
DNA Methylation of Imprinted Genes KCNQ1, KCNQ1OT1, and PHLDA2 in Peripheral Blood Is Associated with the Risk of Breast Cancer

 ,
,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
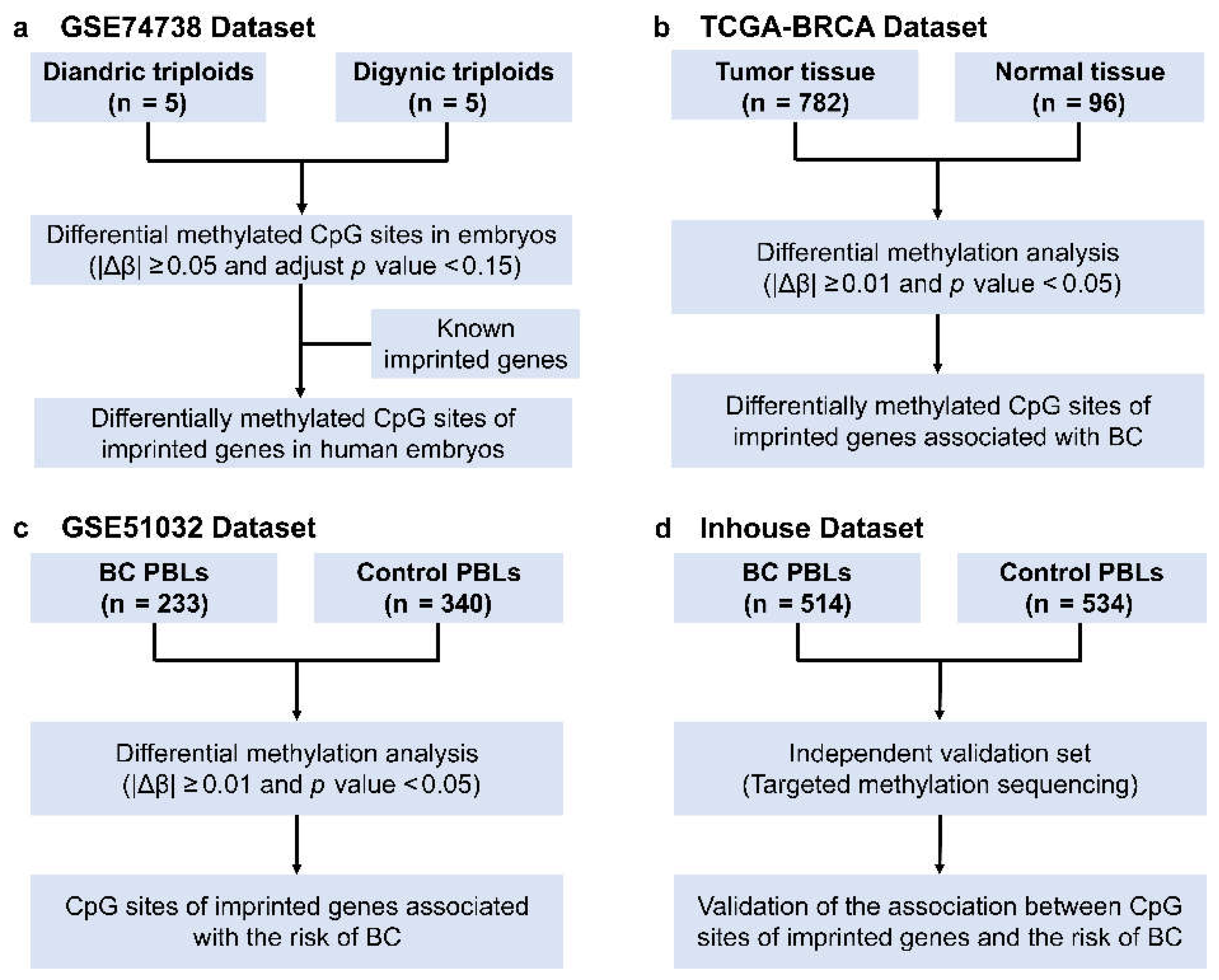
2.1. Public Data Resource
2.2. Selection of CpG Sites of Imprinted Genes Associated with BC Risk
2.3. Inhouse Validation Study
2.4. DNA Extraction and Bisulfite Conversion from Peripheral Blood
2.5. Cell Fractionations from Peripheral Blood Leukocytes
2.6. Quantitative Methylation Analysis
2.7. Statistical Analysis
3. Results
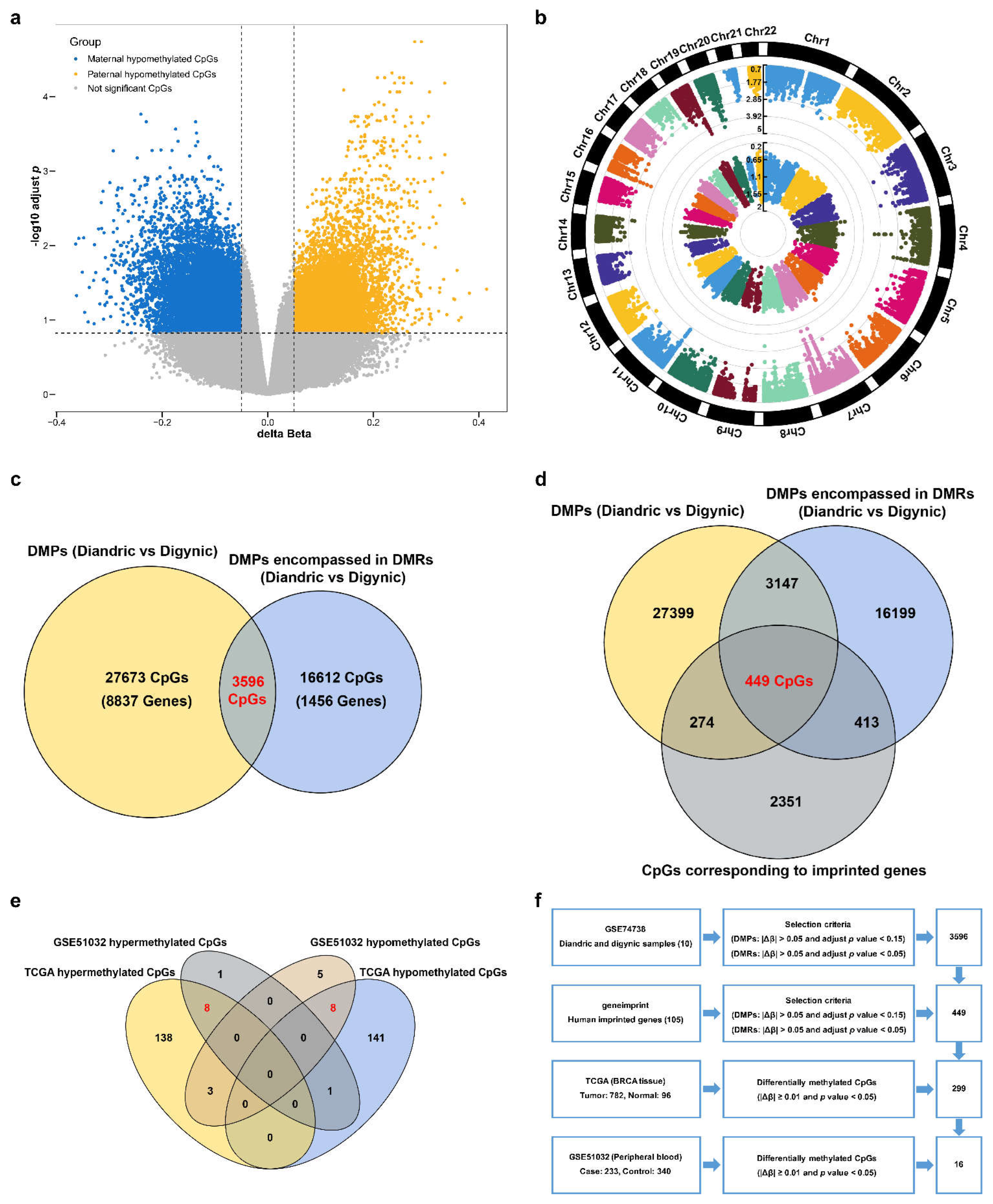
3.1. Discovery of the CpG Sites of Imprinted Genes Associated with BC Risk
3.2. The Methylation of Individual CpG Sites of Selected Imprinted Genes and BC Risk in the Validation Set
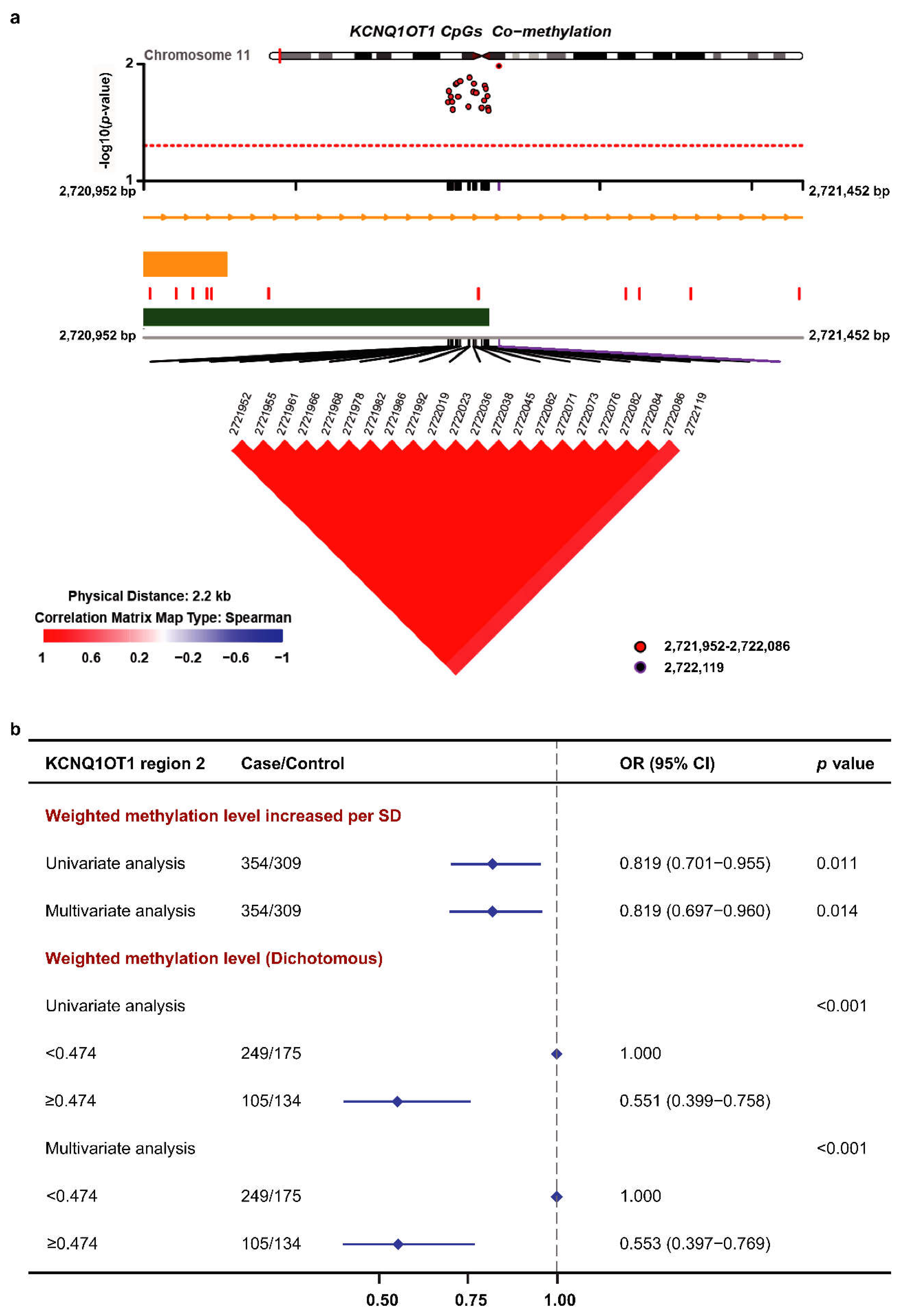
3.3. Methylation of the KCNQ1OT1 Target Region and BC Risk in the Validation Set
3.4. Subgroup Analysis
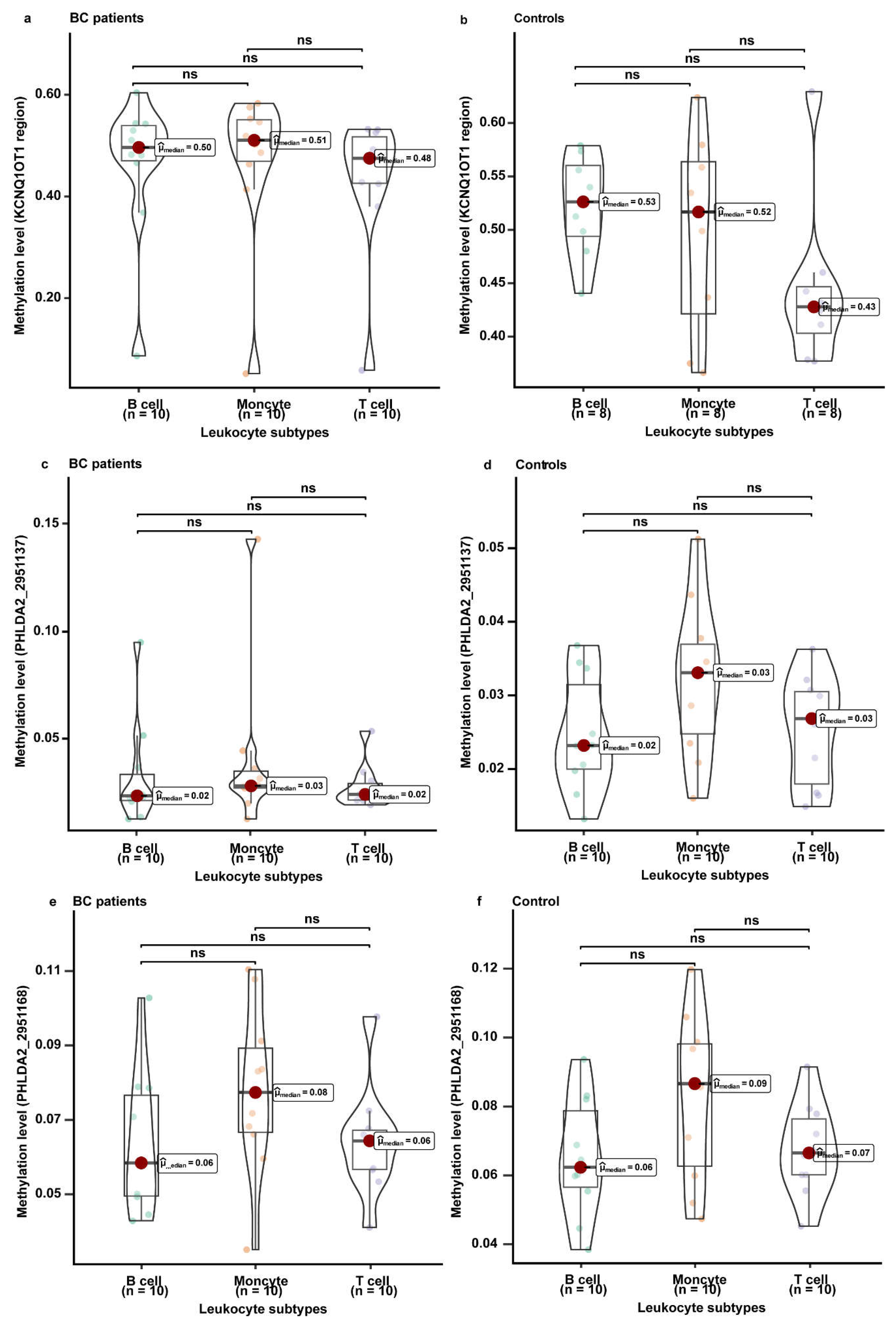
3.5. The Methylation Levels of KCNQ1, KCNQ1OT1, and PHLDA2 in Different Leukocyte Subtypes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Zheng, R.; Baade, P.D.; Zhang, S.; Zeng, H.; Bray, F.; Jemal, A.; Yu, X.Q.; He, J. Cancer statistics in China, 2015. CA Cancer J. Clin. 2016, 66, 115–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collaborative Group on Hormonal Factors in Breast Cancer. Familial breast cancer: Collaborative reanalysis of individual data from 52 epidemiological studies including 58,209 women with breast cancer and 101,986 women without the disease. Lancet 2001, 358, 1389–1399. [Google Scholar] [CrossRef]
- Fackenthal, J.D.; Olopade, O.I. Breast cancer risk associated with BRCA1 and BRCA2 in diverse populations. Nat. Rev. Cancer 2007, 7, 937–948. [Google Scholar] [CrossRef]
- Nathanson, K.L.; Wooster, R.; Weber, B.L. Breast cancer genetics: What we know and what we need. Nat. Med. 2001, 7, 552–556. [Google Scholar] [CrossRef]
