
Homologous Recombination Deficiency Scar: Mutations and Beyond—Implications for Precision Oncology

 ,
,  , and
, and
Abstract
:Simple Summary
Abstract
1. Introduction
2. The Homologous Recombination Repair Pathway
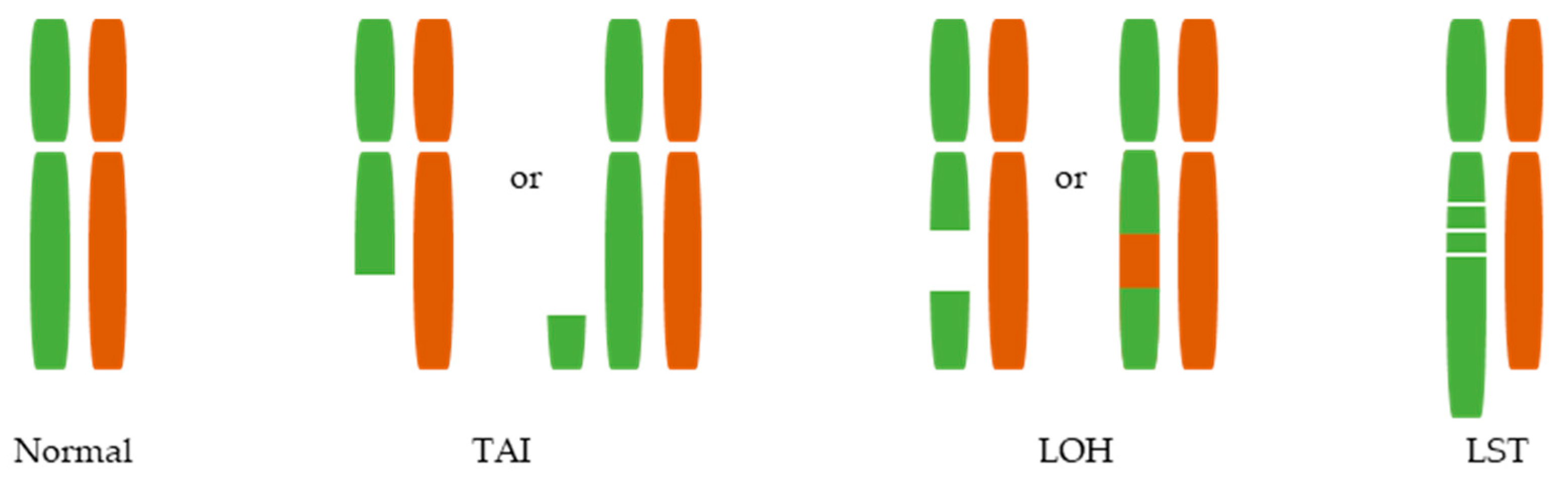
3. Large-Scale Genomic Aberrations Associated with HRD
4. Mutational Signatures of HRD
5. Integrative Models to Predict HRD
6. Limitations of Detecting Mutational Scars
7. HRD Scar in the Clinic: Implications for Precision Oncology
7.1. PARPis
7.2. Platinum-Based and Other DSB-Inducing Agents
7.3. Immunotherapies
8. Conclusions and Future Directions
Author Contributions
Funding
Conflicts of Interest
References
- Lord, C.J.; Ashworth, A. The DNA damage response and cancer therapy. Nature 2012, 481, 287–294. [Google Scholar] [CrossRef] [PubMed]
- Heeke, A.L.; Pishvaian, M.J.; Lynce, F.; Xiu, J.; Brody, J.R.; Chen, W.-J.; Baker, T.M.; Marshall, J.L.; Isaacs, C. Prevalence of Homologous Recombination–Related Gene Mutations Across Multiple Cancer Types. JCO Precis. Oncol. 2018, 2018, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Rose, M.; Burgess, J.T.; O’Byrne, K.; Richard, D.J.; Bolderson, E. PARP Inhibitors: Clinical Relevance, Mechanisms of Action and Tumor Resistance. Front. Cell Dev. Biol. 2020, 8, 564601. [Google Scholar] [CrossRef] [PubMed]
- Telli, M.L.; Timms, K.M.; Reid, J.; Hennessy, B.; Mills, G.B.; Jensen, K.C.; Szallasi, Z.; Barry, W.T.; Winer, E.P.; Tung, N.M.; et al. Homologous Recombination Deficiency (HRD) Score Predicts Response to Platinum-Containing Neoadjuvant Chemotherapy in Patients with Triple-Negative Breast Cancer. Clin. Cancer Res. 2016, 22, 3764–3773. [Google Scholar] [CrossRef] [PubMed]
- Hoppe, M.M.; Sundar, R.; Tan, D.S.P.; Jeyasekharan, A.D. Biomarkers for Homologous Recombination Deficiency in Cancer. JNCI J. Natl. Cancer Inst. 2018, 110, 704–713. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.; Martens, J.W.M.; Van Hoeck, A.; Cuppen, E. Pan-cancer landscape of homologous recombination deficiency. Nat. Commun. 2020, 11, 5584. [Google Scholar] [CrossRef]
- Lord, C.J.; Ashworth, A. BRCAness revisited. Nat. Rev. Cancer 2016, 16, 110–120. [Google Scholar] [CrossRef]
- Abkevich, V.; Timms, K.M.; Hennessy, B.T.; Potter, J.; Carey, M.S.; Meyer, L.A.; Smith-McCune, K.; Broaddus, R.; Lu, K.H.; Chen, J.; et al. Patterns of genomic loss of heterozygosity predict homologous recombination repair defects in epithelial ovarian cancer. Br. J. Cancer 2012, 107, 1776–1782. [Google Scholar] [CrossRef]
- Popova, T.; Manié, E.; Rieunier, G.; Caux-Moncoutier, V.; Tirapo, C.; Dubois, T.; Delattre, O.; Sigal-Zafrani, B.; Bollet, M.; Longy, M.; et al. Ploidy and Large-Scale Genomic Instability Consistently Identify Basal-like Breast Carcinomas with BRCA1/2 Inactivation. Cancer Res. 2012, 72, 5454–5462. [Google Scholar] [CrossRef]
- Grigorova, M.; Staines, J.; Ozdag, H.; Caldas, C.; Edwards, P. Possible causes of chromosome instability: Comparison of chromosomal abnormalities in cancer cell lines with mutations in BRCA1, BRCA2, CHK2 and BUB1. Cytogenet. Genome Res. 2004, 104, 333–340. [Google Scholar] [CrossRef]
- Naim, V.; Rosselli, F. The FANC pathway and BLM collaborate during mitosis to prevent micro-nucleation and chromosome abnormalities. Nat. Cell Biol. 2009, 11, 761–768. [Google Scholar] [CrossRef] [PubMed]
- Nik-Zainal, S.; Davies, H.; Staaf, J.; Ramakrishna, M.; Glodzik, D.; Zou, X.; Martincorena, I.; Alexandrov, L.B.; Martin, S.; Wedge, D.C.; et al. Landscape of somatic mutations in 560 breast cancer whole-genome sequences. Nature 2016, 534, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Scully, R.; Panday, A.; Elango, R.; Willis, N.A. DNA double-strand break repair-pathway choice in somatic mammalian cells. Nat. Rev. Mol. Cell Biol. 2019, 20, 698–714. [Google Scholar] [CrossRef]
- Chang, H.H.Y.; Pannunzio, N.R.; Adachi, N.; Lieber, M.R. Non-homologous DNA end joining and alternative pathways to double-strand break repair. Nat. Rev. Mol. Cell Biol. 2017, 18, 495–506. [Google Scholar] [CrossRef] [PubMed]
- Takata, M.; Sasaki, M.S.; Sonoda, E.; Morrison, C.; Hashimoto, M.; Utsumi, H.; Yamaguchi-Iwai, Y.; Shinohara, A.; Takeda, S. Homologous recombination and non-homologous end-joining pathways of DNA double-strand break repair have overlapping roles in the maintenance of chromosomal integrity in vertebrate cells. EMBO J. 1998, 17, 5497–5508. [Google Scholar] [CrossRef]
- Elbakry, A.; Löbrich, M. Homologous Recombination Subpathways: A Tangle to Resolve. Front. Genet. 2021, 12. [Google Scholar] [CrossRef]
- Sun, Y.; McCorvie, T.J.; Yates, L.A.; Zhang, X. Structural basis of homologous recombination. Exp. 2020, 77, 3–18. [Google Scholar] [CrossRef]
- Rossi, M.J.; DiDomenico, S.F.; Patel, M.; Mazin, A.V. RAD52: Paradigm of Synthetic Lethality and New Developments. Front. Genet. 2021, 12. [Google Scholar] [CrossRef]
- O’Reilly, E.M.; Lee, J.W.; Zalupski, M.; Capanu, M.; Park, J.; Golan, T.; Tahover, E.; Lowery, M.A.; Chou, J.F.; Sahai, V.; et al. Randomized, Multicenter, Phase II Trial of Gemcitabine and Cisplatin with or without Veliparib in Patients with Pancreas Adenocarcinoma and a Germline BRCA/PALB2 Mutation. J. Clin. Oncol. 2020, 38, 1378–1388. [Google Scholar] [CrossRef]
