol. Cancer Ther. 2011, 10, 2320–2329. [Google Scholar] [CrossRef] [Green Version]
- Albert, J.M.; Cao, C.; Kim, K.W.; Willey, C.; Geng, L.; Xiao, D.; Wang, H.; Sandler, A.; Johnson, D.H.; Colevas, A.D.; et al. Inhibition of Poly(ADP-Ribose) Polymerase Enhances Cell Death and Improves Tumor Growth Delay in Irradiated Lung Cancer Models. Clin. Cancer Res. 2007, 13, 3033–3042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tentori, L.; Lacal, P.M.; Muzi, A.; Dorio, A.S.; Leonetti, C.; Scarsella, M.; Ruffini, F.; Xu, W.; Min, W.; Stoppacciaro, A.; et al. Poly(ADP-Ribose) polymerase (PARP) inhibition or PARP-1 gene deletion reduces angiogenesis. Eur. J. Cancer 2007, 43, 2124–2133. [Google Scholar] [CrossRef] [PubMed]
- Appelboom, G.; Detappe, A.; Lopresti, M.; Kunjachan, S.; Mitrasinovic, S.; Goldman, S.; Chang, S.D.; Tillement, O. Stereotactic modulation of blood-brain barrier permeability to enhance drug delivery. Neuro-Oncology 2016, 18, 1601–1609. [Google Scholar] [CrossRef] [PubMed]
- Chalmers, A.J.; Short, S.; Watts, C.; Herbert, C.; Morris, A.; Stobo, J.; Cruickshank, G.; Dunn, L.; Erridge, S.; Godfrey, L.; et al. Phase I clinical trials evaluating olaparib in combination with radiotherapy (RT) and/or temozolomide (TMZ) in glioblastoma patients: Results of OPARATIC and PARADIGM phase I and early results of PARADIGM-2. J. Clin. Oncol. 2018, 36, 2018. [Google Scholar] [CrossRef]
- Lesueur, P.; LeQuesne, J.; Grellard, J.-M.; Dugué, A.; Coquan, E.; Brachet, P.-E.; Geffrelot, J.; Kao, W.; Emery, E.; Berro, D.H.; et al. Phase I/IIa study of concomitant radiotherapy with olaparib and temozolomide in unresectable or partially resectable glioblastoma: OLA-TMZ-RTE-01 trial protocol. BMC Cancer 2019, 19, 198. [Google Scholar] [CrossRef] [PubMed]
- Fulton, B.; Short, S.C.; James, A.; Nowicki, S.; McBain, C.; Jefferies, S.; Kelly, C.; Stobo, J.; Morris, A.; Williamson, A.; et al. PARADIGM-2: Two parallel phase I studies of olaparib and radiotherapy or olaparib and radiotherapy plus temozolomide in patients with newly diagnosed glioblastoma, with treatment stratified by MGMT status. Clin. Transl. Radiat. Oncol. 2018, 8, 12–16. [Google Scholar] [CrossRef] [Green Version]
- Rominiyi, O.; Vanderlinden, A.; Clenton, S.J.; Bridgewater, C.; Al-Tamimi, Y.; Collis, S.J. Tumour treating fields therapy for glioblastoma: Current advances and future directions. Br. J. Cancer 2021, 124, 697–709. [Google Scholar] [CrossRef]
- Sim, H.-W.; McDonald, K.L.; Lwin, Z.; Barnes, E.H.; Rosenthal, M.; Foote, M.C.; Koh, E.-S.; Back, M.; Wheeler, H.; Sulman, E.P.; et al. A randomized phase II trial of veliparib, radiotherapy, and temozolomide in patients with unmethylated MGMT glioblastoma: The VERTU study. Neuro-Oncology 2021, 23, 1736–1749. [Google Scholar] [CrossRef]
- Baxter, P.A.; Su, J.M.; Onar-Thomas, A.; Billups, C.A.; Li, X.-N.; Poussaint, T.Y.; Smith, E.R.; Thompson, P.; Adesina, A.; Ansell, P.; et al. A phase I/II study of veliparib (ABT-888) with radiation and temozolomide in newly diagnosed diffuse pontine glioma: A Pediatric Brain Tumor Consortium study. Neuro-Oncology 2020, 22, 875–885. [Google Scholar] [CrossRef]