- Karsli-Ceppioglu, S.; Dagdemir, A.; Judes, G.; Ngollo, M.; Penault-Llorca, F.; Pajon, A.; Bignon, Y.J.; Bernard-Gallon, D. Epigenetic mechanisms of breast cancer: An update of the current knowledge. Epigenomics 2014, 6, 651–664. [Google Scholar] [CrossRef]
- Reik, W.; Dean, W.; Walter, J. Epigenetic reprogramming in mammalian development. Science 2001, 293, 1089–1093. [Google Scholar] [CrossRef] [Green Version]
- Tucci, V.; Isles, A.R.; Kelsey, G.; Ferguson-Smith, A.C. Genomic Imprinting and Physiological Processes in Mammals. Cell 2019, 176, 952–965. [Google Scholar] [CrossRef] [Green Version]
- Edwards, C.; Ferguson-Smith, A. Mechanisms regulating imprinted genes in clusters. Curr. Opin. Cell Biol. 2007, 19, 281–289. [Google Scholar] [CrossRef]
- Jelinic, P.; Shaw, P. Loss of imprinting and cancer. J. Pathol. 2007, 211, 261–268. [Google Scholar] [CrossRef]
- Feinberg, A.P.; Cui, H.; Ohlsson, R. DNA methylation and genomic imprinting: Insights from cancer into epigenetic mechanisms. Semin. Cancer Biol. 2002, 12, 389–398. [Google Scholar] [CrossRef]
- Falls, J.G.; Pulford, D.J.; Wylie, A.A.; Jirtle, R.L. Genomic imprinting: Implications for human disease. Am. J. Pathol. 1999, 154, 635–647. [Google Scholar] [CrossRef] [Green Version]
- Ennour-Idrissi, K.; Dragic, D.; Issa, E.; Michaud, A.; Chang, S.; Provencher, L.; Durocher, F.; Diorio, C. DNA Methylation and Breast Cancer Risk: An Epigenome-Wide Study of Normal Breast Tissue and Blood. Cancers 2020, 12, 3088. [Google Scholar] [CrossRef] [PubMed]
- Feng, W.; Wang, C.; Liang, C.; Yang, H.; Chen, D.; Yu, X.; Zhao, W.; Geng, D.; Li, S.; Chen, Z.; et al. The Dysregulated Expression of KCNQ1OT1 and Its Interaction with Downstream Factors miR-145/CCNE2 in Breast Cancer Cells. Cell. Physiol. Biochem. 2018, 49, 432–446. [Google Scholar] [CrossRef]