- Tung, N.M.; Robson, M.E.; Ventz, S.; Santa-Maria, C.A.; Nanda, R.; Marcom, P.K.; Shah, P.D.; Ballinger, T.J.; Yang, E.S.; Vinayak, S.; et al. TBCRC 048: Phase II Study of Olaparib for Metastatic Breast Cancer and Mutations in Homologous Recombination-Related Genes. J. Clin. Oncol. 2020, 38, 4274–4282. [Google Scholar] [CrossRef]
- Marshall, C.H.; Sokolova, A.O.; McNatty, A.L.; Cheng, H.H.; Eisenberger, M.A.; Bryce, A.H.; Schweizer, M.T.; Antonarakis, E.S. Differential Response to Olaparib Treatment Among Men with Metastatic Castration-resistant Prostate Cancer Harboring BRCA1 or BRCA2 Versus ATM Mutations. Eur. Urol. 2019, 76, 452–458. [Google Scholar] [CrossRef] [PubMed]
- Shibata, A.; Jeggo, P.A. ATM’s Role in the Repair of DNA Double-Strand Breaks. Genes 2021, 12, 1370. [Google Scholar] [CrossRef] [PubMed]
- Rothblum-Oviatt, C.; Wright, J.; Lefton-Greif, M.A.; McGrath-Morrow, S.A.; Crawford, T.O.; Lederman, H.M. Ataxia telangiectasia: A review. Orphanet J. Rare Dis. 2016, 11, 159. [Google Scholar] [CrossRef] [PubMed]
- Croteau, D.L.; Popuri, V.; Opresko, P.L.; Bohr, V.A. Human RecQ Helicases in DNA Repair, Recombination, and Replication. Annu. Rev. Biochem. 2014, 83, 519–552. [Google Scholar] [CrossRef] [PubMed]
- Yankiwski, V.; Noonan, J.P.; Neff, N.F. The C-terminal domain of the Bloom syndrome DNA helicase is essential for genomic stability. BMC Cell Biol. 2001, 2, 11. [Google Scholar] [CrossRef]
- Kuhn, E.M.; Therman, E. Chromosome breakage and rejoining of sister chromatids in Bloom’s syndrome. Chromosoma 1979, 73, 275–286. [Google Scholar] [CrossRef] [PubMed]
- Saintigny, Y.; Makienko, K.; Swanson, C.; Emond, M.J.; Monnat, R.J., Jr. Homologous Recombination Resolution Defect in Werner Syndrome. Mol. Cell. Biol. 2002, 22, 6971–6978. [Google Scholar] [CrossRef]
- Sharma, R.; Lewis, S.; Wlodarski, M.W. DNA Repair Syndromes and Cancer: Insights Into Genetics and Phenotype Patterns. Front. Pediatr. 2020, 8, 570084. [Google Scholar] [CrossRef]
- Sztupinszki, Z.; Diossy, M.; Krzystanek, M.; Reiniger, L.; Csabai, I.; Favero, F.; Birkbak, N.; Eklund, A.C.; Syed, A.; Szallasi, Z. Migrating the SNP array-based homologous recombination deficiency measures to next generation sequencing data of breast cancer. NPJ Breast Cancer 2018, 4, 16. [Google Scholar] [CrossRef]
- Birkbak, N.J.; Wang, Z.C.; Kim, J.-Y.; Eklund, A.C.; Li, Q.; Tian, R.; Bowman-Colin, C.; Li, Y.; Greene-Colozzi, A.; Iglehart, J.D.; et al. Telomeric Allelic Imbalance Indicates Defective DNA Repair and Sensitivity to DNA-Damaging Agents. Cancer Discov. 2012, 2, 366–375. [Google Scholar] [CrossRef] [Green Version]
- Nichols, C.; Gibson, W.J.; Brown, M.S.; Kosmicki, J.A.; Busanovich, J.P.; Wei, H.; Urbanski, L.M.; Curimjee, N.; Berger, A.C.; Gao, G.F.; et al. Loss of heterozygosity of essential genes represents a widespread class of potential cancer vulnerabilities. Nat. Commun. 2020, 11, 2517. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, M.R.; Bernstein, K.A. RAD-ical New Insights into RAD51 Regulation. Genes 2018, 9, 629. [Google Scholar] [CrossRef] [PubMed]
- Swisher, E.M.; Lin, K.K.; Oza, A.M.; Scott, C.L.; Giordano, H.; Sun, J.; Konecny, G.E.; Coleman, R.L.; Tinker, A.V.; O’Malley, D.M.; et al. Rucaparib in relapsed, platinum-sensitive high-grade ovarian carcinoma (ARIEL2 Part 1): An international, multicentre, open-label, phase 2 trial. Lancet Oncol. 2017, 18, 75–87. [Google Scholar] [CrossRef]
- Manié, E.; Popova, T.; Battistella, A.; Tarabeux, J.; Caux-Moncoutier, V.; Golmard, L.; Smith, N.K.; Mueller, C.R.; Mariani, O.; Sigal-Zafrani, B.; et al. Genomic hallmarks of homologous recombination deficiency in invasive breast carcinomas. Int. J. Cancer 2016, 138, 891–900. [Google Scholar] [CrossRef]
- Watkins, J.A.; Irshad, S.; Grigoriadis, A.; Tutt, A.N.J. Genomic scars as biomarkers of homologous recombination deficiency and drug response in breast and ovarian cancers. Breast Cancer Res. 2014, 16, 211. [Google Scholar] [CrossRef]
- Marquard, A.M.; Eklund, A.C.; Joshi, T.; Krzystanek, M.; Favero, F.; Wang, Z.C.; Richardson, A.L.; Silver, D.P.; Szallasi, Z.; Birkbak, N.J. Pan-cancer analysis of genomic scar signatures associated with homologous recombination deficiency suggests novel indications for existing cancer drugs. Biomark. Res. 2015, 3, 9. [Google Scholar] [CrossRef]
- Timms, K.M.; Abkevich, V.; Hughes, E.; Neff, C.; Reid, J.; Morris, B.; Kalva, S.; Potter, J.; Tran, T.V.; Chen, J.; et al. Association of BRCA1/2defects with genomic scores predictive of DNA damage repair deficiency among breast cancer subtypes. Breast Cancer Res. 2014, 16, 475. [Google Scholar] [CrossRef]
- Lotan, T.L.; Kaur, H.B.; Salles, D.C.; Murali, S.; Schaeffer, E.M.; Lanchbury, J.S.; Isaacs, W.B.; Brown, R.; Richardson, A.L.; Cussenot, O.; et al. Homologous recombination deficiency (HRD) score in germline BRCA2- versus ATM-altered prostate cancer. Mod. Pathol. 2021, 34, 1185–1193. [Google Scholar] [CrossRef]