- Piotrowski, A.; Puduvalli, V.; Wen, P.; Campian, J.; Colman, H.; Pearlman, M.; Butowski, N.; Battiste, J.; Glass, J.; Cloughesy, T.; et al. Actr-Pamiparib in Combination with Radiation Therapy (Rt) And/Or Temozolomide (Tmz) in Patients with Newly Diagnosed Or Recurrent/Refractory (R/R) Glioblastoma (Gbm); Phase 1b/2 Study Update. Neuro-Oncology 2019, 21, vi21–vi22. [Google Scholar] [CrossRef]
- Blakeley, J.O.; Grossman, S.A.; Chi, A.S.; Mikkelsen, T.; Rosenfeld, M.R.; Ahluwalia, M.S.; Nabors, L.B.; Eichler, A.; Garcia-Ribas, I.; Desideri, S.; et al. Phase II Study of Iniparib with Concurrent Chemoradiation in Patients with Newly Diagnosed Glioblastoma. Clin. Cancer Res. 2018, 25, 73–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Shi, Y.; Maag, D.X.; Palma, J.P.; Patterson, M.J.; Ellis, P.A.; Surber, B.W.; Ready, D.B.; Soni, N.B.; Ladror, U.S.; et al. Iniparib Nonselectively Modifies Cysteine-Containing Proteins in Tumor Cells and Is Not a Bona Fide PARP Inhibitor. Clin. Cancer Res. 2012, 18, 510–523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, A.G.; De Lorenzo, S.B.; Flatten, K.S.; Poirier, G.G.; Kaufmann, S.H. Failure of Iniparib to Inhibit Poly(ADP-Ribose) Polymerase In Vitro. Clin. Cancer Res. 2012, 18, 1655–1662. [Google Scholar] [CrossRef] [Green Version]
- Tentori, L.; Leonetti, C.; Scarsella, M.; D’Amati, G.; Vergati, M.; Portarena, I.; Xu, W.; Kalish, V.; Zupi, G.; Zhang, J.; et al. Systemic administration of GPI 15427, a novel poly(ADP-Ribose) polymerase-1 inhibitor, increases the antitumor activity of temozolomide against intracranial melanoma, glioma, lymphoma. Clin. Cancer Res. 2003, 9, 5370–5379. [Google Scholar] [PubMed]
- Russo, A.L.; Kwon, H.-C.; Burgan, W.E.; Carter, D.; Beam, K.; Weizheng, X.; Zhang, J.; Slusher, B.S.; Chakravarti, A.; Tofilon, P.J.; et al. In vitro and In vivo Radiosensitization of Glioblastoma Cells by the Poly(ADP-Ribose) Polymerase Inhibitor E7016. Clin. Cancer Res. 2009, 15, 607–612. [Google Scholar] [CrossRef] [Green Version]
- Clarke, M.J.; Mulligan, E.A.; Grogan, P.T.; Mladek, A.C.; Carlson, B.L.; Schroeder, M.A.; Curtin, N.J.; Lou, Z.; Decker, P.A.; Wu, W.; et al. Effective sensitization of temozolomide by ABT-888 is lost with development of temozolomide resistance in glioblastoma xenograft lines. Mol. Cancer Ther. 2009, 8, 407–414. [Google Scholar] [CrossRef] [Green Version]
- Gupta, S.K.; Mladek, A.C.; Carlson, B.L.; Boakye-Agyeman, F.; Bakken, K.K.; Kizilbash, S.; Schroeder, M.A.; Reid, J.; Sarkaria, J.N. Discordant In Vitro and In Vivo Chemopotentiating Effects of the PARP Inhibitor Veliparib in Temozolomide-Sensitive versus -Resistant Glioblastoma Multiforme Xenografts. Clin. Cancer Res. 2014, 20, 3730–3741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, J.; Yang, K.; Taylor-Harding, B.; Wiedemeyer, W.R.; Buckanovich, R.J. VEGFR3 Inhibition Chemosensitizes Ovarian Cancer Stemlike Cells through Down-Regulation of BRCA1 and BRCA2. Neoplasia 2014, 16, 343–353.e2. [Google Scholar] [CrossRef] [Green Version]