- Rodriguez, B.A.; Weng, Y.I.; Liu, T.M.; Zuo, T.; Hsu, P.Y.; Lin, C.H.; Cheng, A.L.; Cui, H.; Yan, P.S.; Huang, T.H. Estrogen-mediated epigenetic repression of the imprinted gene cyclin-dependent kinase inhibitor 1C in breast cancer cells. Carcinogenesis 2011, 32, 812–821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sunamura, N.; Ohira, T.; Kataoka, M.; Inaoka, D.; Tanabe, H.; Nakayama, Y.; Oshimura, M.; Kugoh, H. Regulation of functional KCNQ1OT1 lncRNA by β-catenin. Sci. Rep. 2016, 6, 20690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moon, H.; Oh, K.; Lee, J.; Lee, M.; Kim, J.; Yoo, T.; Seo, M.; Park, A.; Ryu, H.; Jung, E.; et al. Prognostic and functional importance of the engraftment-associated genes in the patient-derived xenograft models of triple-negative breast cancers. Breast Cancer Res. Treat. 2015, 154, 13–22. [Google Scholar] [CrossRef]
- Liu, X.; Zhang, T.; Li, Y.; Zhang, Y.; Zhang, H.; Wang, X.; Li, L. The Role of Methylation in the CpG Island of the ARHI Promoter Region in Cancers. Adv. Exp. Med. Biol. 2020, 1255, 123–132. [Google Scholar] [CrossRef]
- Fujii, S.; Luo, R.Z.; Yuan, J.; Kadota, M.; Oshimura, M.; Dent, S.R.; Kondo, Y.; Issa, J.P.; Bast, R.C., Jr.; Yu, Y. Reactivation of the silenced and imprinted alleles of ARHI is associated with increased histone H3 acetylation and decreased histone H3 lysine 9 methylation. Hum. Mol. Genet. 2003, 12, 1791–1800. [Google Scholar] [CrossRef] [Green Version]
- Shetty, P.J.; Movva, S.; Pasupuleti, N.; Vedicherlla, B.; Vattam, K.K.; Venkatasubramanian, S.; Ahuja, Y.R.; Hasan, Q. Regulation of IGF2 transcript and protein expression by altered methylation in breast cancer. J. Cancer Res. Clin. Oncol. 2011, 137, 339–345. [Google Scholar] [CrossRef]
- Ito, Y.; Koessler, T.; Ibrahim, A.E.; Rai, S.; Vowler, S.L.; Abu-Amero, S.; Silva, A.L.; Maia, A.T.; Huddleston, J.E.; Uribe-Lewis, S.; et al. Somatically acquired hypomethylation of IGF2 in breast and colorectal cancer. Hum. Mol. Genet. 2008, 17, 2633–2643. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Fujii, S.; Yuan, J.; Luo, R.Z.; Wang, L.; Bao, J.; Kadota, M.; Oshimura, M.; Dent, S.R.; Issa, J.P.; et al. Epigenetic regulation of ARHI in breast and ovarian cancer cells. Ann. N. Y. Acad. Sci. 2003, 983, 268–277. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Bretz, C.L.; Lee, S. Epigenetic instability of imprinted genes in human cancers. Nucleic Acids Res. 2015, 43, 10689–10699. [Google Scholar] [CrossRef] [Green Version]
- Terry, M.B.; Delgado-Cruzata, L.; Vin-Raviv, N.; Wu, H.C.; Santella, R.M. DNA methylation in white blood cells: Association with risk factors in epidemiologic studies. Epigenetics 2011, 6, 828–837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Houseman, E.A.; Kim, S.; Kelsey, K.T.; Wiencke, J.K. DNA Methylation in Whole Blood: Uses and Challenges. Curr. Environ. Health Rep. 2015, 2, 145–154. [Google Scholar] [CrossRef] [PubMed]
- Harrison, K.; Hoad, G.; Scott, P.; Simpson, L.; Horgan, G.W.; Smyth, E.; Heys, S.D.; Haggarty, P. Breast cancer risk and imprinting methylation in blood. Clin. Epigenetics 2015, 7, 92. [Google Scholar] [CrossRef] [Green Version]
- Pu, W.; Wang, C.; Chen, S.; Zhao, D.; Zhou, Y.; Ma, Y.; Wang, Y.; Li, C.; Huang, Z.; Jin, L.; et al. Targeted bisulfite sequencing identified a panel of DNA methylation-based biomarkers for esophageal squamous cell carcinoma (ESCC). Clin. Epigenetics 2017, 9, 129. [Google Scholar] [CrossRef] [Green Version]
- Untergasser, A.; Cutcutache, I.; Koressaar, T.; Ye, J.; Faircloth, B.C.; Remm, M.; Rozen, S.G. Primer3—New capabilities and interfaces. Nucleic Acids Res. 2012, 40, e115. [Google Scholar] [CrossRef] [Green Version]
- Sterne, J.A.; White, I.R.; Carlin, J.B.; Spratt, M.; Royston, P.; Kenward, M.G.; Wood, A.M.; Carpenter, J.R. Multiple imputation for missing data in epidemiological and clinical research: Potential and pitfalls. BMJ 2009, 338, b2393. [Google Scholar] [CrossRef]
- Martin, T.C.; Yet, I.; Tsai, P.C.; Bell, J.T. coMET: Visualisation of regional epigenome-wide association scan results and DNA co-methylation patterns. BMC Bioinform. 2015, 16, 131. [Google Scholar] [CrossRef] [Green Version]
