
How Gut Microbiota Are Shaped by Pattern Recognition Receptors in Colitis and Colorectal Cancer


Abstract
:Simple Summary
Abstract
1. Introduction
2. Nod-like Receptors
3. Nod1 Receptor
4. Nod2 Receptor
5. Nlrp3 Receptor
6. Nlrp6 Receptor
7. Nlrp12 Receptor
8. Aim2 Receptor
9. RIG-I-like Receptor
10. Cyclic GMP-AMP Synthase (cGAS) and Stimulator of Interferon Genes (Sting) Receptor
11. C-Type Lectin-like Receptors
12. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Kudelka, M.R.; Stowell, S.R.; Cummings, R.D.; Neish, A.S. Intestinal epithelial glycosylation in homeostasis and gut microbiota interactions in IBD. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 597–617. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Chen, J.; Wu, B.; He, T.; Xie, L.; Liu, Z. Dock2 affects the host susceptibility to Citrobacter rodentium infection through regulating gut microbiota. Gut Pathog. 2021, 13, 52. [Google Scholar] [CrossRef] [PubMed]
- Eaden, J.A.; Abrams, K.R.; Mayberry, J.F. The risk of colorectal cancer in ulcerative colitis: A meta-analysis. Gut 2001, 48, 526–535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Soerjomataram, I.; Bray, F. Planning for tomorrow: Global cancer incidence and the role of prevention 2020–2070. Nat. Rev. Clin. Oncol. 2021, 18, 663–672. [Google Scholar] [CrossRef] [PubMed]
- Gagniere, J.; Raisch, J.; Veziant, J.; Barnich, N.; Bonnet, R.; Buc, E.; Bringer, M.A.; Pezet, D.; Bonnet, M. Gut microbiota imbalance and colorectal cancer. World J. Gastroenterol. 2016, 22, 501–518. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Li, X.; Liu, S.; Zhang, Y.; Zhang, D. Toll-like Receptors and Inflammatory Bowel Disease. Front. Immunol. 2018, 9, 72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burgueno, J.F.; Abreu, M.T. Epithelial Toll-like receptors and their role in gut homeostasis and disease. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 263–278. [Google Scholar] [CrossRef]
- Mukherjee, T.; Hovingh, E.S.; Foerster, E.G.; Abdel-Nour, M.; Philpott, D.J.; Girardin, S.E. NOD1 and NOD2 in inflammation, immunity and disease. Arch. Biochem. Biophys. 2019, 670, 69–81. [Google Scholar] [CrossRef]
- Krishnan, R.; Rajendran, R.; Jang, Y.; Kim, J.; Yoon, S.; Oh, M.-J. NLRC3 attenuates antiviral immunity and activates inflammasome responses in primary grouper brain cells following nervous necrosis virus infection. Fish Shellfish. Immunol. 2022, 127, 219–227. [Google Scholar] [CrossRef]
- Fekete, T.; Bencze, D.; Biro, E.; Benko, S.; Pazmandi, K. Focusing on the Cell Type Specific Regulatory Actions of NLRX1. Int. J. Mol. Sci. 2021, 22, 1316. [Google Scholar] [CrossRef]
- Duncan, J.A.; Canna, S.W. The NLRC4 Inflammasome. Immunol. Rev. 2018, 281, 115–123. [Google Scholar] [CrossRef]
- Vance, R.E. The NAIP/NLRC4 inflammasomes. Curr. Opin. Immunol. 2015, 32, 84–89. [Google Scholar] [CrossRef] [Green Version]
- Platnich, J.M.; Muruve, D.A. NOD-like receptors and inflammasomes: A review of their canonical and non-canonical signaling pathways. Arch. Biochem. Biophys. 2019, 670, 4–14. [Google Scholar] [CrossRef]
- Fernández-García, V.; González-Ramos, S.; Martín-Sanz, P.; Portillo, F.G.-D.; Laparra, J.M.; Boscá, L. NOD1 in the interplay between microbiota and gastrointestinal immune adaptations. Pharmacol. Res. 2021, 171, 105775. [Google Scholar] [CrossRef]
- McGovern, D.P.; Hysi, P.; Ahmad, T.; van Heel, D.A.; Moffatt, M.F.; Carey, A.; Cookson, W.O.; Jewell, D.P. Association between a complex insertion/deletion polymorphism in NOD1 (CARD4) and susceptibility to inflammatory bowel disease. Hum. Mol. Genet. 2005, 14, 1245–1250. [Google Scholar] [CrossRef] [Green Version]
- Huebner, C.; Ferguson, L.R.; Han, D.Y.; Philpott, M.; Barclay, M.L.; Gearry, R.B.; McCulloch, A.; Demmers, P.S.; Browning, B.L. Nucleotide-binding oligomerization domain containing 1 (NOD1) haplotypes and single nucleotide polymorphisms modify susceptibility to inflammatory bowel diseases in a New Zealand caucasian population: A case-control study. BMC Res. Notes 2009, 2, 52. [Google Scholar] [CrossRef] [Green Version]