- How, J.; Jazaeri, A.; Fellman, B.; Daniels, M.; Penn, S.; Solimeno, C.; Yuan, Y.; Schmeler, K.; Lanchbury, J.; Timms, K.; et al. Modification of Homologous Recombination Deficiency Score Threshold and Association with Long-Term Survival in Epithelial Ovarian Cancer. Cancers 2021, 13, 946. [Google Scholar] [CrossRef]
- Takaya, H.; Nakai, H.; Takamatsu, S.; Mandai, M.; Matsumura, N. Homologous recombination deficiency status-based classification of high-grade serous ovarian carcinoma. Sci. Rep. 2020, 10, 2757. [Google Scholar] [CrossRef] [Green Version]
- Ngoi, N.; Tan, D. The role of homologous recombination deficiency testing in ovarian cancer and its clinical implications: Do we need it? ESMO Open 2021, 6, 100144. [Google Scholar] [CrossRef] [PubMed]
- Forment, J.; Kaidi, A.; Jackson, S.P. Chromothripsis and cancer: Causes and consequences of chromosome shattering. Nat. Cancer 2012, 12, 663–670. [Google Scholar] [CrossRef] [PubMed]
- Ratnaparkhe, M.; Hlevnjak, M.; Kolb, T.; Jauch, A.; Maass, K.K.; Devens, F.; Rode, A.; Hovestadt, V.; Korshunov, A.; Pastorczak, A.; et al. Genomic profiling of Acute lymphoblastic leukemia in ataxia telangiectasia patients reveals tight link between ATM mutations and chromothripsis. Leukemia 2017, 31, 2048–2056. [Google Scholar] [CrossRef] [PubMed]
- Simovic, M.; Ernst, A. Chromothripsis, DNA repair and checkpoints defects. Semin. Cell Dev. Biol. 2022, 123, 110–114. [Google Scholar] [CrossRef]
- Degasperi, A.; Zou, X.; Amarante, T.D.; Martinez-Martinez, A.; Koh, G.C.C.; Dias, J.M.L.; Heskin, L.; Chmelova, L.; Rinaldi, G.; Wang, V.Y.W.; et al. Substitution mutational signatures in whole-genome–sequenced cancers in the UK population. Science 2022, 376, abl9283. [Google Scholar] [CrossRef]
- Steele, C.D.; Abbasi, A.; Islam, S.M.A.; Bowes, A.L.; Khandekar, A.; Haase, K.; Hames-Fathi, S.; Ajayi, D.; Verfaillie, A.; Dhami, P.; et al. Signatures of copy number alterations in human cancer. Nature 2022, 606, 984–991. [Google Scholar] [CrossRef]
- Koh, G.; Degasperi, A.; Zou, X.; Momen, S.; Nik-Zainal, S. Mutational signatures: Emerging concepts, caveats and clinical applications. Nat. Cancer 2021, 21, 619–637. [Google Scholar] [CrossRef]
- Vanderstichele, A.; Busschaert, P.; Olbrecht, S.; Lambrechts, D.; Vergote, I. Genomic signatures as predictive biomarkers of homologous recombination deficiency in ovarian cancer. Eur. J. Cancer 2017, 86, 5–14. [Google Scholar] [CrossRef]
- Alexandrov, L.B.; Kim, J.; Haradhvala, N.J.; Huang, M.N.; Ng, A.W.T.; Wu, Y.; Boot, A.; Covington, K.R.; Gordenin, D.A.; Bergstrom, E.N.; et al. The repertoire of mutational signatures in human cancer. Nature 2020, 578, 94–101. [Google Scholar] [CrossRef]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.J.R.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Børresen-Dale, A.-L.; et al. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef] [Green Version]
- Waddell, N.; Pajic, M.; Patch, A.-M.; Chang, D.K.; Kassahn, K.S.; Bailey, P.; Johns, A.L.; Miller, D.; Nones, K.; Quek, K.; et al. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature 2015, 518, 495–501. [Google Scholar] [CrossRef] [PubMed]
- Bailey, P.; Chang, D.K.; Nones, K.; Johns, A.L.; Patch, A.-M.; Gingras, M.-C.; Miller, D.K.; Christ, A.N.; Bruxner, T.J.C.; Quinn, M.C.; et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 2016, 531, 47–52. [Google Scholar] [CrossRef]
- Patch, A.-M.; Christie, E.L.; Etemadmoghadam, D.; Garsed, D.W.; George, J.; Fereday, S.; Nones, K.; Cowin, P.; Alsop, K.; Bailey, P.J.; et al. Whole–genome characterization of chemoresistant ovarian cancer. Nature 2015, 521, 489–494. [Google Scholar] [CrossRef] [PubMed]
- Alexandrov, L.B.; Nik-Zainal, S.; Siu, H.C.; Leung, S.Y.; Stratton, M.R. A mutational signature in gastric cancer suggests therapeutic strategies. Nat. Commun. 2015, 6, 8683. [Google Scholar] [CrossRef] [PubMed]
- Polak, P.; Kim, J.; Braunstein, L.Z.; Karlic, R.; Haradhavala, N.J.; Tiao, G.; Rosebrock, D.; Livitz, D.; Kübler, K.; Mouw, K.W.; et al. A mutational signature reveals alterations underlying deficient homologous recombination repair in breast cancer. Nat. Genet. 2017, 49, 1476–1486. [Google Scholar] [CrossRef] [PubMed]
- Li, A.; kConFab Investigators; Geyer, F.C.; Blecua, P.; Lee, J.Y.; Selenica, P.; Brown, D.; Pareja, F.; Lee, S.S.K.; Kumar, R.; et al. Homologous recombination DNA repair defects in PALB2-associated breast cancers. NPJ Breast Cancer 2019, 5, 23. [Google Scholar] [CrossRef]
- Póti, Á.; Gyergyák, H.; Németh, E.; Rusz, O.; Tóth, S.; Kovácsházi, C.; Chen, D.; Szikriszt, B.; Spisák, S.; Takeda, S.; et al. Correlation of homologous recombination deficiency induced mutational signatures with sensitivity to PARP inhibitors and cytotoxic agents. Genome Biol. 2019, 20, 240. [Google Scholar] [CrossRef]
- Stok, C.; Kok, Y.P.; Tempel, N.V.D.; Vugt, M.A.T.M.V. Shaping the BRCAness mutational landscape by alternative double-strand break repair, replication stress and mitotic aberrancies. Nucleic Acids Res. 2021, 49, 4239–4257. [Google Scholar] [CrossRef]
- Singh, V.; Rastogi, A.; Hu, X.; Wang, Y.; De, S. Mutational signature SBS8 predominantly arises due to late replication errors in cancer. Commun. Biol. 2020, 3, 421. [Google Scholar] [CrossRef]
