
DEAD-Box RNA Helicases DDX3X and DDX5 as Oncogenes or Oncosuppressors: A Network Perspective


Abstract
:Simple Summary
Abstract
1. Introduction
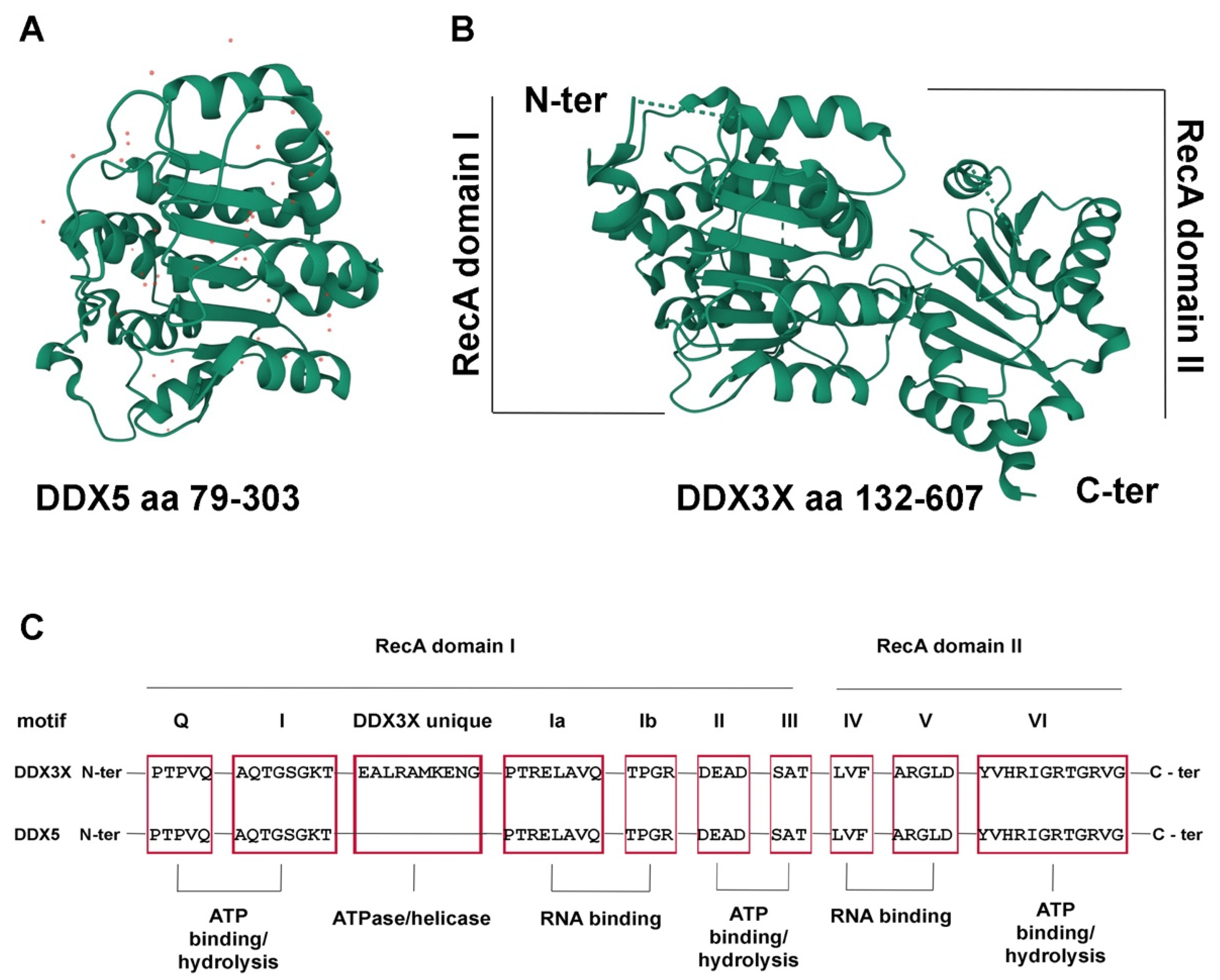
2. The DDX3X RNA Helicase
2.1. Breast Cancer
2.2. Colorectal Cancer
2.3. Lung Cancer
2.4. Hepatocellular Carcinoma
2.5. Prostate Cancer and Ewing Sarcoma
2.6. Oral Squamous Cell Carcinoma and Head and Neck Squamous Cell Carcinoma
2.7. Oncogenic Role of DDX3X in Other Cancers
2.8. Oncosuppressive Role of DDX3X in Other Cancers
3. The DDX5 RNA Helicase
3.1. DDX5: Oncogene or Suppressor?
3.2. Colon and Colorectal Cancer
3.3. Breast Cancer
3.4. Leukemia
3.5. Lung Cancer
3.6. Osteosarcoma
3.7. Prostate Cancer
3.8. Gastric Cancer
3.9. Glioblastoma
3.10. Cervical Cancer
3.11. Endometrial Cancer
3.12. Squamous Cell Carcinoma
3.13. Human Hepatocellular Carcinoma
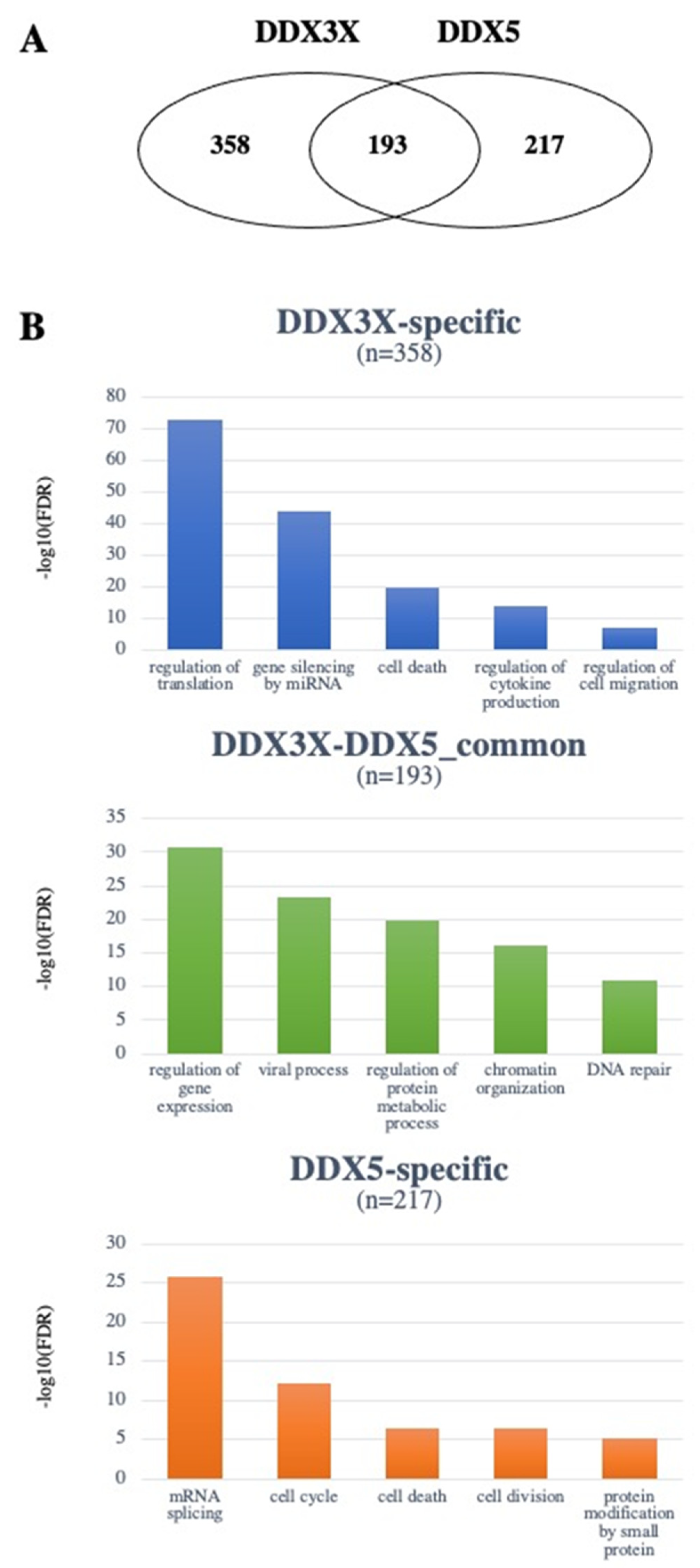
4. The DDX3X and DDX5 Interactome
4.1. DDX3X and DDX5 Share Common Interactors with Known Roles in Tumorigenesis
4.2. Unique Interactors of DDX3X
4.2.1. Breast Cancer
4.2.2. Colorectal Cancer
4.2.3. Oral Squamous Cell Carcinoma
4.2.4. Other Cancers
4.3. Unique Interactors of DDX5
4.3.1. Breast Cancer
4.3.2. Prostate Cancer
4.3.3. Leukemia
4.3.4. Hepatocellular Carcinoma HBV-Related
4.3.5. Glioblastoma
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Rocak, S.; Linder, P. DEAD-Box Proteins: The Driving Forces behind RNA Metabolism. Nat. Rev. Mol. Cell Biol. 2004, 5, 232–241. [Google Scholar] [CrossRef]
- Linder, P.; Jankowsky, E. From Unwinding to Clamping—The DEAD Box RNA Helicase Family. Nat. Rev. Mol. Cell Biol. 2011, 12, 505–516. [Google Scholar] [CrossRef] [Green Version]
- Dutta, S.; Gupta, G.; Choi, Y.W.; Kotaka, M.; Fielding, B.C.; Song, J.; Tan, Y.J. The variable N-terminal region of DDX5 contains structural elements and auto- inhibits its interaction with NS5B of hepatitis C virus. Biochem. J. 2012, 446, 37–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Floor, S.N.; Condon, K.J.; Sharma, D.; Jankowsky, E.; Doudna, J.A. Autoinhibitory Interdomain Interactions and Subfamily-specific Extensions Redefine the Catalytic Core of the Human DEAD-box Protein DDX3. J. Biol. Chem. 2016, 291, 2412–2421. [Google Scholar] [CrossRef] [Green Version]
- Sehnal, D.; Bittrich, S.; Deshpande, M.; Svobodová, R.; Berka, K.; Bazgier, V.; Velankar, S.; Burley, S.K.; Koča, J.; Rose, A.S. Mol* Viewer: Modern web app for 3D visualization and analysis of large biomolecular structures. Nucleic Acids Res. 2021, 49, W431–W437. [Google Scholar] [CrossRef]
- Jankowsky, E. RNA Helicases at Work: Binding and Rearranging. Trends Biochem. Sci. 2011, 36, 19–29. [Google Scholar] [CrossRef] [Green Version]
- Singleton, M.R.; Dillingham, M.S.; Wigley, D.B. Structure and Mechanism of Helicases and Nucleic Acid Translocases. Annu. Rev. Biochem. 2007, 76, 23–50. [Google Scholar] [CrossRef]
- Bourgeois, C.F.; Mortreux, F.; Auboeuf, D. The Multiple Functions of RNA Helicases as Drivers and Regulators of Gene Expression. Nat. Rev. Mol. Cell Biol. 2016, 17, 426–438. [Google Scholar] [CrossRef]
- Riva, V.; Maga, G. From the Magic Bullet to the Magic Target: Exploiting the Diverse Roles of DDX3X in Viral Infections and Tumorigenesis. Future Med. Chem. 2019, 11, 1357–1381. [Google Scholar] [CrossRef]
- Cargill, M.; Venkataraman, R.; Lee, S. DEAD-Box RNA Helicases and Genome Stability. Genes 2021, 12, 1471. [Google Scholar] [CrossRef] [PubMed]
- Naineni, S.K.; Robert, F.; Nagar, B.; Pelletier, J. Targeting DEAD-Box RNA Helicases: The Emergence of Molecular Staples. Wiley Interdiscip. Rev. RNA 2022, e1738. [Google Scholar] [CrossRef] [PubMed]
- Andrisani, O.; Liu, Q.; Kehn, P.; Leitner, W.W.; Moon, K.; Vazquez-Maldonado, N.; Fingerman, I.; Gale, M. Biological Functions of DEAD/DEAH-Box RNA Helicases in Health and Disease. Nat. Immunol. 2022, 23, 354–357. [Google Scholar] [CrossRef]
- Choi, Y.J.; Lee, S.G. The DEAD-Box RNA Helicase DDX3 Interacts with DDX5, Co-Localizes with It in the Cytoplasm during the G2/M Phase of the Cycle, and Affects Its Shuttling during MRNP Export. J. Cell. Biochem. 2012, 113, 985–996. [Google Scholar] [CrossRef] [PubMed]
- Ali, M.A.M. The DEAD-Box Protein Family of RNA Helicases: Sentinels for a Myriad of Cellular Functions with Emerging Roles in Tumorigenesis. Int. J. Clin. Oncol. 2021, 26, 795–825. [Google Scholar] [CrossRef]
- Kim, Y.S.; Lee, S.G.; Park, S.H.; Song, K. Gene Structure of the Human DDX3 and Chromosome Mapping of Its Related Sequences. Mol. Cells 2001, 12, 209–214. [Google Scholar]
- Ali, M.A.M. DEAD-Box RNA Helicases: The Driving Forces behind RNA Metabolism at the Crossroad of Viral Replication and Antiviral Innate Immunity. Virus Res. 2021, 296, 198352. [Google Scholar] [CrossRef] [PubMed]
- Riva, V.; Garbelli, A.; Casiraghi, F.; Arena, F.; Trivisani, C.I.; Gagliardi, A.; Bini, L.; Schroeder, M.; Maffia, A.; Sabbioneda, S.; et al. Novel Alternative Ribonucleotide Excision Repair Pathways in Human Cells by DDX3X and Specialized DNA Polymerases. Nucleic Acids Res. 2020, 48, 11551–11565. [Google Scholar] [CrossRef] [PubMed]