- Robins, H.I.; Zhang, P.; Gilbert, M.R.; Chakravarti, A.; De Groot, J.F.; Grimm, S.A.; Wang, F.; Lieberman, F.S.; Krauze, A.; Trotti, A.M.; et al. A randomized phase I/II study of ABT-888 in combination with temozolomide in recurrent temozolomide resistant glioblastoma: An NRG oncology RTOG group study. J. Neuro-Oncol. 2015, 126, 309–316. [Google Scholar] [CrossRef] [Green Version]
- Gupta, S.K.; Kizilbash, S.H.; Carlson, B.L.; Mladek, A.C.; Boakye-Agyeman, F.; Bakken, K.K.; Pokorny, J.L.; Schroeder, M.A.; Decker, P.A.; Cen, L.; et al. Delineation of MGMTHypermethylation as a Biomarker for Veliparib-Mediated Temozolomide-Sensitizing Therapy of Glioblastoma. J. Natl. Cancer Inst. 2016, 108, djv369. [Google Scholar] [CrossRef] [Green Version]
- Su, J.M.; Thompson, P.; Adesina, A.; Li, X.-N.; Kilburn, L.; Onar-Thomas, A.; Kocak, M.; Chyla, B.; McKeegan, E.; Warren, K.E.; et al. A phase I trial of veliparib (ABT-888) and temozolomide in children with recurrent CNS tumors: A Pediatric Brain Tumor Consortium report†. Neuro-Oncology 2014, 16, 1661–1668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Z.; Cui, P.; Tao, H.; Zhang, S.; Ma, J.; Liu, Z.; Wang, J.; Qian, Y.; Chen, S.; Huang, Z.; et al. The Synergistic Effect of PARP Inhibitors and Immune Checkpoint Inhibitors. Clin. Med. Insights Oncol. 2021, 15. [Google Scholar] [CrossRef] [PubMed]
- Yarchoan, M.; Hopkins, A.; Jaffee, E.M. Tumor Mutational Burden and Response Rate to PD-1 Inhibition. N. Engl. J. Med. 2017, 377, 2500–2501. [Google Scholar] [CrossRef]
- Doroshow, D.B.; Bhalla, S.; Beasley, M.B.; Sholl, L.M.; Kerr, K.M.; Gnjatic, S.; Wistuba, I.I.; Rimm, D.L.; Tsao, M.S.; Hirsch, F.R. PD-L1 as a biomarker of response to immune-checkpoint inhibitors. Nat. Rev. Clin. Oncol. 2021, 18, 345–362. [Google Scholar] [CrossRef] [PubMed]
- Jiao, S.; Xia, W.; Yamaguchi, H.; Wei, Y.; Chen, M.-K.; Hsu, J.-M.; Hsu, J.L.; Yu, W.-H.; Du, Y.; Lee, H.-H.; et al. PARP Inhibitor Upregulates PD-L1 Expression and Enhances Cancer-Associated Immunosuppression. Clin. Cancer Res. 2017, 23, 3711–3720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sato, H.; Niimi, A.; Yasuhara, T.; Permata, T.B.M.; Hagiwara, Y.; Isono, M.; Nuryadi, E.; Sekine, R.; Oike, T.; Kakoti, S.; et al. DNA double-strand break repair pathway regulates PD-L1 expression in cancer cells. Nat. Commun. 2017, 8, 1751. [Google Scholar] [CrossRef] [PubMed]
- Peyraud, F.; Italiano, A. Combined PARP Inhibition and Immune Checkpoint Therapy in Solid Tumors. Cancers 2020, 12, 1502. [Google Scholar] [CrossRef]
- Gieryng, A.; Pszczolkowska, D.; Walentynowicz, K.A.; Rajan, W.D.; Kaminska, B. Immune microenvironment of gliomas. Lab. Investig. 2017, 97, 498–518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yélamos, J.; Moreno-Lama, L.; Jimeno, J.; Ali, S.O. Immunomodulatory Roles of PARP-1 and PARP-2: Impact on PARP-Centered Cancer Therapies. Cancers 2020, 12, 392. [Google Scholar] [CrossRef] [Green Version]