- Hasegawa, M.; Yamazaki, T.; Kamada, N.; Tawaratsumida, K.; Kim, Y.G.; Nunez, G.; Inohara, N. Nucleotide-binding oligomerization domain 1 mediates recognition of Clostridium difficile and induces neutrophil recruitment and protection against the pathogen. J. Immunol. 2011, 186, 4872–4880. [Google Scholar] [CrossRef] [Green Version]
- Zhan, Y.; Seregin, S.S.; Chen, J.; Chen, G.Y. Nod1 Limits Colitis-Associated Tumorigenesis by Regulating IFN-gamma Production. J. Immunol. 2016, 196, 5121–5129. [Google Scholar] [CrossRef] [Green Version]
- Trindade, B.C.; Chen, G.Y. NOD1 and NOD2 in inflammatory and infectious diseases. Immunol. Rev. 2020, 297, 139–161. [Google Scholar] [CrossRef]
- Guo, H.; Gibson, S.A.; Ting, J.P.Y. Gut microbiota, NLR proteins, and intestinal homeostasis. J. Exp. Med. 2020, 217, 20181832. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.Y.; Shaw, M.H.; Redondo, G.; Núñez, G. The innate immune receptor Nod1 protects the intestine from inflammation-induced tumorigenesis. Cancer Res. 2008, 68, 10060–10067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hampe, J.; Cuthbert, A.; Croucher, P.J.P.; Mirza, M.M.; Mascheretti, S.; Fisher, S.; Frenzel, H.; King, K.; Hasselmeyer, A.; MacPherson, A.J.S.; et al. Association between insertion mutation in NOD2 gene and Crohn’s disease in German and British populations. Lancet 2001, 357, 1925–1928. [Google Scholar] [CrossRef]
- Negi, S.; Das, D.K.; Pahari, S.; Nadeem, S.; Agrewala, J.N. Potential Role of Gut Microbiota in Induction and Regulation of Innate Immune Memory. Front. Immunol. 2019, 10, 2441. [Google Scholar] [CrossRef] [Green Version]
- Al Nabhani, Z.; Lepage, P.; Mauny, P.; Montcuquet, N.; Roy, M.; Le Roux, K.; Dussaillant, M.; Berrebi, D.; Hugot, J.P.; Barreau, F. Nod2 Deficiency Leads to a Specific and Transmissible Mucosa-associated Microbial Dysbiosis Which Is Independent of the Mucosal Barrier Defect. J. Crohn’s Colitis 2016, 10, 1428–1436. [Google Scholar] [CrossRef] [Green Version]
- Robertson, S.J.; Geddes, K.; Maisonneuve, C.; Streutker, C.J.; Philpott, D.J. Resilience of the intestinal microbiota following pathogenic bacterial infection is independent of innate immunity mediated by NOD1 or NOD2. Microbes Infect. 2016, 18, 460–471. [Google Scholar] [CrossRef]
- Butera, A.; Di Paola, M.; Pavarini, L.; Strati, F.; Pindo, M.; Sanchez, M.; Cavalieri, D.; Boirivant, M.; De Filippo, C. Nod2 Deficiency in mice is Associated with Microbiota Variation Favouring the Expansion of mucosal CD4+ LAP+ Regulatory Cells. Sci. Rep. 2018, 8, 14241. [Google Scholar] [CrossRef] [Green Version]
- de Souza, P.R.; Guimarães, F.R.; Sales-Campos, H.; Bonfá, G.; Nardini, V.; Chica, J.E.L.; Turato, W.M.; Silva, J.S.; Zamboni, D.S.; Cardoso, C.R.B. Absence of NOD2 receptor predisposes to intestinal inflammation by a deregulation in the immune response in hosts that are unable to control gut dysbiosis. Immunobiology 2018, 223, 577–585. [Google Scholar] [CrossRef]
- Robertson, S.J.; Zhou, J.Y.; Geddes, K.; Rubino, S.J.; Cho, J.H.; Girardin, S.E.; Philpott, D.J. Nod1 and Nod2 signaling does not alter the composition of intestinal bacterial communities at homeostasis. Gut Microbes 2013, 4, 222–231. [Google Scholar] [CrossRef] [Green Version]
- Couturier-Maillard, A.; Secher, T.; Rehman, A.; Normand, S.; De Arcangelis, A.; Haesler, R.; Huot, L.; Grandjean, T.; Bressenot, A.; Delanoye-Crespin, A.; et al. NOD2-mediated dysbiosis predisposes mice to transmissible colitis and colorectal cancer. J. Clin. Invest. 2013, 123, 700–711. [Google Scholar] [CrossRef] [Green Version]