- Mo, J.; Liang, H.; Su, C.; Li, P.; Chen, J.; Zhang, B. DDX3X: Structure, Physiologic Functions and Cancer. Mol. Cancer 2021, 20, 38. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Zhang, D.; Yang, Y.; Wang, X.; Zhao, X.; Zhang, P.; Zhu, H.; Xu, N.; Liang, S.A. Double-edged function of DDX3, as an oncogene or tumor suppressor, in cancer progression (Review). Oncol Rep. 2018, 39, 883–892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cannizzaro, E.; Bannister, A.J.; Han, N.; Alendar, A.; Kouzarides, T. DDX3X RNA Helicase Affects Breast Cancer Cell Cycle Progression by Regulating Expression of KLF4. FEBS Lett. 2018, 592, 2308–2322. [Google Scholar] [CrossRef] [PubMed]
- Botlagunta, M.; Krishnamachary, B.; Vesuna, F.; Winnard, P.T.; Bol, G.M.; Patel, A.H.; Raman, V. Expression of DDX3 Is Directly Modulated by Hypoxia Inducible Factor-1 Alpha in Breast Epithelial Cells. PLoS ONE 2011, 6, e17563. [Google Scholar] [CrossRef]
- Pardeshi, J.; McCormack, N.; Gu, L.; Ryan, C.S.; Schröder, M. DDX3X Functionally and Physically Interacts with Estrogen Receptor-Alpha. Biochim. Biophys. Acta Gene Regul. Mech. 2022, 1865, 194787. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.W.; Lin, P.L.; Wang, L.; Huang, C.C.; Lee, H. The YAP1/SIX2 Axis Is Required for DDX3-Mediated Tumor Aggressiveness and Cetuximab Resistance in KRAS-Wild-Type Colorectal Cancer. Theranostics 2017, 7, 1114–1132. [Google Scholar] [CrossRef]
- Wu, D.W.; Lin, P.L.; Cheng, Y.W.; Huang, C.C.; Wang, L.; Lee, H. DDX3 Enhances Oncogenic KRAS-induced Tumor Invasion in Colorectal Cancer via the Β-catenin/ZEB1 Axis. Oncotarget 2016, 7, 22687–22699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, C.Y.; Lin, T.C.; Lin, Y.F.; Chen, M.H.; Lee, C.H.; Wang, H.Y.; Lee, Y.C.; Liu, Y.P.; Chen, C.L.; Hsiao, M. DDX3 as a Strongest Prognosis Marker and Its Downregulation Promotes Metastasis in Colorectal Cancer. Oncotarget 2015, 6, 18602–18612. [Google Scholar] [CrossRef] [Green Version]
- Bol, G.M.; Vesuna, F.; Xie, M.; Zeng, J.; Aziz, K.; Gandhi, N.; Levine, A.; Irving, A.; Korz, D.; Tantravedi, S.; et al. Targeting DDX3 with a Small Molecule Inhibitor for Lung Cancer Therapy. EMBO Mol. Med. 2015, 7, 648–669. [Google Scholar] [CrossRef] [PubMed]
- Guan, H.; Zhu, T.; Wu, S.; Liu, S.; Liu, B.; Wu, J.; Cai, J.; Zhu, X.; Zhang, X.; Zeng, M.; et al. Long Noncoding RNA LINC00673-v4 Promotes Aggressiveness of Lung Adenocarcinoma via Activating WNT/β-Catenin Signaling. Proc. Natl. Acad. Sci. USA 2019, 116, 14019–14028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, D.W.; Liu, W.S.; Wang, J.; Chen, C.Y.; Cheng, Y.W.; Lee, H. Reduced P21(WAF1/CIP1) via Alteration of P53-DDX3 Pathway Is Associated with Poor Relapse-Free Survival in Early-Stage Human Papillomavirus-Associated Lung Cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2011, 17, 1895–1905. [Google Scholar] [CrossRef] [Green Version]
- Wu, D.W.; Lee, M.C.; Wang, J.; Chen, C.Y.; Cheng, Y.W.; Lee, H. DDX3 Loss by P53 Inactivation Promotes Tumor Malignancy via the MDM2/Slug/E-Cadherin Pathway and Poor Patient Outcome in Non-Small-Cell Lung Cancer. Oncogene 2014, 33, 1515–1526. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.S.; Chao, C.C.; Su, T.L.; Yeh, S.H.; Chen, D.S.; Chen, C.T.; Chen, P.J.; Jou, Y.S. Diverse Cellular Transformation Capability of Overexpressed Genes in Human Hepatocellular Carcinoma. Biochem. Biophys. Res. Commun. 2004, 315, 950–958. [Google Scholar] [CrossRef] [PubMed]
- Chang, P.C.; Chi, C.W.; Chau, G.Y.; Li, F.Y.; Tsai, Y.H.; Wu, J.C.; Wu Lee, Y.H. DDX3, a DEAD Box RNA Helicase, Is Deregulated in Hepatitis Virus-Associated Hepatocellular Carcinoma and Is Involved in Cell Growth Control. Oncogene 2006, 25, 1991–2003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chao, C.H.; Chen, C.M.; Cheng, P.L.; Shih, J.W.; Tsou, A.P.; Wu Lee, Y.H. DDX3, a DEAD Box RNA Helicase with Tumor Growth-Suppressive Property and Transcriptional Regulation Activity of the P21waf1/Cip1 Promoter, Is a Candidate Tumor Suppressor. Cancer Res. 2006, 66, 6579–6588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.K.; Mai, R.T.; Huang, H.-D.; Chou, C.H.; Chang, Y.A.; Chang, Y.W.; You, L.R.; Chen, C.M.; Lee, Y.H.W. DDX3 Represses Stemness by Epigenetically Modulating Tumor-Suppressive MiRNAs in Hepatocellular Carcinoma. Sci. Rep. 2016, 6, 28637. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Shen, G.-H.; Xie, J.-M.; Li, B.; Gao, Q.-G. Rottlerin Upregulates DDX3 Expression in Hepatocellular Carcinoma. Biochem. Biophys. Res. Commun. 2018, 495, 1503–1509. [Google Scholar] [CrossRef]