- Ramos, R.; Climans, S.A.; Adile, A.; Ghiassi, P.; Baker, S.; Phillips, M.J.; Zadeh, G.; Chen, E.X.; Mason, W.P. Combination olaparib and durvalumab for patients with recurrent IDH-mutated gliomas. J. Clin. Oncol. 2021, 39, e14026. [Google Scholar] [CrossRef]
- Ward, P.S.; Thompson, C.B. Metabolic Reprogramming: A Cancer Hallmark Even Warburg Did Not Anticipate. Cancer Cell 2012, 21, 297–308. [Google Scholar] [CrossRef] [Green Version]
- Cairns, R.A.; Mak, T.W. Oncogenic Isocitrate Dehydrogenase Mutations: Mechanisms, Models, and Clinical Opportunities. Cancer Discov. 2013, 3, 730–741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, Y.; Kwintkiewicz, J.; Liu, Y.; Tech, K.; Frady, L.N.; Su, Y.-T.; Bautista, W.; Moon, S.I.; MacDonald, J.; Ewend, M.G.; et al. Chemosensitivity of IDH1-Mutated Gliomas Due to an Impairment in PARP1-Mediated DNA Repair. Cancer Res. 2017, 77, 1709–1718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, F.; Bian, K.; Tang, Q.; Fedeles, B.I.; Singh, V.; Humulock, Z.T.; Essigmann, J.M.; Li, D. Oncometabolites d- and l-2-Hydroxyglutarate Inhibit the AlkB Family DNA Repair Enzymes under Physiological Conditions. Chem. Res. Toxicol. 2017, 30, 1102–1110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, P.; Wu, J.; Ma, S.; Zhang, L.; Yao, J.; Hoadley, K.; Wilkerson, M.D.; Perou, C.; Guan, K.-L.; Ye, D.; et al. Oncometabolite D-2-Hydroxyglutarate Inhibits ALKBH DNA Repair Enzymes and Sensitizes IDH Mutant Cells to Alkylating Agents. Cell Rep. 2015, 13, 2353–2361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, C.; Sule, A.; Beckta, J.; Sundaram, R.; Bindra, R. Exploiting inherent DNA damage repair defects in IDH1/2 mutated gliomas with the CNS penetrant PARP inhibitor, pamiparib. Cancer Res. 2020, 80, 1389. [Google Scholar]
- Ducray, F.; Sanson, M.; Chinot, O.L.; Fontanilles, M.; Rivoirard, R.; Thomas-Maisonneuve, L.; Cartalat, S.; Tabouret, E.; Bonneville-Levard, A.; Darlix, A.; et al. Olaparib in recurrent IDH-mutant high-grade glioma (OLAGLI). J. Clin. Oncol. 2021, 39, 2007. [Google Scholar] [CrossRef]
- Patel, M.; Nowsheen, S.; Maraboyina, S.; Xia, F. The role of poly(ADP-Ribose) polymerase inhibitors in the treatment of cancer and methods to overcome resistance: A review. Cell Biosci. 2020, 10, 1–12. [Google Scholar] [CrossRef]
- Demény, M.; Virág, L. The PARP Enzyme Family and the Hallmarks of Cancer Part Cell Intrinsic Hallmarks. Cancers 2021, 13, 2042. [Google Scholar] [CrossRef]
- Johnson, S.F.; Cruz, C.; Greifenberg, A.K.; Dust, S.; Stover, D.; Chi, D.; Primack, B.; Cao, S.; Bernhardy, A.J.; Coulson, R.; et al. CDK12 Inhibition Reverses De Novo and Acquired PARP Inhibitor Resistance in BRCA Wild-Type and Mutated Models of Triple-Negative Breast Cancer. Cell Rep. 2016, 17, 2367–2381. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
| PARP Inhibitor | Indication | Evidence |
|---|---|---|
| Olaparib (AZD2281) | Newly diagnosed advanced ovarian cancer: maintenance treatment after a response to platinum chemotherapy; germline or somatic BRCA mutation | SOLO-1 [13,14] olaparib (n = 260) vs. placebo (n = 131) PFS HR 0.33 (95% CI 0.25–0.43) |