- Jiang, W.; Wang, X.; Zeng, B.; Liu, L.; Tardivel, A.; Wei, H.; Han, J.; MacDonald, H.R.; Tschopp, J.; Tian, Z.; et al. Recognition of gut microbiota by NOD2 is essential for the homeostasis of intestinal intraepithelial lymphocytes. J. Exp. Med. 2013, 210, 2465–2476. [Google Scholar] [CrossRef]
- Al Nabhani, Z.; Montcuquet, N.; Roy, M.; Dussaillant, M.; Hugot, J.P.; Barreau, F. Complementary Roles of Nod2 in Hematopoietic and Nonhematopoietic Cells in Preventing Gut Barrier Dysfunction Dependent on MLCK Activity. Inflamm. Bowel Dis 2017, 23, 1109–1119. [Google Scholar] [CrossRef] [Green Version]
- Amendola, A.; Butera, A.; Sanchez, M.; Strober, W.; Boirivant, M. Nod2 deficiency is associated with an increased mucosal immunoregulatory response to commensal microorganisms. Mucosal Immunol. 2014, 7, 391–404. [Google Scholar] [CrossRef] [Green Version]
- Ramanan, D.; Tang, M.S.; Bowcutt, R.; Loke, P.; Cadwell, K. Bacterial sensor Nod2 prevents inflammation of the small intestine by restricting the expansion of the commensal Bacteroides vulgatus. Immunity 2014, 41, 311–324. [Google Scholar] [CrossRef] [Green Version]
- Karki, R.; Man, S.M.; Kanneganti, T.D. Inflammasomes and Cancer. Cancer Immunol. Res. 2017, 5, 94–99. [Google Scholar] [CrossRef] [Green Version]
- Zaki, M.H.; Boyd, K.L.; Vogel, P.; Kastan, M.B.; Lamkanfi, M.; Kanneganti, T.D. The NLRP3 inflammasome protects against loss of epithelial integrity and mortality during experimental colitis. Immunity 2010, 32, 379–391. [Google Scholar] [CrossRef] [Green Version]
- Mak’Anyengo, R.; Duewell, P.; Reichl, C.; Horth, C.; Lehr, H.A.; Fischer, S.; Clavel, T.; Denk, G.; Hohenester, S.; Kobold, S.; et al. Nlrp3-dependent IL-1beta inhibits CD103+ dendritic cell differentiation in the gut. JCI Insight 2018, 3, e96322. [Google Scholar] [CrossRef]
- Hirota, S.A.; Ng, J.; Lueng, A.; Khajah, M.; Parhar, K.; Li, Y.; Lam, V.; Potentier, M.S.; Ng, K.; Bawa, M.; et al. NLRP3 inflammasome plays a key role in the regulation of intestinal homeostasis. Inflamm. Bowel Dis. 2011, 17, 1359–1372. [Google Scholar] [CrossRef]
- Qu, S.; Fan, L.; Qi, Y.; Xu, C.; Hu, Y.; Chen, S.; Liu, W.; Liu, W.; Si, J. Akkermansia muciniphila Alleviates Dextran Sulfate Sodium (DSS)-Induced Acute Colitis by NLRP3 Activation. Microbiol. Spectr. 2021, 9, e0073021. [Google Scholar] [CrossRef]
- Shao, X.; Sun, S.; Zhou, Y.; Wang, H.; Yu, Y.; Hu, T.; Yao, Y.; Zhou, C. Bacteroides fragilis restricts colitis-associated cancer via negative regulation of the NLRP3 axis. Cancer Lett. 2021, 523, 170–181. [Google Scholar] [CrossRef]
- Yao, X.; Zhang, C.; Xing, Y.; Xue, G.; Zhang, Q.; Pan, F.; Wu, G.; Hu, Y.; Guo, Q.; Lu, A.; et al. Remodelling of the gut microbiota by hyperactive NLRP3 induces regulatory T cells to maintain homeostasis. Nat. Commun. 2017, 8, 1896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bouskra, D.; Brezillon, C.; Berard, M.; Werts, C.; Varona, R.; Boneca, I.G.; Eberl, G. Lymphoid tissue genesis induced by commensals through NOD1 regulates intestinal homeostasis. Nature 2008, 456, 507–510. [Google Scholar] [CrossRef] [PubMed]
- Theivanthiran, B.; Batra, S.; Balamayooran, G.; Cai, S.; Kobayashi, K.; Flavell, R.A.; Jeyaseelan, S. NOD2 signaling contributes to host defense in the lungs against Escherichia coli infection. Infect. Immun. 2012, 80, 2558–2569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mukherjee, S.; Kumar, R.; Tsakem Lenou, E.; Basrur, V.; Kontoyiannis, D.L.; Ioakeimidis, F.; Mosialos, G.; Theiss, A.L.; Flavell, R.A.; Venuprasad, K. Deubiquitination of NLRP6 inflammasome by Cyld critically regulates intestinal inflammation. Nat. Immunol. 2020, 21, 626–635. [Google Scholar] [CrossRef]
- Mamantopoulos, M.; Ronchi, F.; Van Hauwermeiren, F.; Vieira-Silva, S.; Yilmaz, B.; Martens, L.; Saeys, Y.; Drexler, S.K.; Yazdi, A.S.; Raes, J.; et al. Nlrp6- and ASC-Dependent Inflammasomes Do Not Shape the Commensal Gut Microbiota Composition. Immunity 2017, 47, 339–348.e334. [Google Scholar] [CrossRef] [Green Version]