- Xie, M.; Vesuna, F.; Tantravedi, S.; Bol, G.M.; Van Voss, M.R.H.; Nugent, K.; Malek, R.; Gabrielson, K.; Van Diest, P.J.; Tran, P.T.; et al. RK-33 Radiosensitizes Prostate Cancer Cells by Blocking the RNA Helicase DDX3. Cancer Res. 2016, 76, 6340–6350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilky, B.A.; Kim, C.; McCarty, G.; Montgomery, E.A.; Kammers, K.; Devine, L.R.; Cole, R.N.; Raman, V.; Loeb, D.M. RNA Helicase DDX3: A Novel Therapeutic Target in Ewing Sarcoma. Oncogene 2016, 35, 2574–2583. [Google Scholar] [CrossRef] [PubMed]
- Vellky, J.E.; Ricke, E.A.; Huang, W.; Ricke, W.A. Expression and Localization of DDX3 in Prostate Cancer Progression and Metastasis. Am. J. Pathol. 2019, 189, 1256–1267. [Google Scholar] [CrossRef] [PubMed]
- Heerma van Voss, M.R.; van Kempen, P.M.W.; Noorlag, R.; van Diest, P.J.; Willems, S.M.; Raman, V. DDX3 Has Divergent Roles in Head and Neck Squamous Cell Carcinomas in Smoking versus Non-Smoking Patients. Oral Dis. 2015, 21, 270–271. [Google Scholar] [CrossRef]
- Lee, C.H.; Lin, S.H.; Yang, S.F.; Yang, S.M.; Chen, M.K.; Lee, H.; Ko, J.L.; Chen, C.J.; Yeh, K.T. Low/Negative Expression of DDX3 Might Predict Poor Prognosis in Non-Smoker Patients with Oral Cancer. Oral Dis. 2014, 20, 76–83. [Google Scholar] [CrossRef]
- Chen, H.H.; Yu, H.I.; Rudy, R.; Lim, S.L.; Chen, Y.F.; Wu, S.H.; Lin, S.C.; Yang, M.H.; Tarn, W.Y. DDX3 Modulates the Tumor Microenvironment via Its Role in Endoplasmic Reticulum-Associated Translation. iScience 2021, 24, 103086. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.H.; Yu, H.I.; Yang, M.H.; Tarn, W.Y. DDX3 Activates CBC-EIF3-Mediated Translation of UORF-Containing Oncogenic MRNAs to Promote Metastasis in HNSCC. Cancer Res. 2018, 78, 4512–4523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, T.C. DDX3X Multifunctionally Modulates Tumor Progression and Serves as a Prognostic Indicator to Predict Cancer Outcomes. Int. J. Mol. Sci. 2020, 21, 281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hueng, D.Y.; Tsai, W.C.; Chiou, H.Y.C.; Feng, S.W.; Lin, C.; Li, Y.F.; Huang, L.C.; Lin, M.H. DDX3X Biomarker Correlates with Poor Survival in Human Gliomas. Int. J. Mol. Sci. 2015, 16, 15578–15591. [Google Scholar] [CrossRef] [Green Version]
- Tantravedi, S.; Vesuna, F.; Winnard, P.T.; Martin, A.; Lim, M.; Eberhart, C.G.; Berlinicke, C.; Raabe, E.; van Diest, P.J.; Raman, V. Targeting DDX3 in Medulloblastoma Using the Small Molecule Inhibitor RK-33. Transl. Oncol. 2019, 12, 96–105. [Google Scholar] [CrossRef] [PubMed]
- Pugh, T.J.; Weeraratne, S.D.; Archer, T.C.; Pomeranz Krummel, D.A.; Auclair, D.; Bochicchio, J.; Carneiro, M.O.; Carter, S.L.; Cibulskis, K.; Erlich, R.L.; et al. Medulloblastoma Exome Sequencing Uncovers Subtype-Specific Somatic Mutations. Nature 2012, 488, 106–110. [Google Scholar] [CrossRef]
- Valentin-Vega, Y.A.; Wang, Y.D.; Parker, M.; Patmore, D.M.; Kanagaraj, A.; Moore, J.; Rusch, M.; Finkelstein, D.; Ellison, D.W.; Gilbertson, R.J.; et al. Cancer-Associated DDX3X Mutations Drive Stress Granule Assembly and Impair Global Translation. Sci. Rep. 2016, 6, 25996. [Google Scholar] [CrossRef] [PubMed]
- Miao, X.; Yang, Z.; Xiong, L.; Zou, Q.; Yuan, Y.; Li, J.; Liang, L.; Chen, M.; Chen, S. Nectin-2 and DDX3 Are Biomarkers for Metastasis and Poor Prognosis of Squamous Cell/Adenosquamous Carcinomas and Adenocarcinoma of Gallbladder. Int. J. Clin. Exp. Pathol. 2013, 6, 179–190. [Google Scholar]
- Liang, S.; Yang, Z.; Li, D.; Miao, X.; Yang, L.; Zou, Q.; Yuan, Y. The Clinical and Pathological Significance of Nectin-2 and DDX3 Expression in Pancreatic Ductal Adenocarcinomas. Dis. Markers 2015, 2015, 379568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, Y.; Xu, B.; Zhao, Y.; Yang, S.; Wang, S.; Ma, L.; Dong, L. DEAD-Box Helicase 3 X-Linked Promotes Metastasis by Inducing Epithelial-Mesenchymal Transition via P62/Sequestosome-1. Dig. Dis. Sci. 2021, 66, 3893–3902. [Google Scholar] [CrossRef] [PubMed]
- Phung, B.; Cieśla, M.; Sanna, A.; Guzzi, N.; Beneventi, G.; Cao Thi Ngoc, P.; Lauss, M.; Cabrita, R.; Cordero, E.; Bosch, A.; et al. The X-Linked DDX3X RNA Helicase Dictates Translation Reprogramming and Metastasis in Melanoma. Cell Rep. 2019, 27, 3573–3586.E7. [Google Scholar] [CrossRef] [Green Version]
- Alkallas, R.; Lajoie, M.; Moldoveanu, D.; Hoang, K.V.; Lefrançois, P.; Lingrand, M.; Ahanfeshar-Adams, M.; Watters, K.; Spatz, A.; Zippin, J.H.; et al. Multi-Omic Analysis Reveals Significantly Mutated Genes and DDX3X as a Sex-Specific Tumor Suppressor in Cutaneous Melanoma. Nat. Cancer 2020, 1, 635–652. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Gu, Z.H.; Yan, Z.X.; Zhao, X.; Xie, Y.Y.; Zhang, Z.G.; Pan, C.M.; Hu, Y.; Cai, C.P.; Dong, Y.; et al. Exome Sequencing Identifies Somatic Mutations of DDX3X in Natural Killer/T-Cell Lymphoma. Nat. Genet. 2015, 47, 1061–1066. [Google Scholar] [CrossRef] [PubMed]
- Kizhakeyil, A.; Zaini, N.B.M.; Poh, Z.S.; Wong, B.H.S.; Loh, X.; Ng, A.S.; Low, Z.S.; Prasannan, P.; Gong, C.; Tan, M.G.K.; et al. DDX3X Loss Is an Adverse Prognostic Marker in Diffuse Large B-Cell Lymphoma and Is Associated with Chemoresistance in Aggressive Non-Hodgkin Lymphoma Subtypes. Mol. Cancer 2021, 20, 134. [Google Scholar] [CrossRef] [PubMed]