- Schultz, M.D.; Schmitz, R.J.; Ecker, J.R. ‘Leveling’ the playing field for analyses of single-base resolution DNA methylomes. Trends Genet. 2012, 28, 583–585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bevers, T.B.; Helvie, M.; Bonaccio, E.; Calhoun, K.E.; Daly, M.B.; Farrar, W.B.; Garber, J.E.; Gray, R.; Greenberg, C.C.; Greenup, R.; et al. Breast Cancer Screening and Diagnosis, Version 3.2018, NCCN Clinical Practice Guidelines in Oncology. J. Natl. Compr. Cancer Netw. 2018, 16, 1362–1389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vennin, C.; Spruyt, N.; Robin, Y.M.; Chassat, T.; Le Bourhis, X.; Adriaenssens, E. The long non-coding RNA 91H increases aggressive phenotype of breast cancer cells and up-regulates H19/IGF2 expression through epigenetic modifications. Cancer Lett. 2017, 385, 198–206. [Google Scholar] [CrossRef] [PubMed]
- Scelfo, R.A.; Schwienbacher, C.; Veronese, A.; Gramantieri, L.; Bolondi, L.; Querzoli, P.; Nenci, I.; Calin, G.A.; Angioni, A.; Barbanti-Brodano, G.; et al. Loss of methylation at chromosome 11p15.5 is common in human adult tumors. Oncogene 2002, 21, 2564–2572. [Google Scholar] [CrossRef] [Green Version]
- Collette, J.; Le Bourhis, X.; Adriaenssens, E. Regulation of Human Breast Cancer by the Long Non-Coding RNA H19. Int. J. Mol. Sci. 2017, 18, 2319. [Google Scholar] [CrossRef] [Green Version]
- Ferguson-Smith, A.C.; Surani, M.A. Imprinting and the epigenetic asymmetry between parental genomes. Science 2001, 293, 1086–1089. [Google Scholar] [CrossRef]
- Li, E.; Zhang, Y. DNA methylation in mammals. Cold Spring Harb. Perspect. Biol. 2014, 6, a019133. [Google Scholar] [CrossRef]
- Iacobuzio-Donahue, C.A. Epigenetic changes in cancer. Annu. Rev. Pathol. 2009, 4, 229–249. [Google Scholar] [CrossRef]
- Braakhuis, B.; Tabor, M.; Kummer, J.; Leemans, C.; Brakenhoff, R. A genetic explanation of Slaughter’s concept of field cancerization: Evidence and clinical implications. Cancer Res. 2003, 63, 1727–1730. [Google Scholar]