- Nik-Zainal, S.; Morganella, S. Mutational Signatures in Breast Cancer: The Problem at the DNA Level. Clin. Cancer Res. 2017, 23, 2617–2629. [Google Scholar] [CrossRef] [Green Version]
- Nik-Zainal, S.; Alexandrov, L.B.; Wedge, D.C.; Van Loo, P.; Greenman, C.D.; Raine, K.; Jones, D.; Hinton, J.; Marshall, J.; Stebbings, L.A.; et al. Mutational Processes Molding the Genomes of 21 Breast Cancers. Cell 2012, 149, 979–993. [Google Scholar] [CrossRef] [PubMed]
- Davies, H.; Glodzik, D.; Morganella, S.; Yates, L.R.; Staaf, J.; Zou, X.; Ramakrishna, M.; Martin, S.; Boyault, S.; Sieuwerts, A.M.; et al. HRDetect is a predictor of BRCA1 and BRCA2 deficiency based on mutational signatures. Nat. Med. 2017, 23, 517–525. [Google Scholar] [CrossRef] [PubMed]
- Gulhan, D.C.; Lee, J.J.-K.; Melloni, G.E.M.; Cortés-Ciriano, I.; Park, P.J. Detecting the mutational signature of homologous recombination deficiency in clinical samples. Nat. Genet. 2019, 51, 912–919. [Google Scholar] [CrossRef] [PubMed]
- Degasperi, A.; Amarante, T.D.; Czarnecki, J.; Shooter, S.; Zou, X.; Glodzik, D.; Morganella, S.; Nanda, A.S.; Badja, C.; Koh, G.; et al. A practical framework and online tool for mutational signature analyses show intertissue variation and driver dependencies. Nat. Cancer 2020, 1, 249–263. [Google Scholar] [CrossRef] [PubMed]
- Coleman, R.L.; Oza, A.M.; Lorusso, D.; Aghajanian, C.; Oaknin, A.; Dean, A.; Colombo, N.; Weberpals, J.I.; Clamp, A.; Scambia, G.; et al. Rucaparib maintenance treatment for recurrent ovarian carcinoma after response to platinum therapy (ARIEL3): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2017, 390, 1949–1961. [Google Scholar] [CrossRef]
- Timms, K.M.; Mills, G.B.; Perry, M.; Gutin, A.; Lanchbury, J.; Brown, R. Comparison of genomic instability test scores used for predicting PARP activity in ovarian cancer. J. Clin. Oncol. 2020, 38, 1586. [Google Scholar] [CrossRef]
- Golan, T.; O’Kane, G.M.; Denroche, R.E.; Raitses-Gurevich, M.; Grant, R.C.; Holter, S.; Wang, Y.; Zhang, A.; Jang, G.H.; Stossel, C.; et al. Genomic Features and Classification of Homologous Recombination Deficient Pancreatic Ductal Adenocarcinoma. Gastroenterology 2021, 160, 2119–2132.e9. [Google Scholar] [CrossRef]
- Chopra, N.; Tovey, H.; Pearson, A.; Cutts, R.; Toms, C.; Proszek, P.; Hubank, M.; Dowsett, M.; Dodson, A.; Daley, F.; et al. Homologous recombination DNA repair deficiency and PARP inhibition activity in primary triple negative breast cancer. Nat. Commun. 2020, 11, 2662. [Google Scholar] [CrossRef]
- Sztupinszki, Z.; Diossy, M.; Borcsok, J.; Prosz, A.; Cornelius, N.; Kjeldsen, M.K.; Mirza, M.R.; Szallasi, Z. Comparative Assessment of Diagnostic Homologous Recombination Deficiency–Associated Mutational Signatures in Ovarian Cancer. Clin. Cancer Res. 2021, 27, 5681–5687. [Google Scholar] [CrossRef]
- Priestley, P.; Baber, J.; Lolkema, M.P.; Steeghs, N.; de Bruijn, E.; Shale, C.; Duyvesteyn, K.; Haidari, S.; van Hoeck, A.; Onstenk, W.; et al. Pan-cancer whole-genome analyses of metastatic solid tumours. Nature 2019, 575, 210–216. [Google Scholar] [CrossRef] [Green Version]
- Maura, F.; Degasperi, A.; Nadeu, F.; Leongamornlert, D.; Davies, H.; Moore, L.; Royo, R.; Ziccheddu, B.; Puente, X.S.; Avet-Loiseau, H.; et al. A practical guide for mutational signature analysis in hematological malignancies. Nat. Commun. 2019, 10, 2969. [Google Scholar] [CrossRef]
- Sakai, W.; Swisher, E.M.; Karlan, B.Y.; Agarwal, M.K.; Higgins, J.; Friedman, C.; Villegas, E.; Jacquemont, C.; Farrugia, D.J.; Couch, F.J.; et al. Secondary mutations as a mechanism of cisplatin resistance in BRCA2-mutated cancers. Nature 2008, 451, 1116–1120. [Google Scholar] [CrossRef] [PubMed]
- Kondrashova, O.; Nguyen, M.; Shield-Artin, K.; Tinker, A.V.; Teng, N.N.H.; Harrell, M.I.; Kuiper, M.J.; Ho, G.Y.; Barker, H.; Jasin, M.; et al. Secondary Somatic Mutations Restoring RAD51C and RAD51D Associated with Acquired Resistance to the PARP Inhibitor Rucaparib in High-Grade Ovarian Carcinoma. Cancer Discov. 2017, 7, 984–998. [Google Scholar] [CrossRef] [PubMed]
- Goodall, J.; Mateo, J.; Yuan, W.; Mossop, H.; Porta, N.; Miranda, S.; Perez-Lopez, R.; Dolling, D.; Robinson, D.R.; Sandhu, S.; et al. Circulating Cell-Free DNA to Guide Prostate Cancer Treatment with PARP Inhibition. Cancer Discov. 2017, 7, 1006–1017. [Google Scholar] [CrossRef] [PubMed]
- Wagener-Ryczek, S.; Merkelbach-Bruse, S.; Siemanowski, J. Biomarkers for Homologous Recombination Deficiency in Cancer. J. Pers. Med. 2021, 11, 612. [Google Scholar] [CrossRef] [PubMed]
- Guffanti, F.; Alvisi, M.F.; Anastasia, A.; Ricci, F.; Chiappa, M.; Llop-Guevara, A.; Serra, V.; Fruscio, R.; Degasperi, A.; Nik-Zainal, S.; et al. Basal expression of RAD51 foci predicts olaparib response in patient-derived ovarian cancer xenografts. Br. J. Cancer 2022, 126, 120–128. [Google Scholar] [CrossRef]
- Cruz, C.; Castroviejo-Bermejo, M.; Gutiérrez-Enríquez, S.; Llop-Guevara, A.; Ibrahim, Y.; Gris-Oliver, A.; Bonache, S.; Morancho, B.; Bruna, A.; Rueda, O.; et al. RAD51 foci as a functional biomarker of homologous recombination repair and PARP inhibitor resistance in germline BRCA-mutated breast cancer. Ann. Oncol. 2018, 29, 1203–1210. [Google Scholar] [CrossRef]