- Iggo, R.; Gough, A.; Xu, W.; Lane, D.P.; Spurr, N.K. Chromosome mapping of the human gene encoding the 68-kDa nuclear antigen (p68) by using the polymerase chain reaction. Proc. Natl. Acad. Sci. USA 1989, 86, 6211–6214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Y.; Liu, Z.R. The ATPase, RNA Unwinding, and RNA Binding Activities of Recombinant P68 RNA Helicase. J. Biol. Chem. 2002, 277, 12810–12815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, F.; Fountzilas, C.; Puzanov, I.; Attwood, K.M.; Morrison, C.; Ling, X. Multiple Functions of the DEAD-Box RNA Helicase, DDX5 (P68), Make DDX5 a Superior Oncogenic Biomarker and Target for Targeted Cancer Therapy. Am. J. Cancer Res. 2021, 11, 5190–5213. [Google Scholar]
- Yang, L.; Lin, C.; Liu, Z.R. Phosphorylations of DEAD Box P68 RNA Helicase Are Associated with Cancer Development and Cell Proliferation. Mol. Cancer Res. MCR 2005, 3, 355–363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, L.; Lin, C.; Sun, S.Y.; Zhao, S.; Liu, Z.R. A Double Tyrosine Phosphorylation of P68 RNA Helicase Confers Resistance to TRAIL-Induced Apoptosis. Oncogene 2007, 26, 6082–6092. [Google Scholar] [CrossRef] [Green Version]
- Janknecht, R. Multi-Talented DEAD-Box Proteins and Potential Tumor Promoters: P68 RNA Helicase (DDX5) and Its Paralog, P72 RNA Helicase (DDX17). Am. J. Transl. Res. 2010, 2, 223–234. [Google Scholar] [PubMed]
- Dey, H.; Liu, Z.R. Phosphorylation of P68 RNA Helicase by P38 MAP Kinase Contributes to Colon Cancer Cells Apoptosis Induced by Oxaliplatin. BMC Cell Biol. 2012, 13, 27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, T.Y.; Cao, L.; Yang, Z.C.; Li, Y.S.; Tan, L.; Ran, X.Z.; Shi, C.M. P68 RNA Helicase as a Molecular Target for Cancer Therapy. J. Exp. Clin. Cancer Res. 2014, 33, 64. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, M.; Ghosh, M.K. DEAD Box RNA Helicases: Crucial Regulators of Gene Expression and Oncogenesis. Front. Biosci. (Landmark Ed) 2016, 21, 225–250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarkar, M.; Khare, V.; Guturi, K.K.N.; Das, N.; Ghosh, M.K. The DEAD Box Protein P68: A Crucial Regulator of AKT/FOXO3a Signaling Axis in Oncogenesis. Oncogene 2015, 34, 5843–5856. [Google Scholar] [CrossRef] [PubMed]
- Khare, V.; Tabassum, S.; Chatterjee, U.; Chatterjee, S.; Ghosh, M.K. RNA Helicase P68 Deploys β-Catenin in Regulating RelA/P65 Gene Expression: Implications in Colon Cancer. J. Exp. Clin. Cancer Res. 2019, 38, 330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, N.; Jiang, M.; Han, Y.; Liu, H.; Chu, Y.; Liu, H.; Cao, J.; Hou, Q.; Zhao, Y.; Xu, B.; et al. O-GlcNAcylation Promotes Colorectal Cancer Progression by Regulating Protein Stability and Potential Catcinogenic Function of DDX5. J. Cell. Mol. Med. 2019, 23, 1354–1362. [Google Scholar] [CrossRef] [PubMed]
- Dai, L.; Pan, G.; Liu, X.; Huang, J.; Jiang, Z.; Zhu, X.; Gan, X.; Xu, Q.; Tan, N. High Expression of ALDOA and DDX5 Are Associated with Poor Prognosis in Human Colorectal Cancer. Cancer Manag. Res. 2018, 10, 1799–1806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, K.; Zhao, G.; Yuan, H.; Zhang, J.; Li, Q.; Gong, D.; Lin, P. Upregulated Expression of DDX5 Predicts Recurrence and Poor Prognosis in Breast Cancer. Pathol. Res. Pract. 2022, 229, 153736. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Huang, J.; Hu, Z. RNA Helicase DDX5 Regulates MicroRNA Expression and Contributes to Cytoskeletal Reorganization in Basal Breast Cancer Cells. Mol. Cell. Proteom. MCP 2012, 11, M111.011932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guturi, N.K.N.; Sarkar, M.; Bhowmik, A.; Das, N.; Ghosh, K.K. DEAD-Box Protein P68 Is Regulated by β-Catenin/Transcription Factor 4 to Maintain a Positive Feedback Loop in Control of Breast Cancer Progression. Breast Cancer Res. BCR 2014, 16, 496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazurek, A.; Park, Y.; Miething, C.; Wilkinson, J.E.; Gillis, J.; Lowe, S.W.; Vakoc, C.R.; Stillman, B. Acquired Dependence of Acute Myeloid Leukemia on the DEAD-Box RNA Helicase DDX5. Cell Rep. 2014, 7, 1887–1899. [Google Scholar] [CrossRef] [Green Version]
- Lin, S.; Tian, L.; Shen, H.; Gu, Y.; Li, J.L.; Chen, Z.; Sun, X.; James You, M.; Wu, L. DDX5 Is a Positive Regulator of Oncogenic NOTCH1 Signaling in T Cell Acute Lymphoblastic Leukemia. Oncogene 2013, 32, 4845–4853. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; You, Y.Q.; Ma, Y.X.; Kang, Y.H.; Wu, T.; Wu, X.J.; Hu, X.X.; Meng, Q.H.; Huang, Y.; Zhang, N.; et al. DDX5-Targeting Fully Human Monoclonal Autoantibody Inhibits Proliferation and Promotes Differentiation of Acute Promyelocytic Leukemia Cells by Increasing ROS Production. Cell Death Dis. 2020, 11, 552. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Luo, Z.; Zhou, L.; Li, X.; Jiang, T.; Fu, E. DDX5 Promotes Proliferation and Tumorigenesis of Non-Small-Cell Lung Cancer Cells by Activating β-Catenin Signaling Pathway. Cancer Sci. 2015, 106, 1303–1312. [Google Scholar] [CrossRef]