- Barrow, T.M.; Barault, L.; Ellsworth, R.E.; Harris, H.R.; Binder, A.M.; Valente, A.L.; Shriver, C.D.; Michels, K.B. Aberrant methylation of imprinted genes is associated with negative hormone receptor status in invasive breast cancer. Int. J. Cancer 2015, 137, 537–547. [Google Scholar] [CrossRef] [Green Version]
- Martin, G. Epigenetic drift in aging identical twins. Proc. Natl. Acad. Sci. USA 2005, 102, 10413–10414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Talens, R.; Boomsma, D.; Tobi, E.; Kremer, D.; Jukema, J.; Willemsen, G.; Putter, H.; Slagboom, P.; Heijmans, B. Variation, patterns, and temporal stability of DNA methylation: Considerations for epigenetic epidemiology. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2010, 24, 3135–3144. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristics | Case (%) | Control (%) | p |
|---|---|---|---|
| Number of Participants | 514 | 534 | |
| Age (year) a,b | 52.0 ± 9.35 | 52.1 ± 10.76 | 0.908 |
| <50 | 215 (41.8) | 237 (44.4) | 0.003 |
| 50– | 192 (37.4) | 147 (27.5) | |
| 60– | 89 (17.3) | 122 (22.8) | |
| ≥70 | 18 (3.5) | 28 (5.3) | |
| BMI (kg/m2) a,b | 23.9 ± 3.65 | 23.7 ± 4.10 | 0.367 |
| <18.5 | 15 (2.9) | 27 (5.1) | 0.325 |
| 18.5– | 272 (52.9) | 272 (50.9) | |
| 24– | 134 (26.1) | 133 (24.9) | |
| ≥28 | 93 (18.1) | 102 (19.1) | |
| Ethnicity b | 0.139 | ||
| Han | 502 (97.7) | 513 (96.1) | |
| Other | 12 (2.3) | 21 (3.9) | |
| Location b | 0.440 | ||
| City | 417 (81.1) | 443 (83.0) | |
| Rural area | 97 (18.9) | 91 (17.0) | |
| Family history of other cancers b | <0.001 | ||
| No | 392 (76.3) | 454 (85.0) | |
| Yes | 122 (23.7) | 80 (15.0) | |
| Family history of breast cancer b | <0.001 | ||
| No | 473 (92.0) | 526 (98.5) | |
| Yes | 41 (8.0) | 8 (1.5) |
| CpG_Position | Δβ | p (t-Test) | Univariate Analysis | Multivariate Analysis | ||
|---|---|---|---|---|---|---|
| OR (95% CI) | p | OR (95% CI) a | p | |||