- Seregin, S.S.; Golovchenko, N.; Schaf, B.; Chen, J.; Pudlo, N.A.; Mitchell, J.; Baxter, N.T.; Zhao, L.; Schloss, P.D.; Martens, E.C.; et al. NLRP6 Protects Il10(-/-) Mice from Colitis by Limiting Colonization of Akkermansia muciniphila. Cell Rep. 2017, 19, 733–745. [Google Scholar] [CrossRef]
- Lemire, P.; Robertson, S.J.; Maughan, H.; Tattoli, I.; Streutker, C.J.; Platnich, J.M.; Muruve, D.A.; Philpott, D.J.; Girardin, S.E. The NLR Protein NLRP6 Does Not Impact Gut Microbiota Composition. Cell Rep. 2017, 21, 3653–3661. [Google Scholar] [CrossRef] [Green Version]
- Henao-Mejia, J.; Elinav, E.; Jin, C.; Hao, L.; Mehal, W.Z.; Strowig, T.; Thaiss, C.A.; Kau, A.L.; Eisenbarth, S.C.; Jurczak, M.J.; et al. Inflammasome-mediated dysbiosis regulates progression of NAFLD and obesity. Nature 2012, 482, 179–185. [Google Scholar] [CrossRef] [Green Version]
- Elinav, E.; Strowig, T.; Kau, A.L.; Henao-Mejia, J.; Thaiss, C.A.; Booth, C.J.; Peaper, D.R.; Bertin, J.; Eisenbarth, S.C.; Gordon, J.I.; et al. NLRP6 inflammasome is a regulator of colonic microbial ecology and risk for colitis. Cell 2011, 145, 745–757. [Google Scholar] [CrossRef] [Green Version]
- Zaki, M.H.; Vogel, P.; Malireddi, R.K.; Body-Malapel, M.; Anand, P.K.; Bertin, J.; Green, D.R.; Lamkanfi, M.; Kanneganti, T.D. The NOD-like receptor NLRP12 attenuates colon inflammation and tumorigenesis. Cancer Cell 2011, 20, 649–660. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Wilson, J.E.; Koenigsknecht, M.J.; Chou, W.C.; Montgomery, S.A.; Truax, A.D.; Brickey, W.J.; Packey, C.D.; Maharshak, N.; Matsushima, G.K.; et al. NLRP12 attenuates colon inflammation by maintaining colonic microbial diversity and promoting protective commensal bacterial growth. Nat. Immunol. 2017, 18, 541–551. [Google Scholar] [CrossRef]
- Ratsimandresy, R.A.; Indramohan, M.; Dorfleutner, A.; Stehlik, C. The AIM2 inflammasome is a central regulator of intestinal homeostasis through the IL-18/IL-22/STAT3 pathway. Cell. Mol. Immunol. 2017, 14, 127–142. [Google Scholar] [CrossRef]
- Man, S.M.; Zhu, Q.; Zhu, L.; Liu, Z.; Karki, R.; Malik, A.; Sharma, D.; Li, L.; Malireddi, R.K.; Gurung, P.; et al. Critical Role for the DNA Sensor AIM2 in Stem Cell Proliferation and Cancer. Cell 2015, 162, 45–58. [Google Scholar] [CrossRef] [Green Version]
- Zhu, H.; Xu, W.Y.; Hu, Z.; Zhang, H.; Shen, Y.; Lu, S.; Wei, C.; Wang, Z.G. RNA virus receptor Rig-I monitors gut microbiota and inhibits colitis-associated colorectal cancer. J. Exp. Clin. Cancer Res. 2017, 36, 2. [Google Scholar] [CrossRef] [Green Version]
- Canesso, M.C.C.; Lemos, L.; Neves, T.C.; Marim, F.M.; Castro, T.B.R.; Veloso, É.S.; Queiroz, C.P.; Ahn, J.; Santiago, H.C.; Martins, F.S.; et al. The cytosolic sensor STING is required for intestinal homeostasis and control of inflammation. Mucosal Immunol. 2018, 11, 820–834. [Google Scholar] [CrossRef] [Green Version]
- Ahn, J.; Son, S.; Oliveira, S.C.; Barber, G.N. STING-Dependent Signaling Underlies IL-10 Controlled Inflammatory Colitis. Cell Rep. 2017, 21, 3873–3884. [Google Scholar] [CrossRef] [Green Version]
- Kamiya, T.; Tang, C.; Kadoki, M.; Oshima, K.; Hattori, M.; Saijo, S.; Adachi, Y.; Ohno, N.; Iwakura, Y. β-Glucans in food modify colonic microflora by inducing antimicrobial protein, calprotectin, in a Dectin-1-induced-IL-17F-dependent manner. Mucosal Immunol. 2018, 11, 763–773. [Google Scholar] [CrossRef]
- Wang, T.; Pan, D.; Zhou, Z.; You, Y.; Jiang, C.; Zhao, X.; Lin, X. Dectin-3 Deficiency Promotes Colitis Development due to Impaired Antifungal Innate Immune Responses in the Gut. PLoS Pathog. 2016, 12, e1005662. [Google Scholar] [CrossRef] [Green Version]
- Zheng, D.; Kern, L.; Elinav, E. The NLRP6 inflammasome. Immunology 2021, 162, 281–289. [Google Scholar] [CrossRef]
- Levy, M.; Thaiss, C.A.; Zeevi, D.; Dohnalova, L.; Zilberman-Schapira, G.; Mahdi, J.A.; David, E.; Savidor, A.; Korem, T.; Herzig, Y.; et al. Microbiota-Modulated Metabolites Shape the Intestinal Microenvironment by Regulating NLRP6 Inflammasome Signaling. Cell 2015, 163, 1428–1443. [Google Scholar] [CrossRef] [Green Version]