- Xing, Z.; Russon, M.P.; Utturkar, S.M.; Tran, E.J. The RNA Helicase DDX5 Supports Mitochondrial Function in Small Cell Lung Cancer. J. Biol. Chem. 2020, 295, 8988–8998. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Zhang, C.; Wang, X. Long Noncoding RNA DLEU1 Aggravates Osteosarcoma Carcinogenesis via Regulating the MiR-671-5p/DDX5 Axis. Artif. Cells Nanomed. Biotechnol. 2019, 47, 3322–3328. [Google Scholar] [CrossRef] [Green Version]
- You, Z.; Liu, C.; Wang, C.; Ling, Z.; Wang, Y.; Wang, Y.; Zhang, M.; Chen, S.; Xu, B.; Guan, H.; et al. LncRNA CCAT1 Promotes Prostate Cancer Cell Proliferation by Interacting with DDX5 and MIR-28-5P. Mol. Cancer Ther. 2019, 18, 2469–2479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, B.; Mehra, R.; Dhanasekaran, S.M.; Yu, J.; Menon, A.; Lonigro, R.J.; Wang, X.; Gong, Y.; Wang, L.; Shankar, S.; et al. A Fluorescence in Situ Hybridization Screen for E26 Transformation-Specific Aberrations: Identification of DDX5-ETV4 Fusion Protein in Prostate Cancer. Cancer Res. 2008, 68, 7629–7637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, C.; Li, D.Q.; Li, N.; Chen, L.; Li, S.S.; Yang, Y.; Hou, M.X.; Xie, M.J.; Zheng, Z.D. DDX5 Promotes Gastric Cancer Cell Proliferation in Vitro and in Vivo through MTOR Signaling Pathway. Sci. Rep. 2017, 7, 42876. [Google Scholar] [CrossRef] [PubMed]
- Sha, M.; Lin, M.; Wang, J.; Ye, J.; Xu, J.; Xu, N.; Huang, J. Long Non-Coding RNA MIAT Promotes Gastric Cancer Growth and Metastasis through Regulation of MiR-141/DDX5 Pathway. J. Exp. Clin. Cancer Res. 2018, 37, 58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, R.; Jiao, Z.; Li, R.; Yue, H.; Chen, L. P68 RNA Helicase Promotes Glioma Cell Proliferation in Vitro and in Vivo via Direct Regulation of NF-ΚB Transcription Factor P50. Neuro-Oncology 2012, 14, 1116–1124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, T.; Cao, L.; Dong, X.; Wu, F.; De, W.; Huang, L.; Wan, Q. LINC01116 Promotes Tumor Proliferation and Neutrophil Recruitment via DDX5-Mediated Regulation of IL-1β in Glioma Cell. Cell Death Dis. 2020, 11, 302. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Bao, H.B.; Du, W.Z.; Chen, X.F.; Liu, H.L.; Han, D.Y.; Wang, L.G.; Wu, J.N.; Wang, C.L.; Yang, M.C.; et al. P68 RNA Helicase Promotes Invasion of Glioma Cells through Negatively Regulating DUSP5. Cancer Sci. 2019, 110, 107–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, M.Y.; Liu, J.Q.; Chen, D.P.; Li, Z.Y.; Qi, B.; Yin, W.J.; He, L. P68 Prompts the Epithelial-Mesenchymal Transition in Cervical Cancer Cells by Transcriptionally Activating the TGF-β1 Signaling Pathway. Oncol. Lett. 2018, 15, 2111–2116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, C.; Wang, L.; Jiang, Q.; Zhang, J.; Zhu, L.; Lin, L.; Jiang, H.; Lin, D.; Xiao, Y.; Fang, W.; et al. Hepatoma-Derived Growth Factor and DDX5 Promote Carcinogenesis and Progression of Endometrial Cancer by Activating β-Catenin. Front. Oncol. 2019, 9, 211. [Google Scholar] [CrossRef] [Green Version]
- Beier, H.U.; Maune, S.; Meyer, J.E.; Gorogh, T. Overexpression of P68 MRNA in Head and Neck Squamous Cell Carcinoma Cells. Anticancer Res. 2006, 26, 1941–1946. [Google Scholar]
- Ma, L.; Zhao, X.; Wang, S.; Zheng, Y.; Yang, S.; Hou, Y.; Zou, B.; Dong, L. Decreased Expression of DEAD-Box Helicase 5 Inhibits Esophageal Squamous Cell Carcinomas by Regulating Endoplasmic Reticulum Stress and Autophagy. Biochem. Biophys. Res. Commun. 2020, 533, 1449–1456. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Zhang, Y.; Zhu, X.; Chen, C.; Zhang, C.; Xia, Y.; Zhao, Y.; Andrisani, O.; Kong, L. DEAD Box Protein 5 Inhibits Liver Tumorigenesis by Stimulating Autophagy via Interaction with P62/SQSTM1. Hepatology 2019, 69, 1046–1063. [Google Scholar] [CrossRef] [PubMed]
- Croce, C.M. Causes and Consequences of MicroRNA Dysregulation in Cancer. Nat. Rev. Genet. 2009, 10, 704–714. [Google Scholar] [CrossRef] [PubMed]
- Fuller-Pace, F.V. DEAD Box RNA Helicase Functions in Cancer. RNA Biol. 2013, 10, 121–132. [Google Scholar] [CrossRef] [Green Version]
- Kitagawa, N.; Ojima, H.; Shirakihara, T.; Shimizu, H.; Kokubu, A.; Urushidate, T.; Totoki, Y.; Kosuge, T.; Miyagawa, S.; Shibata, T. Downregulation of the MicroRNA Biogenesis Components and Its Association with Poor Prognosis in Hepatocellular Carcinoma. Cancer Sci. 2013, 104, 543–551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Xing, Z.; Mani, S.K.K.; Bancel, B.; Durantel, D.; Zoulim, F.; Tran, E.J.; Merle, P.; Andrisani, O. RNA Helicase DEAD Box Protein 5 Regulates Polycomb Repressive Complex 2/Hox Transcript Antisense Intergenic RNA Function in Hepatitis B Virus Infection and Hepatocarcinogenesis. Hepatology 2016, 64, 1033–1048. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.C.; Juan, C.W.; Chen, K.Y.; Chang, Y.C.; Lee, J.C.; Chang, M.C. Upregulation of RPA2 Promotes NF-ΚB Activation in Breast Cancer by Relieving the Antagonistic Function of Menin on NF-ΚB-Regulated Transcription. Carcinogenesis 2017, 38, 196–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, Y.; Liu, G.; Wang, H.; Tian, Z.; Cai, Z.; Zhang, F.; Luo, Y.; Wang, S.; Guo, G.; Wang, X.; et al. Valproic Acid Sensitizes Breast Cancer Cells to Hydroxyurea through Inhibiting RPA2 Hyperphosphorylation-Mediated DNA Repair Pathway. DNA Repair 2017, 58, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Oh, S.; Oh, C.; Yoo, K.H. Functional Roles of CTCF in Breast Cancer. BMB Rep. 2017, 50, 445–453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akhtar, M.S.; Akhter, N.; Najm, M.Z.; Deo, S.V.S.; Shukla, N.K.; Almalki, S.S.R.; Alharbi, R.A.; Sindi, A.A.A.; Alruwetei, A.; Ahmad, A.; et al. Association of Mutation and Low Expression of the CTCF Gene with Breast Cancer Progression. Saudi Pharm. J. 2020, 28, 607–614. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.; Pelletier, J. The Biology of DHX9 and Its Potential as a Therapeutic Target. Oncotarget 2016, 7, 42716–42739. [Google Scholar] [CrossRef] [Green Version]
- Koirala, P.; Huang, J.; Ho, T.T.; Wu, F.; Ding, X.; Mo, Y.Y. LncRNA AK023948 Is a Positive Regulator of AKT. Nat. Commun. 2017, 8, 14422. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.F.; Zhang, Z.; Zhang, M.; Chen, Y.S.; Song, J.; Hou, P.F.; Yong, H.M.; Zheng, J.N.; Bai, J. CUL1 Promotes Breast Cancer Metastasis through Regulating EZH2-Induced the Autocrine Expression of the Cytokines CXCL8 and IL11. Cell Death Dis. 2019, 10, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fay, M.J.; Longo, K.A.; Karathanasis, G.A.; Shope, D.M.; Mandernach, C.J.; Leong, J.R.; Hicks, A.; Pherson, K.; Husain, A. Analysis of CUL-5 Expression in Breast Epithelial Cells, Breast Cancer Cell Lines, Normal Tissues and Tumor Tissues. Mol. Cancer 2003, 2, 40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kong, X.; Li, J.; Li, Y.; Duan, W.; Qi, Q.; Wang, T.; Yang, Q.; Du, L.; Mao, H.; Wang, C. A Novel Long Non-Coding RNA AC073352.1 Promotes Metastasis and Angiogenesis via Interacting with YBX1 in Breast Cancer. Cell Death Dis. 2021, 12, 670. [Google Scholar] [CrossRef] [PubMed]
- Slattery, M.L.; Lundgreen, A.; Herrick, J.S.; Wolff, R.K. Genetic Variation in RPS6KA1, RPS6KA2, RPS6KB1, RPS6KB2, and PDK1 and Risk of Colon or Rectal Cancer. Mutat. Res. 2011, 706, 13–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ilozumba, M.N.; Yao, S.; Llanos, A.A.M.; Omilian, A.R.; Zhang, W.; Datta, S.; Hong, C.-C.; Davis, W.; Khoury, T.; Bandera, E.V.; et al. MTOR Pathway Gene Expression in Association with Race and Clinicopathological Characteristics in Black and White Breast Cancer Patients. Discov. Oncol. 2022, 13, 34. [Google Scholar] [CrossRef] [PubMed]
- Salmans, M.L.; Zhao, F.; Andersen, B. The Estrogen-Regulated Anterior Gradient 2 (AGR2) Protein in Breast Cancer: A Potential Drug Target and Biomarker. Breast Cancer Res. 2013, 15, 204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kereh, D.S.; Pieter, J.; Hamdani, W.; Haryasena, H.; Sampepajung, D.; Prihantono, P. Correlation of AGR2 Expression with the Incidence of Metastasis in Luminal Breast Cancer. Breast Dis. 2021, 40, S103–S107. [Google Scholar] [CrossRef]
- Warowicka, A.; Broniarczyk, J.; Węglewska, M.; Kwaśniewski, W.; Goździcka-Józefiak, A. Dual Role of YY1 in HPV Life Cycle and Cervical Cancer Development. Int. J. Mol. Sci. 2022, 23, 3453. [Google Scholar] [CrossRef]
- Li, Y.; Liu, Q.; McGrail, D.J.; Dai, H.; Li, K.; Lin, S. CHD4 Mutations Promote Endometrial Cancer Stemness by Activating TGF-Beta Signaling. Am. J. Cancer Res. 2018, 8, 903–914. [Google Scholar] [PubMed]
- Schatz, N.; Brändlein, S.; Rückl, K.; Hensel, F.; Vollmers, H.P. Diagnostic and Therapeutic Potential of a Human Antibody Cloned from a Cancer Patient That Binds to a Tumor-Specific Variant of Transcription Factor TAF15. Cancer Res. 2010, 70, 398–408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, X.; Zhang, H.; Tang, S.; Zheng, X.; Jiang, L. Bioinformatics Analysis to Reveal Potential Differentially Expressed Long Non-Coding RNAs and Genes Associated with Tumour Metastasis in Lung Adenocarcinoma. OncoTargets Ther. 2020, 13, 3197–3207. [Google Scholar] [CrossRef] [Green Version]
- Nho, S.H.; Yoon, G.; Seo, J.H.; Oh, H.N.; Cho, S.S.; Kim, H.; Choi, H.W.; Shim, J.H.; Chae, J. Il Licochalcone H Induces the Apoptosis of Human Oral Squamous Cell Carcinoma Cells via Regulation of Matrin 3. Oncol. Rep. 2019, 41, 333–340. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.O.; Pioro, E.P.; Boehringer, A.; Chia, R.; Feit, H.; Renton, A.E.; Pliner, H.A.; Abramzon, Y.; Marangi, G.; Winborn, B.J.; et al. Mutations in the Matrin 3 Gene Cause Familial Amyotrophic Lateral Sclerosis. Nat. Neurosci. 2014, 17, 664–666. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Zhang, L.; Wang, D.; Bao, Y.; Liu, C.; Xu, Z.; Huang, W.; Cheng, C. Regulation of Glioma Cells Migration by DYRK2. Neurochem. Res. 2017, 42, 3093–3102. [Google Scholar] [CrossRef] [PubMed]