| KCNQ1OT1_2721952 | −0.011 | 0.022 | 0.834 (0.714–0.973) | 0.021 | 0.835 (0.711–0.978) | 0.026 |
| KCNQ1OT1_2721955 | −0.012 | 0.017 | 0.829 (0.709–0.966) | 0.017 | 0.828 (0.705–0.970) | 0.020 |
| KCNQ1OT1_2721961 | −0.012 | 0.019 | 0.831 (0.712–0.969) | 0.019 | 0.828 (0.705–0.971) | 0.021 |
| KCNQ1OT1_2721966 | −0.012 | 0.021 | 0.834 (0.714–0.972) | 0.021 | 0.834 (0.710–0.977) | 0.025 |
| KCNQ1OT1_2721968 | −0.011 | 0.025 | 0.838 (0.717–0.977) | 0.025 | 0.836 (0.712–0.979) | 0.027 |
| KCNQ1OT1_2721978 | −0.012 | 0.015 | 0.825 (0.706–0.962) | 0.015 | 0.823 (0.701–0.965) | 0.017 |
| KCNQ1OT1_2721982 | −0.012 | 0.015 | 0.825 (0.706–0.962) | 0.015 | 0.822 (0.700–0.963) | 0.016 |
| KCNQ1OT1_2721986 | −0.012 | 0.019 | 0.831 (0.711–0.969) | 0.019 | 0.828 (0.705–0.971) | 0.021 |
| KCNQ1OT1_2721992 | −0.012 | 0.014 | 0.824 (0.705–0.961) | 0.014 | 0.820 (0.698–0.961) | 0.015 |
| KCNQ1OT1_2722019 | −0.011 | 0.023 | 0.836 (0.716–0.975) | 0.023 | 0.836 (0.712–0.979) | 0.027 |
| KCNQ1OT1_2722023 | −0.012 | 0.013 | 0.822 (0.704–0.959) | 0.013 | 0.820 (0.698–0.961) | 0.015 |
| KCNQ1OT1_2722036 | −0.012 | 0.017 | 0.829 (0.710–0.967) | 0.017 | 0.829 (0.706–0.971) | 0.021 |
| KCNQ1OT1_2722038 | −0.012 | 0.015 | 0.825 (0.706–0.962) | 0.015 | 0.823 (0.701–0.964) | 0.017 |
| KCNQ1OT1_2722045 | −0.012 | 0.018 | 0.829 (0.710–0.967) | 0.018 | 0.828 (0.705–0.970) | 0.020 |
| KCNQ1OT1_2722062 | −0.011 | 0.024 | 0.837 (0.717–0.976) | 0.024 | 0.835 (0.711–0.979) | 0.027 |
| KCNQ1OT1_2722071 | −0.011 | 0.020 | 0.833 (0.713–0.971) | 0.020 | 0.832 (0.708–0.975) | 0.023 |
| KCNQ1OT1_2722073 | −0.012 | 0.015 | 0.826 (0.707–0.963) | 0.015 | 0.823 (0.701–0.965) | 0.017 |
| KCNQ1OT1_2722076 | −0.012 | 0.016 | 0.828 (0.708–0.965) | 0.016 | 0.824 (0.701–0.965) | 0.017 |
| KCNQ1OT1_2722082 | −0.012 | 0.019 | 0.831 (0.711–0.969) | 0.019 | 0.829 (0.705–0.971) | 0.021 |
| KCNQ1OT1_2722084 | −0.011 | 0.024 | 0.837 (0.717–0.976) | 0.024 | 0.834 (0.710–0.977) | 0.025 |
| KCNQ1OT1_2722086 | −0.011 | 0.025 | 0.838 (0.718–0.977) | 0.025 | 0.833 (0.709–0.976) | 0.025 |