- Mamantopoulos, M.; Ronchi, F.; McCoy, K.D.; Wullaert, A. Inflammasomes make the case for littermate-controlled experimental design in studying host-microbiota interactions. Gut Microbes 2018, 9, 374–381. [Google Scholar] [CrossRef] [Green Version]
- Lau, J.M.-F.; Dombrowski, Y. The innate immune receptor NLRP12 maintains intestinal homeostasis by regulating microbiome diversity. Cell. Mol. Immunol. 2017, 15, 193–195. [Google Scholar] [CrossRef] [Green Version]
- He, L.; Liu, Y.; Lai, W.; Tian, H.; Chen, L.; Xie, L.; Liu, Z. DNA sensors, crucial receptors to resist pathogens, are deregulated in colorectal cancer and associated with initiation and progression of the disease. J. Cancer 2020, 11, 893–905. [Google Scholar] [CrossRef] [Green Version]
- Hu, S.; Peng, L.; Kwak, Y.T.; Tekippe, E.M.; Pasare, C.; Malter, J.S.; Hooper, L.V.; Zaki, M.H. The DNA Sensor AIM2 Maintains Intestinal Homeostasis via Regulation of Epithelial Antimicrobial Host Defense. Cell Rep. 2015, 13, 1922–1936. [Google Scholar] [CrossRef] [Green Version]
- Leite, J.A.; Pessenda, G.; Guerra-Gomes, I.C.; de Santana, A.K.M.; Andre Pereira, C.; Ribeiro Campos Costa, F.; Ramos, S.G.; Simoes Zamboni, D.; Caetano Faria, A.M.; Candido de Almeida, D.; et al. The DNA Sensor AIM2 Protects against Streptozotocin-Induced Type 1 Diabetes by Regulating Intestinal Homeostasis via the IL-18 Pathway. Cells 2020, 9, 959. [Google Scholar] [CrossRef]
- Plantamura, E.; Dzutsev, A.; Chamaillard, M.; Djebali, S.; Moudombi, L.; Boucinha, L.; Grau, M.; Macari, C.; Bauché, D.; Dumitrescu, O.; et al. MAVS deficiency induces gut dysbiotic microbiota conferring a proallergic phenotype. Proc. Natl. Acad. Sci. USA 2018, 115, 10404–10409. [Google Scholar] [CrossRef] [Green Version]
- Fischer, J.C.; Bscheider, M.; Eisenkolb, G.; Lin, C.C.; Wintges, A.; Otten, V.; Lindemans, C.A.; Heidegger, S.; Rudelius, M.; Monette, S.; et al. RIG-I/MAVS and STING signaling promote gut integrity during irradiation- and immune-mediated tissue injury. Sci. Transl. Med. 2017, 9, eaag2513. [Google Scholar] [CrossRef] [Green Version]
- Hopfner, K.P.; Hornung, V. Molecular mechanisms and cellular functions of cGAS-STING signalling. Nat. Rev. Mol. Cell Biol. 2020, 21, 501–521. [Google Scholar] [CrossRef]
- Martin, G.R.; Blomquist, C.M.; Henare, K.L.; Jirik, F.R. Stimulator of interferon genes (STING) activation exacerbates experimental colitis in mice. Sci. Rep. 2019, 9, 14281. [Google Scholar] [CrossRef] [Green Version]
- Goyal, S.; Castrillon-Betancur, J.C.; Klaile, E.; Slevogt, H. The Interaction of Human Pathogenic Fungi With C-Type Lectin Receptors. Front. Immunol. 2018, 9, 1261. [Google Scholar] [CrossRef] [Green Version]
- Aziz, F.; Khan, I.; Shukla, S.; Dey, D.K.; Yan, Q.; Chakraborty, A.; Yoshitomi, H.; Hwang, S.K.; Sonwal, S.; Lee, H.; et al. Partners in crime: The Lewis Y antigen and fucosyltransferase IV in Helicobacter pylori-induced gastric cancer. Pharmacol. Ther. 2022, 232, 107994. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.Y. Regulation of the gut microbiome by inflammasomes. Free Radic. Biol. Med. 2017, 105, 35–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaki, M.H.; Lamkanfi, M.; Kanneganti, T.D. The Nlrp3 inflammasome: Contributions to intestinal homeostasis. Trends Immunol. 2011, 32, 171–179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, L.; Chen, Y.; Wu, Y.; Xu, Y.; Zhang, Z.; Liu, Z. Nucleic acid sensing pattern recognition receptors in the development of colorectal cancer and colitis. Cell Mol. Life Sci. 2017, 74, 2395–2411. [Google Scholar] [CrossRef] [PubMed]