- Agnihotri, S.; Golbourn, B.; Huang, X.; Remke, M.; Younger, S.; Cairns, R.A.; Chalil, A.; Smith, C.A.; Krumholtz, S.L.; Mackenzie, D.; et al. PINK1 Is a Negative Regulator of Growth and the Warburg Effect in Glioblastoma. Cancer Res. 2016, 76, 4708–4719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vu, L.P.; Prieto, C.; Amin, E.M.; Chhangawala, S.; Krivtsov, A.; Calvo-Vidal, M.N.; Chou, T.; Chow, A.; Minuesa, G.; Park, S.M.; et al. Functional Screen of MSI2 Interactors Identifies an Essential Role for SYNCRIP in Myeloid Leukemia Stem Cells. Nat. Genet. 2017, 49, 866–875. [Google Scholar] [CrossRef] [Green Version]
- Attia, H.R.M.; Ibrahim, M.H.; El-Aziz, S.H.A.; Hassan, N.M.; Osman, R.A.; Hagag, H.A.; Yassa, M.E.; Abdelrahman, A.H.; Salama, I.I.; Sobeih, M.E. ITGA4 Gene Methylation Status in Chronic Lymphocytic Leukemia. Futur. Sci. OA 2020, 6, FSO583. [Google Scholar] [CrossRef] [PubMed]
- Adler, D.; Offermann, A.; Braun, M.; Menon, R.; Syring, I.; Nowak, M.; Halbach, R.; Vogel, W.; Ruiz, C.; Zellweger, T.; et al. MED12 Overexpression Is a Frequent Event in Castration-Resistant Prostate Cancer. Endocr. Relat. Cancer 2014, 21, 663–675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Yang, L.; Shi, J.; Lu, Y.; Chen, X.; Yang, Z. The Oncogenic Role of CENPA in Hepatocellular Carcinoma Development: Evidence from Bioinformatic Analysis. Biomed. Res. Int. 2020, 2020, 3040839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, F.; Fang, E.; Mei, H.; Chen, Y.; Li, H.; Li, D.; Song, H.; Wang, J.; Hong, M.; Xiao, W.; et al. Cis-Acting Circ-CTNNB1 Promotes β-Catenin Signaling and Cancer Progression via DDX3-Mediated Transactivation of YY1. Cancer Res. 2019, 79, 557–571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeon, M.; You, D.; Bae, S.Y.; Kim, S.W.; Nam, S.J.; Kim, H.H.; Kim, S.; Lee, J.E. Dimerization of EGFR and HER2 Induces Breast Cancer Cell Motility through STAT1-Dependent ACTA2 Induction. Oncotarget 2016, 8, 50570–50581. [Google Scholar] [CrossRef] [Green Version]
- Hu, T.; Wei, H.; Xiao, E.; Long, M. Mechanism and Effect of Stress Granule Formation in Cancer and Its Potential Roles in Breast Cancer Therapy. Genes Dis. 2022, 3, 659–667. [Google Scholar] [CrossRef] [PubMed]
- Gupta, N.; Badeaux, M.; Liu, Y.; Naxerova, K.; Sgroi, D.; Munn, L.L.; Jain, R.K.; Garkavtsev, I. Stress Granule-Associated Protein G3BP2 Regulates Breast Tumor Initiation. Proc. Natl. Acad. Sci. USA 2017, 114, 1033–1038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, H.; Wang, W.; Mo, S.; Liu, Q.; Chen, X.; Chen, R.; Zhang, Y.; Zou, K.; Ye, M.; He, X.; et al. Long Non-Coding RNA SNHG14 Induces Trastuzumab Resistance of Breast Cancer via Regulating PABPC1 Expression through H3K27 Acetylation. J. Cell. Mol. Med. 2018, 22, 4935–4947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Copsey, A.C.; Cooper, S.; Parker, R.; Lineham, E.; Lapworth, C.; Jallad, D.; Sweet, S.; Morley, S.J. The Helicase, DDX3X, Interacts with Poly(A)-Binding Protein 1 (PABP1) and Caprin-1 at the Leading Edge of Migrating Fibroblasts and Is Required for Efficient Cell Spreading. Biochem. J. 2017, 474, 3109–3120. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Sheng, C.; Yin, Y.; Wen, S.; Yang, G.; Cheng, Z.; Zhu, Q. PABPC1 Interacts with AGO2 and Is Responsible for the MicroRNA Mediated Gene Silencing in High Grade Hepatocellular Carcinoma. Cancer Lett. 2015, 367, 49–57. [Google Scholar] [CrossRef] [PubMed]
- Feng, C.; Han, Y.H.; Qi, N.; Li, J.; Sheng, Q.H.; Liu, Y.; Yang, L.L. Functional Implications of PABPC1 in the Development of Ovarian Cancer. Open Med. 2021, 16, 805–815. [Google Scholar] [CrossRef] [PubMed]
- Yeh, C.H.; Bellon, M.; Nicot, C. FBXW7: A Critical Tumor Suppressor of Human Cancers. Mol. Cancer 2018, 17, 115. [Google Scholar] [CrossRef] [PubMed]
- Lai, M.-C.; Chang, W.-C.; Shieh, S.-Y.; Tarn, W.-Y. DDX3 Regulates Cell Growth through Translational Control of Cyclin E1. Mol. Cell. Biol. 2010, 30, 5444–5453. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Kokabee, L.; Kokabee, M.; Conklin, D.S. Bruton’s Tyrosine Kinase and Its Isoforms in Cancer. Front. Cell Dev. Biol. 2021, 9, 668996. [Google Scholar] [CrossRef]
- George, J.; Li, Y.; Kadamberi, I.P.; Parashar, D.; Tsaih, S.W.; Gupta, P.; Geethadevi, A.; Chen, C.; Ghosh, C.; Sun, Y.; et al. RNA-Binding Protein FXR1 Drives CMYC Translation by Recruiting EIF4F Complex to the Translation Start Site. Cell Rep. 2021, 37, 109934. [Google Scholar] [CrossRef] [PubMed]
- Jeon, S.-A.; Kim, D.W.; Lee, D.-B.; Cho, J.-Y. NEDD4 Plays Roles in the Maintenance of Breast Cancer Stem Cell Characteristics. Front. Oncol. 2020, 10, 1680. [Google Scholar] [CrossRef] [PubMed]