- Birkelbach, M.; Ferraiolo, N.; Gheorghiu, L.; Pfäffle, H.N.; Daly, B.; Ebright, M.I.; Spencer, C.; O’Hara, C.; Whetstine, J.R.; Benes, C.H.; et al. Detection of Impaired Homologous Recombination Repair in NSCLC Cells and Tissues. J. Thorac. Oncol. 2013, 8, 279–286. [Google Scholar] [CrossRef]
- Meijer, T.G.; Nguyen, L.; Van Hoeck, A.; Sieuwerts, A.M.; Verkaik, N.S.; Ladan, M.M.; Ruigrok-Ritstier, K.; van Deurzen, C.H.M.; van de Werken, H.J.G.; Lips, E.H.; et al. Functional RECAP (REpair CAPacity) assay identifies homologous recombination deficiency undetected by DNA-based BRCAness tests. Oncogene 2022, 41, 3498–3506. [Google Scholar] [CrossRef]
- Verhagen, C.V.; Vossen, D.M.; Borgmann, K.; Hageman, F.; Grénman, R.; Verwijs-Janssen, M.; Mout, L.; Kluin, R.J.; Nieuwland, M.; Severson, T.M.; et al. Fanconi anemia and homologous recombination gene variants are associated with functional DNA repair defects in vitro and poor outcome in patients with advanced head and neck squamous cell carcinoma. Oncotarget 2018, 9, 18198–18213. [Google Scholar] [CrossRef] [Green Version]
- Helleday, T. The underlying mechanism for the PARP and BRCA synthetic lethality: Clearing up the misunderstandings. Mol. Oncol. 2011, 5, 387–393. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Aoyagi-Scharber, M.; Wang, B. Trapping Poly(ADP-Ribose) Polymerase. J. Pharmacol. Exp. Ther. 2015, 353, 446–457. [Google Scholar] [CrossRef] [PubMed]
- Tomao, F.; Bardhi, E.; Di Pinto, A.; Sassu, C.M.; Biagioli, E.; Petrella, M.C.; Palaia, I.; Muzii, L.; Colombo, N.; Panici, P.B. Parp inhibitors as maintenance treatment in platinum sensitive recurrent ovarian cancer: An updated meta-analysis of randomized clinical trials according to BRCA mutational status. Cancer Treat. Rev. 2019, 80, 101909. [Google Scholar] [CrossRef]
- Chen, S.-H.; Chang, J.-Y. New Insights into Mechanisms of Cisplatin Resistance: From Tumor Cell to Microenvironment. Int. J. Mol. Sci. 2019, 20, 4136. [Google Scholar] [CrossRef] [PubMed]
- Patsouris, A.; Diop, K.; Tredan, O.; Nenciu, D.; Gonçalves, A.; Arnedos, M.; Sablin, M.-P.; Jézéquel, P.; Jimenez, M.; Droin, N.; et al. Rucaparib in patients presenting a metastatic breast cancer with homologous recombination deficiency, without germline BRCA1/2 mutation. Eur. J. Cancer 2021, 159, 283–295. [Google Scholar] [CrossRef] [PubMed]
- Ray-Coquard, I.; Pautier, P.; Pignata, S.; Pérol, D.; González-Martín, A.; Berger, R.; Fujiwara, K.; Vergote, I.; Colombo, N.; Mäenpää, J.; et al. Olaparib plus Bevacizumab as First-Line Maintenance in Ovarian Cancer. N. Engl. J. Med. 2019, 381, 2416–2428. [Google Scholar] [CrossRef]
- Moore, K.N.; Secord, A.A.; Geller, M.A.; Miller, D.S.; Cloven, N.; Fleming, G.F.; Wahner Hendrickson, A.E.; Azodi, M.; DiSilvestro, P.; Oza, A.M.; et al. Niraparib monotherapy for late-line treatment of ovarian cancer (QUADRA): A multicentre, open-label, single-arm, phase 2 trial. Lancet Oncol. 2019, 20, 636–648. [Google Scholar] [CrossRef]
- Byrski, T.; Huzarski, T.; Dent, R.; Gronwald, J.; Zuziak, D.; Cybulski, C.; Kladny, J.; Gorski, B.; Lubinski, J.; Narod, S.A. Response to neoadjuvant therapy with cisplatin in BRCA1-positive breast cancer patients. Breast Cancer Res. Treat. 2009, 115, 359–363. [Google Scholar] [CrossRef]
- Rabik, C.A.; Dolan, M.E. Molecular mechanisms of resistance and toxicity associated with platinating agents. Cancer Treat. Rev. 2007, 33, 9–23. [Google Scholar] [CrossRef]
- Basourakos, S.P.; Li, L.; Aparicio, A.M.; Corn, P.G.; Kim, J.; Thompson, T.C. Combination Platinum-based and DNA Damage Response-targeting Cancer Therapy: Evolution and Future Directions. Curr. Med. Chem. 2017, 24, 1586–1606. [Google Scholar] [CrossRef]
- Bhattacharyya, A.; Ear, U.S.; Koller, B.H.; Weichselbaum, R.R.; Bishop, D.K. The Breast Cancer Susceptibility Gene BRCA1 Is Required for Subnuclear Assembly of Rad51 and Survival following Treatment with the DNA Cross-linking Agent Cisplatin. J. Biol. Chem. 2000, 275, 23899–23903. [Google Scholar] [CrossRef] [PubMed]
- Alli, E.; Sharma, V.B.; Hartman, A.-R.; Lin, P.S.; McPherson, L.; Ford, J.M. Enhanced sensitivity to cisplatin and gemcitabine in Brca1-deficient murine mammary epithelial cells. BMC Pharmacol. 2011, 11, 7. [Google Scholar] [CrossRef] [PubMed]
- Gronwald, J.; Byrski, T.; Lubinski, J.; A Narod, S. Cisplatin in breast cancer treatment in BRCA1 carriers. Hered. Cancer Clin. Pract. 2012, 10, A17. [Google Scholar] [CrossRef]
- Park, W.; Chen, J.; Chou, J.F.; Varghese, A.M.; Yu, K.H.; Wong, W.; Capanu, M.; Balachandran, V.; McIntyre, C.A.; El Dika, I.; et al. Genomic Methods Identify Homologous Recombination Deficiency in Pancreas Adenocarcinoma and Optimize Treatment Selection. Clin. Cancer Res. 2020, 26, 3239–3247. [Google Scholar] [CrossRef] [PubMed]
- Silver, D.P.; Richardson, A.L.; Eklund, A.C.; Wang, Z.C.; Szallasi, Z.; Li, Q.; Juul, N.; Leong, C.-O.; Calogrias, D.; Buraimoh, A.; et al. Efficacy of Neoadjuvant Cisplatin in Triple-Negative Breast Cancer. J. Clin. Oncol. 2010, 28, 1145–1153. [Google Scholar] [CrossRef]