- Hummel, S.; Veltman, K.; Cichon, C.; Sonnenborn, U.; Schmidt, M.A. Differential targeting of the E-Cadherin/beta-Catenin complex by gram-positive probiotic lactobacilli improves epithelial barrier function. Appl. Environ. Microbiol. 2012, 78, 1140–1147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fabrega, M.J.; Rodriguez-Nogales, A.; Garrido-Mesa, J.; Algieri, F.; Badia, J.; Gimenez, R.; Galvez, J.; Baldoma, L. Intestinal Anti-inflammatory Effects of Outer Membrane Vesicles from Escherichia coli Nissle 1917 in DSS-Experimental Colitis in Mice. Front. Microbiol. 2017, 8, 1274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, F.; Cao, H.; Cover, T.L.; Whitehead, R.; Washington, M.K.; Polk, D.B. Soluble proteins produced by probiotic bacteria regulate intestinal epithelial cell survival and growth. Gastroenterology 2007, 132, 562–575. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez-Rodriguez, I.; Sanchez, B.; Ruiz, L.; Turroni, F.; Ventura, M.; Ruas-Madiedo, P.; Gueimonde, M.; Margolles, A. Role of extracellular transaldolase from Bifidobacterium bifidum in mucin adhesion and aggregation. Appl. Environ. Microbiol. 2012, 78, 3992–3998. [Google Scholar] [CrossRef] [Green Version]
- Gryp, T.; Huys, G.R.B.; Joossens, M.; Van Biesen, W.; Glorieux, G.; Vaneechoutte, M. Isolation and Quantification of Uremic Toxin Precursor-Generating Gut Bacteria in Chronic Kidney Disease Patients. Int. J. Mol. Sci. 2020, 21, 1986. [Google Scholar] [CrossRef] [Green Version]
- Dey, D.K.; Kang, S.C. Weissella confusa DD_A7 pre-treatment to zebrafish larvae ameliorates the inflammation response against Escherichia coli O157:H7. Microbiol. Res. 2020, 237, 126489. [Google Scholar] [CrossRef]
- Wu, X.; Wu, Y.; He, L.; Wu, L.; Wang, X.; Liu, Z. Effects of the intestinal microbial metabolite butyrate on the development of colorectal cancer. J. Cancer 2018, 9, 2510–2517. [Google Scholar] [CrossRef]
- Javanshir, N.; Hosseini, G.N.G.; Sadeghi, M.; Esmaeili, R.; Satarikia, F.; Ahmadian, G.; Allahyari, N. Evaluation of the Function of Probiotics, Emphasizing the Role of their Binding to the Intestinal Epithelium in the Stability and their Effects on the Immune System. Biol. Proced. Online 2021, 23, 23. [Google Scholar] [CrossRef]
- Matsubara, V.H.; Ishikawa, K.H.; Ando-Suguimoto, E.S.; Bueno-Silva, B.; Nakamae, A.E.M.; Mayer, M.P.A. Probiotic Bacteria Alter Pattern-Recognition Receptor Expression and Cytokine Profile in a Human Macrophage Model Challenged with Candida albicans and Lipopolysaccharide. Front. Microbiol. 2017, 8, 2280. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
| PRRs | Possible Mechanisms | Microbiota Changes |
|---|---|---|
| Nod1 | Nod1 is a cytoplasmic receptor that can recognize iE-DAP, activate NF-κB and MAPK signals, regulate AMPs, proinflammatory cytokines, autophagy, and acquired immunity to enhance epithelial barrier function, promote intestinal homeostasis, and resist the invasion of pathogens that may cause colitis. Nod1 knockout enhances the damage and apoptotic responses of intestinal epithelial cells, enhances intestinal epithelial permeability, reduces epithelial barrier, and increases bacterial abundance [42], thereby increasing susceptibility to colitis. However, antibiotic treatment of Nod1 knockout mice significantly inhibits the formation of intestinal tumors, suggesting that gut microbiota plays an important role in the development of colorectal associated tumors; the specific microbial species have not been elucidated [19,22]. | The number of bacteria has increased, but the specific species are not characterized. |