| KCNQ1OT1_2722119 | −0.031 | 0.009 | 0.814 (0.693–0.951) | 0.010 | 0.834 (0.707–0.979) | 0.028 |
| Subgroup | KCNQ1OT1 Region 2 | Univariate Analysis | Multivariate Analysis | ||
|---|---|---|---|---|---|
| OR (95% CI) | p | OR (95% CI) a | p | ||
| Age (year) | |||||
| <50 | <0.474 | 1.000 | <0.001 | 1.000 | <0.001 |
| ≥0.474 | 0.428 (0.265–0.687) | 0.432 (0.263–0.703) | |||
| ≥50 | <0.474 | 1.000 | 0.122 | 1.000 | 0.172 |
| ≥0.474 | 0.701 (0.447–1.099) | 0.728 (0.460–1.150) | |||
| Molecular subtype | |||||
| Luminal A | <0.474 | 1.000 | <0.001 | 1.000 | <0.001 |
| ≥0.474 | 0.449 (0.282–0.702) | 0.446 (0.276–0.706) | |||
| Luminal B | <0.474 | 1.000 | 0.085 | 1.000 | 0.107 |
| ≥0.474 | 0.696 (0.458–1.048) | 0.698 (0.450–1.080) | |||
| HER-2 | <0.474 | 1.000 | 0.061 | 1.000 | 0.042 |
| ≥0.474 | 0.484 (0.216–1.004) | 0.424 (0.176–0.936) | |||
| Basal-like | <0.474 | 1.000 | 0.315 | 1.000 | 0.420 |
| ≥0.474 | 0.653 (0.272–1.465) | 0.705 (0.289–1.614) | |||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fu, J.; Zhang, L.; Li, D.; Tian, T.; Wang, X.; Sun, H.; Ge, A.; Liu, Y.; Zhang, X.; Huang, H.; et al. DNA Methylation of Imprinted Genes KCNQ1, KCNQ1OT1, and PHLDA2 in Peripheral Blood Is Associated with the Risk of Breast Cancer. Cancers 2022, 14, 2652. https://doi.org/10.3390/cancers14112652
Fu J, Zhang L, Li D, Tian T, Wang X, Sun H, Ge A, Liu Y, Zhang X, Huang H, et al. DNA Methylation of Imprinted Genes KCNQ1, KCNQ1OT1, and PHLDA2 in Peripheral Blood Is Associated with the Risk of Breast Cancer. Cancers. 2022; 14(11):2652. https://doi.org/10.3390/cancers14112652
Chicago/Turabian StyleFu, Jinming, Lei Zhang, Dapeng Li, Tian Tian, Xuan Wang, Hongru Sun, Anqi Ge, Yupeng Liu, Xianyu Zhang, Hao Huang, and et al. 2022. "DNA Methylation of Imprinted Genes KCNQ1, KCNQ1OT1, and PHLDA2 in Peripheral Blood Is Associated with the Risk of Breast Cancer" Cancers 14, no. 11: 2652. https://doi.org/10.3390/cancers14112652