- Telli, M.L.; Jensen, K.C.; Vinayak, S.; Kurian, A.W.; Lipson, J.A.; Flaherty, P.J.; Timms, K.; Abkevich, V.; Schackmann, E.A.; Wapnir, I.L.; et al. Phase II Study of Gemcitabine, Carboplatin, and Iniparib As Neoadjuvant Therapy for Triple-Negative and BRCA1/2 Mutation–Associated Breast Cancer with Assessment of a Tumor-Based Measure of Genomic Instability: PrECOG 0105. J. Clin. Oncol. 2015, 33, 1895–1901. [Google Scholar] [CrossRef]
- Kaklamani, V.G.; Jeruss, J.S.; Hughes, E.; Siziopikou, K.P.; Timms, K.M.; Gutin, A.; Abkevich, V.; Sangale, Z.; Solimeno, C.; Brown, K.L.; et al. Phase II neoadjuvant clinical trial of carboplatin and eribulin in women with triple negative early-stage breast cancer (NCT01372579). Breast Cancer Res. Treat. 2015, 151, 629–638. [Google Scholar] [CrossRef]
- Mayer, E.L.; Abramson, V.; Jankowitz, R.; Falkson, C.; Marcom, P.K.; Traina, T.; Carey, L.; Rimawi, M.; Specht, J.; Miller, K.; et al. TBCRC 030: A phase II study of preoperative cisplatin versus paclitaxel in triple-negative breast cancer: Evaluating the homologous recombination deficiency (HRD) biomarker. Ann. Oncol. 2020, 31, 1518–1525. [Google Scholar] [CrossRef]
- Tacconi, E.; Badie, S.; De Gregoriis, G.; Reisländer, T.; Lai, X.; Porru, M.; Folio, C.; Moore, J.; Kopp, A.; Torres, J.B.; et al. Chlorambucil targets BRCA 1/2-deficient tumours and counteracts PARP inhibitor resistance. EMBO Mol. Med. 2019, 11, e9982. [Google Scholar] [CrossRef]
- Moynahan, M.E.; Cui, T.Y.; Jasin, M. Homology-directed dna repair, mitomycin-c resistance, and chromosome stability is restored with correction of a Brca1 mutation. Cancer Res. 2001, 61, 4842–4850. [Google Scholar]
- Liao, G.; Jiang, Z.; Yang, Y.; Zhang, C.; Jiang, M.; Zhu, J.; Xu, L.; Xie, A.; Yan, M.; Zhang, Y.; et al. Combined homologous recombination repair deficiency and immune activation analysis for predicting intensified responses of anthracycline, cyclophosphamide and taxane chemotherapy in triple-negative breast cancer. BMC Med. 2021, 19, 190. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Barlow, W.; Godwin, A.; Pathak, H.; Isakova, K.; Williams, D.; Timms, K.; Hartman, A.; Wenstrup, R.; Linden, H.; et al. Impact of homologous recombination deficiency biomarkers on outcomes in patients with triple-negative breast cancer treated with adjuvant doxorubicin and cyclophosphamide (SWOG S9313). Ann. Oncol. 2018, 29, 654–660. [Google Scholar] [CrossRef] [PubMed]
- Hunter, F.; Wouters, B.; Wilson, W.R. Hypoxia-activated prodrugs: Paths forward in the era of personalised medicine. Br. J. Cancer 2016, 114, 1071–1077. [Google Scholar] [CrossRef] [PubMed]
- Peeters, S.G.; Zegers, C.M.; Biemans, R.; Lieuwes, N.G.; van Stiphout, R.G.; Yaromina, A.; Sun, J.D.; Hart, C.P.; Windhorst, A.D.; van Elmpt, W.; et al. TH-302 in Combination with Radiotherapy Enhances the Therapeutic Outcome and Is Associated with Pretreatment [18F]HX4 Hypoxia PET Imaging. Clin. Cancer Res. 2015, 21, 2984–2992. [Google Scholar] [CrossRef]
- Spiegelberg, L.; Houben, R.; Niemans, R.; de Ruysscher, D.; Yaromina, A.; Theys, J.; Guise, C.P.; Smaill, J.B.; Patterson, A.V.; Lambin, P.; et al. Hypoxia-activated prodrugs and (lack of) clinical progress: The need for hypoxia-based biomarker patient selection in phase III clinical trials. Clin. Transl. Radiat. Oncol. 2019, 15, 62–69. [Google Scholar] [CrossRef]
- Yaromina, A.; Granzier, M.; Biemans, R.; Lieuwes, N.; van Elmpt, W.; Shakirin, G.; Dubois, L.; Lambin, P. A novel concept for tumour targeting with radiation: Inverse dose-painting or targeting the “Low Drug Uptake Volume”. Radiother. Oncol. 2017, 124, 513–520. [Google Scholar] [CrossRef]
- van der Wiel, A.M.; Jackson-Patel, V.; Niemans, R.; Yaromina, A.; Liu, E.; Marcus, D.; Mowday, A.M.; Lieuwes, N.G.; Biemans, R.; Lin, X.; et al. Selectively Targeting Tumor Hypoxia with the Hypoxia-Activated Prodrug CP-506. Mol. Cancer Ther. 2021, 20, 2372–2383. [Google Scholar] [CrossRef]
- Hunter, F.W.; Hsu, H.-L.; Su, J.; Pullen, S.M.; Wilson, W.R.; Wang, J. Dual Targeting of Hypoxia and Homologous Recombination Repair Dysfunction in Triple-Negative Breast Cancer. Mol. Cancer Ther. 2014, 13, 2501–2514. [Google Scholar] [CrossRef]
- Solivio, M.J.; Stornetta, A.; Gilissen, J.; Villalta, P.W.; Deschoemaeker, S.; Heyerick, A.; Dubois, L.; Balbo, S. In Vivo Identification of Adducts from the New Hypoxia-Activated Prodrug CP-506 Using DNA Adductomics. Chem. Res. Toxicol. 2022, 35, 275–282. [Google Scholar] [CrossRef]
- van Wilpe, S.; Tolmeijer, S.; Koornstra, R.; de Vries, I.; Gerritsen, W.; Ligtenberg, M.; Mehra, N. Homologous Recombination Repair Deficiency and Implications for Tumor Immunogenicity. Cancers 2021, 13, 2249. [Google Scholar] [CrossRef]
- McGranahan, N.; Furness, A.J.S.; Rosenthal, R.; Ramskov, S.; Lyngaa, R.; Saini, S.K.; Jamal-Hanjani, M.; Wilson, G.A.; Birkbak, N.J.; Hiley, C.T.; et al. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science 2016, 351, 1463–1469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, C.; Zhang, Z.; Tang, X.; Zhang, X.; Chen, Y.; Hu, T.; Zhang, H.; Guan, M.; Zhang, X.; Wu, Z. Pan-cancer analysis reveals homologous recombination deficiency score as a predictive marker for immunotherapy responders. Hum. Cell 2021, 35, 199–213. [Google Scholar] [CrossRef] [PubMed]