| Nod2 | Nod2 is a cytoplasmic receptor that can recognize MDP and recruit RIP2, thereby activating NF-κB and MAPK signaling pathways. Nod2 also plays an important role by promoting the production of antibacterial compounds (including defensin) in Paneth cells. After Nod2 knockout, the production of AMPs decreases, and the tumor necrosis factor α and interferon γ increase, resulting in the overexpression of MLCK. MLCK phosphorylates Ser19 and Thr18 of the myosin light chain, changing its spatial conformation, promoting the contraction of actin and myosin filaments, and opening epithelial cell contacts, thus increasing the permeability of the intestinal epithelial mucosa. Nod2 knockout results in reduced goblet cells in colon mucosa, decreased expression of MUC2 and phospholipase A2, impaired intestinal epithelial barrier function, bacterial translocation and imbalance, and increased susceptibility to colitis and CRC [28,32,33,34,43]. One study showed that gut microbiota richness increases after Nod2 knockout. For example, Firmicutes, Corynebacteriaceae, Bdellovibrionaceae, and Deferribacteriaceae were increased [27]. | Firmicutes↑ Proteobacteria↑ Deferribacteres phyla↑ Lachnospiraceae↑ Ruminococcaceae↑ Corynebacteriaceae (Actinobacteria)↑ Bdellovibrionaceae (Proteobacteria)↑ Deferribacteriaceae (Deferribacteres)↑ |
| Nlrp3 | Nlrp3 is a cytoplasmic receptor that promotes IL-18 production by non-hematopoietic cells, thereby maintaining gut microbiota homeostasis and the integrity of the intestinal epithelial barrier. After Nlrp3 deletion, the expression of IL-1β, IL-18, IL-10, and TGF-β is decreased, the secretion of antibacterial products is decreased, and the expression of β defensin altered, resulting in the destruction of the intestinal integrity, and altered gut microbiota (Rikenellaceae, Enterobacteriaceae, Mycobacterium, Clostridium are increased, Bacteroidaceae is decreased) [37,38]. These mice are more likely to develop CRC and have more severe disease manifestations. Nlrp3R258W mice have increased Treg cell numbers by increasing IL-1β production and AMPs secretion to reshape gut microbiota composition (such as decreased Actinobacteria, Verrucomicrobia, and Akkermansia, while Lactobacillus are increased). Their resistance to colitis and CRC is increased [41]. | Rikenellaceae ↑ Enterobacteriaceae ↑ Clostridium ↑ Lactobacillus ↑ Bacteroidaceae ↓ Verrucomicrobia ↓ Akkermansia ↓ |
| Nlrp6 | Nlrp6 is a cytoplasmic receptor that is highly expressed in intestinal epithelial cells and myeloid-derived immune cells, such as dendritic cells, macrophages, and monocytes [44]. It can regulate mucus secretion of goblet cells in response to bacterial invasion and also regulate microbial colonization by regulating AMPs secretion. After Nlrp6 knockout, secretion of mucus in goblet cells is decreased, secretion of IL-18 is decreased, the ability of intestinal epithelial cells to recover after DSS-induced injury is weakened, and the expression of intestinal downstream AMPs genes is altered, thus increasing the susceptibility to colitis and related tumors. Nlrp6 knockout mice have changes in gut microbiota (Prevotellaceae, TM7, Bacteroidetes, Proteobacteria are increased, while Firmicutes are decreased); this change could not be repeated in the same littermate control experiment and contradictory results were identified, indicating that unidentified genetic or environmental factors may play a critical role [45,46,47,48,49]. Therefore, the effect of Nlrp6 on the relative abundance of gut microbiota needs to be further verified. | Prevotellaceae
↑
TM7 ↑ Akkermansia muciniphila ↑ Firmicutes ↓ |
| Nlrp12 | Nlrp12 is a cytoplasmic receptor that is primarily expressed in DCs and neutrophils [50]. It can inhibit intestinal inflammation by inhibiting both typical and atypical NF-κB and excessive immune signaling. The activation of NF-κB, ERK, and STAT3 is increased after Nlrp12 deletion, and the diversity and composition of gut microbiota are altered (Bacteroidales, Clostridiales, and Lachnospiraceae are decreased, while Erysipelotrichaceae is increased) and dyshomeostasis of the intestinal environment occurs, thereby increasing host susceptibility to colitis and related tumors [51]. | Bacteroides ↓ Clostridia ↓ Lachnospiraceae ↓ Erysipelotrichaceae ↑ |
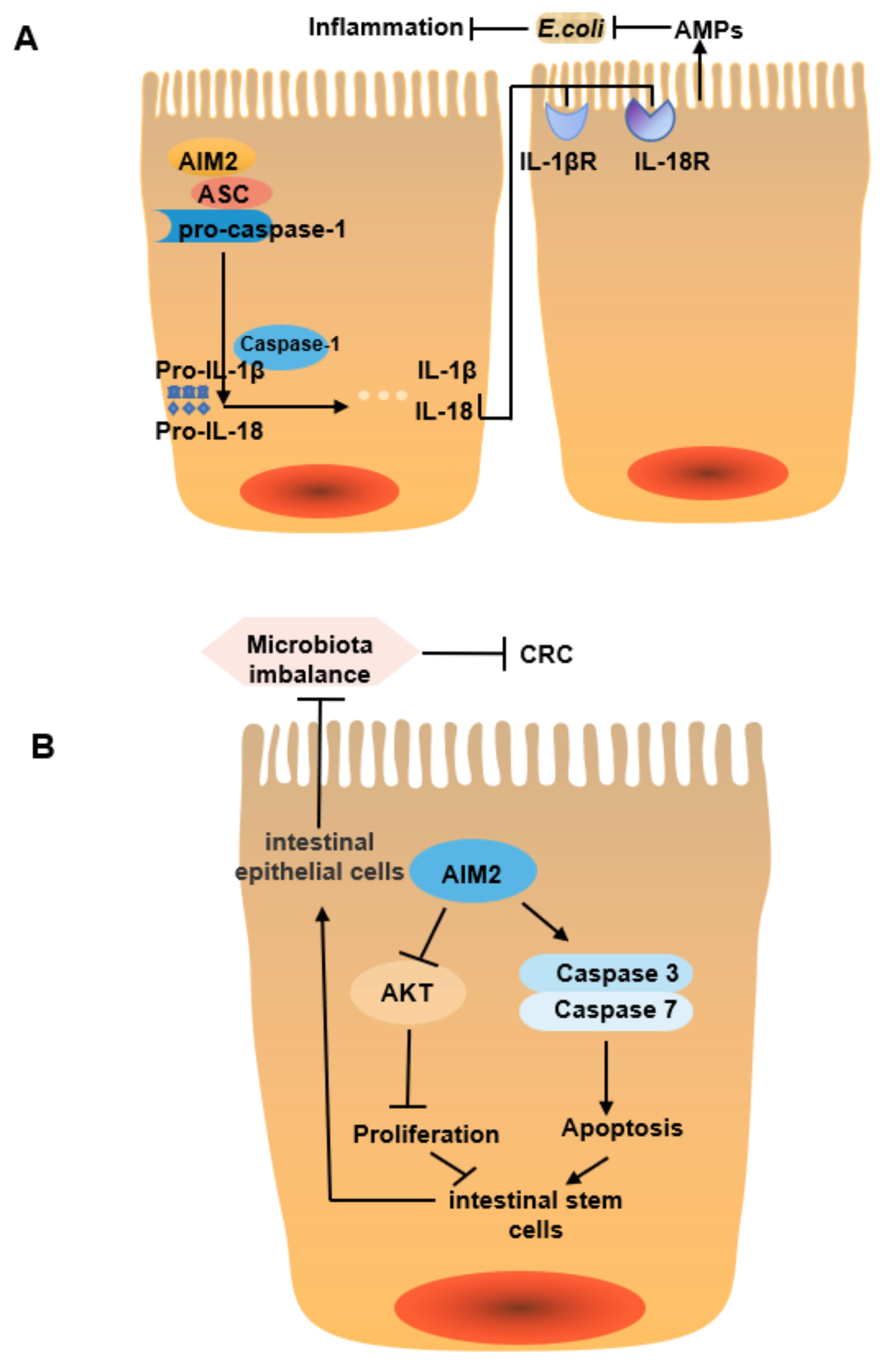