| Recurrent ovarian cancer: maintenance treatment (in combination with bevacizumab) after a response to platinum chemotherapy; underlying HRD | PAOLA-1 [15] bevacizumab/olaparib (n = 537) vs. bevacizumab/placebo (n = 269) overall: PFS HR 0.59 (95% CI 0.49–0.72) HRD subgroup: PFS HR 0.33 (95% CI 0.25–0.45) | |
| Recurrent ovarian cancer: maintenance treatment after a response to platinum chemotherapy | SOLO-2 [16,17] olaparib (n = 196) vs. placebo (n = 99) PFS HR 0.30 (95% CI 0.22–0.41) OS HR 0.74 (95% CI 0.54–1.00) | |
| Study 19 [18,19] olaparib (n = 136) vs. placebo (n = 129) PFS HR 0.35 (95% CI 0.25–0.49) OS HR 0.73 (95% CI 0.55–0.95) | ||
| Recurrent ovarian cancer: germline BRCA mutation; three or more prior chemotherapy regimens | Pooled analysis [20] olaparib (n = 205 in primary efficacy population) response rate 31% (95% CI 25–38%) | |
| Recurrent HER2-negative breast cancer: germline BRCA mutation | OlympiAD [21] olaparib (n = 205) vs. physician’s choice (n = 97) PFS HR 0.58 (95% CI 0.43–0.80) | |
| Newly diagnosed advanced pancreatic cancer: maintenance treatment after a response to platinum chemotherapy; germline BRCA mutation | POLO [22] olaparib (n = 92) vs. placebo (n = 62) PFS HR 0.53 (95% CI 0.35–0.82) | |
| Recurrent prostate cancer: underlying HRD; prior enzalutamide/abiraterone | PROfound [23] olaparib (n = 256) vs. physician’s choice (n = 131) overall: PFS HR 0.49 (95% CI 0.38–0.63) BRCA subgroup: PFS HR 0.34 (95% CI 0.25–0.47) | |
| Rucaparib (AG014699) | Recurrent ovarian cancer: maintenance treatment after a response to platinum chemotherapy | ARIEL3 [24,25] rucaparib (n = 375) vs. placebo (n = 189) overall: PFS HR 0.36 (95% CI 0.30–0.45) HRD subgroup: PFS HR 0.32 (95% CI 0.24–0.42) BRCA subgroup: PFS HR 0.23 (95% CI 0.16–0.34) |
| Recurrent ovarian cancer: germline or somatic BRCA mutation; two or more prior chemotherapy regimens | Study 10 [26] rucaparib (n = 42 in phase II expansion) response rate 60% (95% CI 43–74%) | |
| ARIEL2 [27] rucaparib (n = 40 in BRCA-mutant subgroup) response rate 80% (95% CI 64–91%) | ||
| Recurrent prostate cancer: germline or somatic BRCA mutation; prior androgen receptor-directed therapy/taxane chemotherapy | TRITON2 [28] rucaparib (n = 62 in primary efficacy population) response rate 44% (95% CI 31–57%) | |
| Niraparib (MK4827) | Newly diagnosed advanced ovarian cancer: maintenance treatment after a response to platinum chemotherapy | PRIMA [30] niraparib (n = 487) vs. placebo (n = 246) overall: PFS HR 0.62 (95% CI 0.50–0.76) HRD subgroup: PFS HR 0.43 (95% CI 0.31–0.59) |
| Recurrent ovarian cancer: maintenance treatment after a response to platinum chemotherapy | NOVA [31,32] niraparib (n = 372) vs. placebo (n = 181) germline BRCA subgroup: PFS HR 0.27 (95% CI 0.17–0.41) non germline BRCA subgroup: PFS HR 0.45 (95% CI 0.34–0.61) | |