- Jenzer, M.; Keß, P.; Nientiedt, C.; Endris, V.; Kippenberger, M.; Leichsenring, J.; Stögbauer, F.; Haimes, J.; Mishkin, S.; Kudlow, B.; et al. The BRCA2 mutation status shapes the immune phenotype of prostate cancer. Cancer Immunol. Immunother. 2019, 68, 1621–1633. [Google Scholar] [CrossRef] [PubMed]
- Morse, C.B.; Toukatly, M.N.; Kilgore, M.R.; Agnew, K.J.; Bernards, S.S.; Norquist, B.M.; Pennington, K.P.; Garcia, R.L.; Liao, J.B.; Swisher, E.M. Tumor infiltrating lymphocytes and homologous recombination deficiency are independently associated with improved survival in ovarian carcinoma. Gynecol. Oncol. 2019, 153, 217–222. [Google Scholar] [CrossRef] [PubMed]
- Kaur, H.B.; Vidotto, T.; Mendes, A.A.; Salles, D.C.; Isaacs, W.B.; Antonarakis, E.S.; Lotan, T.L. Association between pathogenic germline mutations in BRCA2 and ATM and tumor-infiltrating lymphocytes in primary prostate cancer. Cancer Immunol. Immunother. 2021, 71, 943–951. [Google Scholar] [CrossRef] [PubMed]
- van Vugt, M.A.; Parkes, E.E. When breaks get hot: Inflammatory signaling in BRCA1/2-mutant cancers. Trends Cancer 2022, 8, 174–189. [Google Scholar] [CrossRef] [PubMed]
- Nolan, E.; Savas, P.; Policheni, A.N.; Darcy, P.K.; Vaillant, F.; Mintoff, C.P.; Dushyanthen, S.; Mansour, M.; Pang, J.-M.B.; Fox, S.B.; et al. Combined immune checkpoint blockade as a therapeutic strategy for BRCA1 -mutated breast cancer. Sci. Transl. Med. 2017, 9, eaal4922. [Google Scholar] [CrossRef]
- Wen, W.X.; Leong, C.-O. Association of BRCA1- and BRCA2-deficiency with mutation burden, expression of PD-L1/PD-1, immune infiltrates, and T cell-inflamed signature in breast cancer. PLoS ONE 2019, 14, e0215381. [Google Scholar] [CrossRef]
- Strickland, K.C.; Howitt, B.E.; Shukla, S.A.; Rodig, S.; Ritterhouse, L.L.; Liu, J.F.; Garber, J.E.; Chowdhury, D.; Wu, C.J.; D’Andrea, A.D.; et al. Association and prognostic significance of BRCA1/2-mutation status with neoantigen load, number of tumor-infiltrating lymphocytes and expression of PD-1/PD-L1 in high grade serous ovarian cancer. Oncotarget 2016, 7, 13587–13598. [Google Scholar] [CrossRef]
- Parkes, E.E.; Walker, S.M.; Taggart, L.E.; McCabe, N.; Knight, L.A.; Wilkinson, R.; McCloskey, K.D.; Buckley, N.E.; Savage, K.I.; Salto-Tellez, M.; et al. Activation of STING-Dependent Innate Immune Signaling By S-Phase-Specific DNA Damage in Breast Cancer. JNCI J. Natl. Cancer Inst. 2016, 109, djw199. [Google Scholar] [CrossRef]
- Sato, H.; Niimi, A.; Yasuhara, T.; Permata, T.B.M.; Hagiwara, Y.; Isono, M.; Nuryadi, E.; Sekine, R.; Oike, T.; Kakoti, S.; et al. DNA double-strand break repair pathway regulates PD-L1 expression in cancer cells. Nat. Commun. 2017, 8, 1751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teo, M.Y.; Seier, K.; Ostrovnaya, I.; Regazzi, A.M.; Kania, B.E.; Moran, M.M.; Cipolla, C.K.; Bluth, M.J.; Chaim, J.; Al-Ahmadie, H.; et al. Alterations in DNA Damage Response and Repair Genes as Potential Marker of Clinical Benefit From PD-1/PD-L1 Blockade in Advanced Urothelial Cancers. J. Clin. Oncol. 2018, 36, 1685–1694. [Google Scholar] [CrossRef]
- Hugo, W.; Zaretsky, J.M.; Sun, L.; Song, C.; Moreno, B.H.; Hu-Lieskovan, S.; Berent-Maoz, B.; Pang, J.; Chmielowski, B.; Cherry, G.; et al. Genomic and Transcriptomic Features of Response to Anti-PD-1 Therapy in Metastatic Melanoma. Cell 2016, 165, 35–44. [Google Scholar] [CrossRef] [PubMed]
- George, J.; Alsop, K.; Etemadmoghadam, D.; Hondow, H.; Mikeska, T.; Dobrovic, A.; Defazio, A.; Smyth, G.K.; Levine, D.A.; Mitchell, G.; et al. Nonequivalent Gene Expression and Copy Number Alterations in High-Grade Serous Ovarian Cancers with BRCA1 and BRCA2 Mutations. Clin. Cancer Res. 2013, 19, 3474–3484. [Google Scholar] [CrossRef]
- Samstein, R.M.; Krishna, C.; Ma, X.; Pei, X.; Lee, K.-W.; Makarov, V.; Kuo, F.; Chung, J.; Srivastava, R.M.; Purohit, T.A.; et al. Mutations in BRCA1 and BRCA2 differentially affect the tumor microenvironment and response to checkpoint blockade immunotherapy. Nat. Cancer 2021, 1, 1188–1203. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Name | Phase | Cancer Type | Patients | HRD Assay | BRCA1/2 Mutation | Total HRD | Aims and Results | Study Identifier | Associated Publications |
|---|---|---|---|---|---|---|---|---|---|
| PrECOG 0105 | 2 | Breast | 80 | LOH score | 24.2% | 81.5% | Assessment of the safety and efficacy of iniparib in combination with gemcitabine and carboplatin. Mean LOH score was higher in responders vs. nonresponders, an association that remained significant when only BRCA1/2 wild-type tumors were considered. | NCT00813956 | Telli et al., J. Clin. Oncol. 2015. |
| NU 10B07 | 2 | Breast | 30 | HRD score | 10.0% | 46.2% | Evaluation of safety of carboplatin and eribulin in breast cancer patients and use of HRD score as biomarker of response. Combination of carboplatin and eribulin was safe and HRD score could predict outcome regardless of BRCA1/2 mutational status. | NCT01372579 | Kaklamani et al., Breast Cancer Res. Treat. 2015. |
| ENGOT-OV16/NOVA | 3 | Ovarian | 594 | Myriad myChoice® CDx | 36.7% | 78.5% | Evaluation the efficacy of niraparib in platinum-sensitive ovarian cancer. Niraparib treatment resulted in significant longer progression-free survival in patients with BRCA1/2 mutation and positive HRD classification. | NCT01847274 | Mirza et al., NEJM 2016. |