| Aim2 | Aim2 is a cytosolic innate immune receptor that can inhibit colitis and related CRC by regulating intestinal stem cell proliferation and gut microbiota composition. Specifically, Aim2 mediates IL-18 secretion, which in turn regulates IL-22 secretion and increases the release of AMPs such as Reg3β and Reg3γ to maintain intestinal homeostasis. After Aim2 knockout, intestinal microbial composition is changed significantly (the abundance of Akkermansia, Muciniphila, and Anaeroplasma is increased, while the abundance of Anaerostipes, Bifidobacterium, Flexispira, Prevotella, and Paraprevotella is decreased), AKT activation and transcription of genes related to cell proliferation increased, further increasing the host susceptibility to colitis and related tumors [52,53]. | Akkermansia muciniphila ↑ Odoribacter ↑ Anaeroplasma ↑ Anaerostipes ↓ Flexispira ↓ Paraprevotella ↓ Bifidobacterium ↓ Prevotella ↓ Fusobacterium ↓ |
| RIG-I | RIG-I is a cytoplasmic RNA sensor. After Rig-I knockout, IgA secretion is decreased, STAT3 phosphorylation is decreased, and Reg3γ secretion is decreased, inducing a gut microbiota disorder, leading to colitis and related tumors. The species richness and diversity of the gut microbiota are increased in Rig-I−/− mice, but the bacterial species have not been elucidated. However, antibiotic-treated Rig-I−/− mice remain susceptible to colitis, suggesting that gut microbiota disorders are not the primary underlying cause of susceptibility [54]. Therefore, the role of gut microbiota in CRC needs to be further verified. | The species richness and diversity of intestinal microbiota are increased, but the bacterial species have not been characterized. |
| cGAS/Sting | cGAS/Sting is a cytoplasmic DNA sensor that can be activated by CDNs or cytoplasm dsDNA to recruit TBK1, phosphorylate IRF3 and NF-κB, and induce IFN-Ⅰ production. In Sting−/− mouse, intestinal mucosa secretion and IgA production are decreased, cup cells are decreased, the secretion of IL-1β and IL-10 is decreased, and the gut microbiota is altered (Allobacolum, Bifidobacterium, and Actinomycetes are decreased, while Disulfovibrio and Proteus are increased), increasing the susceptibility to DSS-induced colitis [55,56]. | Disulfovibrio↑ Proteus↑ Allobacolum ↓ Bifidobacterium ↓ Actinomycetes ↓ |
| CLRs | CLRs are localized at the cell surface that recognizes carbohydrates on the surface of fungal pathogens in a Ca2+-dependent manner to initiate an antifungal immune response. Loss of CLRs leads to the decreased activation of NF-κB, decreased production of cytokines such as IL-6, TNF-α, and Th17, decreased production of AMPs such as S100A8 and S100A9, decreased repair ability of intestinal epithelial tissue, decreased phagocytosis and bactericidal ability of macrophages, and altered gut microbiota (for example, Lactobacillus murinus and Lactobacillus Johnsonii Gal-2 are increased in the intestinal tract after Dectin-1 deletion, and Candida Tropicalis is increased in the intestinal tract after Dectin-3 deficiency.), further aggravating DSS-induced colitis [57,58]. | Dectin-1−/−: Lactobacillus murinus ↑ Lactobacillus johnsonii GAL-2 ↑ Dectin-3−/−: Candida tropicalis ↑ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qing, F.; Xie, T.; Xie, L.; Guo, T.; Liu, Z. How Gut Microbiota Are Shaped by Pattern Recognition Receptors in Colitis and Colorectal Cancer. Cancers 2022, 14, 3821. https://doi.org/10.3390/cancers14153821
Qing F, Xie T, Xie L, Guo T, Liu Z. How Gut Microbiota Are Shaped by Pattern Recognition Receptors in Colitis and Colorectal Cancer. Cancers. 2022; 14(15):3821. https://doi.org/10.3390/cancers14153821
Chicago/Turabian StyleQing, Furong, Tao Xie, Lu Xie, Tianfu Guo, and Zhiping Liu. 2022. "How Gut Microbiota Are Shaped by Pattern Recognition Receptors in Colitis and Colorectal Cancer" Cancers 14, no. 15: 3821. https://doi.org/10.3390/cancers14153821