| Recurrent ovarian cancer: underlying HRD; three or more prior chemotherapy regimens | QUADRA [33] niraparib (n = 47 in primary efficacy population) response rate 28% (95% CI 16–43%) | |
| Talazoparib (BMN673) | Recurrent HER2-negative breast cancer: germline BRCA mutation | EMBRACA [34] talazoparib (n = 287) vs. physician’s choice (n = 144) PFS HR 0.54 (95% CI 0.41–0.71) |
| Clinical Trial | Phase | Study Population | Intervention | Status |
|---|---|---|---|---|
| PARP Inhibition with Radiotherapy or Tumor Treating Fields | ||||
| NCT03212742 [55] (OLA-TMZ-RTE-01) | 1/2a | Newly diagnosed glioblastoma | Radiotherapy with olaparib/temozolomide, then olaparib/temozolomide | Recruiting |
| CRUKD/16/010 [56] (PARADIGM-2) | 1 | Newly diagnosed glioblastoma | MGMT-methylated cohort: Radiotherapy with olaparib/temozolomide, then olaparib/temozolomide MGMT-unmethylated cohort: Radiotherapy with olaparib, then olaparib | Recruiting |
| NCT05076513 | 0 “trigger” | Newly diagnosed glioblastoma (Cohort A) | Radiotherapy with niraparib, then niraparib | Recruiting |
| NCT04221503 | 2 | Recurrent glioblastoma | Tumor treating fields with niraparib | Recruiting |
| NCT03581292 | 2 | Newly diagnosed high-grade glioma; H3 K27M-wildtype; BRAFV600E-wildtype; children/young adults | Radiotherapy with veliparib, then veliparib/temozolomide | Completed accrual |
| PARP Inhibition with Chemotherapy or Antiangiogenics | ||||
| NCT02974621 | 2 | Recurrent glioblastoma | Olaparib/cediranib vs. bevacizumab | Completed accrual |
| NCT02152982 (Alliance A071102) | 2/3 | Newly diagnosed glioblastoma; MGMT-methylated | Veliparib/temozolomide | Completed accrual |
| NCT04552977 | 2 | Recurrent glioblastoma | Fluzoparil/temozolomide | Not yet open |
| NCT04910022 | 1/2 | Recurrent glioblastoma | NMS-03305293/temozolomide | Not yet open |
| PARP Inhibition in IDH-Mutant Glioma | ||||
| NCT03212274 (ETCTN-10129) | 2 | Recurrent IDH-mutant glioma, cholangiocarcinoma or other solid tumor | Olaparib | Recruiting |
| NCT05076513 | 0 “trigger” | Recurrent IDH-mutant glioma (Cohort B) | Niraparib | Recruiting |
| NCT04740190 (TAC-GReD) | 2 | Recurrent high-grade glioma; DNA damage repair deficiency e.g., IDH-mutant, PTEN-mutant, BRCAness signature | Radiotherapy with talazoparib/carboplatin | Recruiting |
| NCT03914742 (ABTC-1801) | 1/2 | Recurrent IDH-mutant glioma | Pamiparib/temozolomide | Recruiting |
| NCT03749187 (PNOC-017) | 1 | Recurrent IDH-mutant glioma; children/young adults | Pamiparib/temozolomide | Recruiting |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sim, H.-W.; Galanis, E.; Khasraw, M. PARP Inhibitors in Glioma: A Review of Therapeutic Opportunities. Cancers 2022, 14, 1003. https://doi.org/10.3390/cancers14041003
Sim H-W, Galanis E, Khasraw M. PARP Inhibitors in Glioma: A Review of Therapeutic Opportunities. Cancers. 2022; 14(4):1003. https://doi.org/10.3390/cancers14041003
Chicago/Turabian StyleSim, Hao-Wen, Evanthia Galanis, and Mustafa Khasraw. 2022. "PARP Inhibitors in Glioma: A Review of Therapeutic Opportunities" Cancers 14, no. 4: 1003. https://doi.org/10.3390/cancers14041003