| ARIEL2 | 2 | Ovarian, fallopia, peritoneal | 206 | LOH score | 19.6% | 59.2% | Evaluation of LOH as biomarker of response to rucaparib. Patients without BRCA1/2 mutation but high LOH score responded better to rucaparib. | NCT01891344 | Swischer et al., Lancet Oncol. 2017. |
| Study 19 | 2 | Ovarian | 53 | HRD score | 60.3% | 69.8% | Characterization of long-term and short-term responders to olaparib. BRCA1/2 status and a high HRD score were associated with long-term response to olaparib. | NCT00753545 | Lheureux et al., Clin. Cancer Res. 2017. |
| SWOG S9313 | 2 | Breast | 425 | HRD score | 29.3% | 67.3% | Evaluation of the combination of anthracycline and cyclophosphamide in breast cancer patients and use of HRD as biomarker of response. HRD-positivity was associated with better response to anthracycline and cyclophosphamide combination. | Int0137 | Sharma et al., Ann. Oncol. 2018. |
| M10-976 | 1 | Ovarian, fallopia, peritoneal | 60 | HRD score | 43.3% | 56.7% | Assess the safety of a novel PARPi, ABT-767. Patients with a mutation in BRCA1/2 or with a HRD score ≥42 responded better to ABT-767. | NCT01339650 | Van der Biessen et al., Invest. New Drugs 2018 |
| QUADRA | 2 | Ovarian, fallopia, peritoneal | 463 | Myriad myChoice® CDx | 18.7% | 47.9% | Assessment of safety of niraparib in patients with ovarian, fallopian, or peritoneal cancer. | NCT02354586 | Moore et al., Lancet Oncol. 2019. |
| PAOLA-1 | 3 | Ovarian, fallopia, peritoneal | 806 | Myriad myChoice® CDx | 30.0% | 48.0% | Assessment of efficacy of the combination of olaparib and bevacizumab in ovarian cancer. Significant benefit of addition of olaparib was observed in patients with a high HRD score, regardless of BRCA1/2 mutations. | NCT02477644 | Ray-Coquard et al., NEJM 2019. |
| LIGHT | 2 | Ovarian, fallopia, peritoneal | 272 | Myriad myChoice® CDx | 37.0% | 62.2% | Evaluation of safety of olaparib in patients with ovarian, fallopian, or peritoneal cancer and use of HRD score as biomarker of response. Patients with BRCA1/2 mutations and positive HRD score responded better to olaparib. | NCT02983799 | Cadoo et al., 2020 ASCO Annu. Meet. 2020. |
| GeparOla | 2 | Breast | 107 | HRD score | 56.2% | 99.7% | Investigation of the combination of paclitaxel and olaparib (PO) in HER2-negative HRD breast cancer patients. PO was safe and resulted in higher pathologic complete response. | NCT02789332 | Fasching et al., Ann. Oncol. 2021. |
| RUBY | 2 | Breast | 40 | LOH score HRDetect | 12.5% | 100.0% | Assessment of efficacy of rucaparib in breast HER2-positive HRD breast cancer and use of LOH and HRDetect as biomarkers of response. A positive HRD score was an inclusion criterion. A subset of patients with high LOH and HRDetect scores without BRCA1/2 mutations benefited from rucaparib treatment. | NCT02505048 | Patsouris et al., EJC 2021. |
| TBCRC | 2 | Breast | 138 | Myriad myChoice® CDx | 6.7% | 71.2% | Assessment of a correlation between HRD score and response to cisplatin or paclitaxel. Tumors with a HRD score ≥33 were classified as HRD instead of ≥42. HRD was not predictive of response to either cisplatin or paclitaxel. | NCT01982448 | Mayer et al., Ann. Oncol. 2021. |
| Lung-MAP S1900A | 2 | Lung | 59 | LOH score | 38.9% | 100.0% | Assessment of efficacy of rucaparib in lung cancer patients with HRD. A positive LOH score was an inclusion criterion. The degree of LOH did not predict response to rucaparib. | NCT03377556 | Riess et al., 2021 ASCO Annu. Meet. 2021. |
| JBCRG-22 | 2 | Breast | 99 | HRD score | N/A | 46.5% | Investigation of clinical usefulness of combination of eribulin and carboplatin or paclitaxel. HRD patients (≥42) responded significantly better to combination therapy. | UMIN000023162 | Masuda et al., Breast Cancer Res. Treat. 2021. |
| Meet-URO 12 | 2 | Urothelial | 58 | FoundationOne® CDx | 12.8% | 44.7% | Evaluation of niraparib in combination with best supportive care (BSC). Addition of niraparib did not improve progression-free survival | NCT03945084 | Vignani et al., 2022 ASCO Genitourin. Cancers Symp. 2022. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
van der Wiel, A.M.A.; Schuitmaker, L.; Cong, Y.; Theys, J.; Van Hoeck, A.; Vens, C.; Lambin, P.; Yaromina, A.; Dubois, L.J. Homologous Recombination Deficiency Scar: Mutations and Beyond—Implications for Precision Oncology. Cancers 2022, 14, 4157. https://doi.org/10.3390/cancers14174157
van der Wiel AMA, Schuitmaker L, Cong Y, Theys J, Van Hoeck A, Vens C, Lambin P, Yaromina A, Dubois LJ. Homologous Recombination Deficiency Scar: Mutations and Beyond—Implications for Precision Oncology. Cancers. 2022; 14(17):4157. https://doi.org/10.3390/cancers14174157
Chicago/Turabian Stylevan der Wiel, Alexander M. A., Lesley Schuitmaker, Ying Cong, Jan Theys, Arne Van Hoeck, Conchita Vens, Philippe Lambin, Ala Yaromina, and Ludwig J. Dubois. 2022. "Homologous Recombination Deficiency Scar: Mutations and Beyond—Implications for Precision Oncology" Cancers 14, no. 17: 4157. https://doi.org/10.3390/cancers14174157