- Heinecke, J.L.; Ridnour, L.A.; Cheng, R.Y.S.; Switzer, C.H.; Lizardo, M.M.; Khanna, C.; Glynn, S.A.; Hussain, S.P.; Young, H.A.; Ambs, S.; et al. Tumor Microenvironment-Based Feed-Forward Regulation of NOS2 in Breast Cancer Progression. Proc. Natl. Acad. Sci. USA 2014, 111, 6323–6328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, E.J.; Kang, J.H.; Kim, Y.J.; Kim, S.; Lee, S.J. ICAM-1 Promotes Cancer Progression by Regulating SRC Activity as an Adapter Protein in Colorectal Cancer. Cell Death Dis. 2022, 13, 417. [Google Scholar] [CrossRef] [PubMed]
- Jin, W. Regulation of Src Family Kinases during Colorectal Cancer Development and Its Clinical Implications. Cancers 2020, 12, 1339. [Google Scholar] [CrossRef] [PubMed]
- Duan, L.; Wu, R.; Ye, L.; Wang, H.; Yang, X.; Zhang, Y.; Chen, X.; Zuo, G.; Zhang, Y.; Weng, Y.; et al. S100A8 and S100A9 Are Associated with Colorectal Carcinoma Progression and Contribute to Colorectal Carcinoma Cell Survival and Migration via Wnt/β-Catenin Pathway. PLoS ONE 2013, 8, e62092. [Google Scholar] [CrossRef]
- Ohkura, S.; Kondoh, N.; Hada, A.; Arai, M.; Yamazaki, Y.; Sindoh, M.; Takahashi, M.; Matsumoto, I.; Yamamoto, M. Differential Expression of the Keratin-4, -13, -14, -17 and Transglutaminase 3 Genes during the Development of Oral Squamous Cell Carcinoma from Leukoplakia. Oral Oncol. 2005, 41, 607–613. [Google Scholar] [CrossRef] [PubMed]
- Khanom, R.; Nguyen, C.T.K.; Kayamori, K.; Zhao, X.; Morita, K.; Miki, Y.; Katsube, K.I.; Yamaguchi, A.; Sakamoto, K. Keratin 17 Is Induced in Oral Cancer and Facilitates Tumor Growth. PLoS ONE 2016, 11, e0161163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Srivastava, S.S.; Alam, H.; Patil, S.J.; Shrinivasan, R.; Raikundalia, S.; Chaudhari, P.R.; Vaidya, M.M. Keratin 5/14-mediated Cell Differentiation and Transformation Are Regulated by TAp63 and Notch-1 in Oral Squamous Cell Carcinoma-derived Cells. Oncol. Rep. 2018, 39, 2393–2401. [Google Scholar] [CrossRef]
- Frohwitter, G.; Buerger, H.; van Diest, P.J.; Korsching, E.; Kleinheinz, J.; Fillies, T. Cytokeratin and Protein Expression Patterns in Squamous Cell Carcinoma of the Oral Cavity Provide Evidence for Two Distinct Pathogenetic Pathways. Oncol. Lett. 2016, 12, 107–113. [Google Scholar] [CrossRef] [Green Version]
- Han, W.; Hu, C.; Fan, Z.J.; Shen, G.L. Transcript Levels of Keratin 1/5/6/14/15/16/17 as Potential Prognostic Indicators in Melanoma Patients. Sci. Rep. 2021, 11, 1023. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Mao, Y.; Zhao, Y.; He, Y. DDX3X Promotes the Biogenesis of a Subset of MiRNAs and the Potential Roles They Played in Cancer Development. Sci. Rep. 2016, 6, 32739. [Google Scholar] [CrossRef]
- Yeung, T.L.; Tsai, C.C.; Leung, C.S.; Yeung, C.L.A.; Thompson, M.S.; Lu, K.H.; Freedman, R.S.; Birrer, M.J.; Wong, K.K.; Mok, S.C. ISG15 Promotes ERK1 ISGylation, CD8+ T Cell Activation and Suppresses Ovarian Cancer Progression. Cancers 2018, 10, 464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loureiro, M.E.; Zorzetto-Fernandes, A.L.; Radoshitzky, S.; Chi, X.; Dallari, S.; Marooki, N.; Lèger, P.; Foscaldi, S.; Harjono, V.; Sharma, S.; et al. DDX3 Suppresses Type I Interferons and Favors Viral Replication during Arenavirus Infection. PLoS Pathog. 2018, 14, e1007125. [Google Scholar] [CrossRef] [PubMed]
- Darb-Esfahani, S.; Sinn, B.V.; Rudl, M.; Sehouli, J.; Braicu, I.; Dietel, M.; Denkert, C. Interferon-Stimulated Gene, 15 KDa (ISG15) in Ovarian High-Grade Serous Carcinoma: Prognostic Impact and Link to NF-ΚB Pathway. Int. J. Gynecol. Pathol. 2014, 33, 16–22. [Google Scholar] [CrossRef]
- Minakawa, M.; Sone, T.; Takeuchi, T.; Yokosawa, H. Regulation of the Nuclear Factor (NF)-KappaB Pathway by ISGylation. Biol. Pharm. Bull. 2008, 31, 2223–2227. [Google Scholar] [CrossRef] [Green Version]
- Uno, M.; Saitoh, Y.; Mochida, K.; Tsuruyama, E.; Kiyono, T.; Imoto, I.; Inazawa, J.; Yuasa, Y.; Kubota, T.; Yamaoka, S. NF-ΚB Inducing Kinase, a Central Signaling Component of the Non-Canonical Pathway of NF-ΚB, Contributes to Ovarian Cancer Progression. PLoS ONE 2014, 9, e88347. [Google Scholar] [CrossRef] [PubMed]
- Hufnagel, D.H.; Wilson, A.J.; Saxon, J.; Blackwell, T.S.; Watkins, J.; Khabele, D.; Crispens, M.A.; Yull, F.E.; Beeghly-Fadiel, A. Expression of P52, a Non-Canonical NF-KappaB Transcription Factor, Is Associated with Poor Ovarian Cancer Prognosis. Biomark. Res. 2020, 8, 45. [Google Scholar] [CrossRef] [PubMed]
- Xiang, N.; He, M.; Ishaq, M.; Gao, Y.; Song, F.; Guo, L.; Ma, L.; Sun, G.; Liu, D.; Guo, D.; et al. The DEAD-Box RNA Helicase DDX3 Interacts with NF-ΚB Subunit P65 and Suppresses P65-Mediated Transcription. PLoS ONE 2016, 11, e0164471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, X.; Harvey, S.E.; Zheng, R.; Lyu, J.; Grzeskowiak, C.L.; Powell, E.; Piwnica-Worms, H.; Scott, K.L.; Cheng, C. The RNA-Binding Protein AKAP8 Suppresses Tumor Metastasis by Antagonizing EMT-Associated Alternative Splicing. Nat. Commun. 2020, 11, 486. [Google Scholar] [CrossRef] [PubMed]
- Akileswaran, L.; Taraska, J.W.; Sayer, J.A.; Gettemy, J.M.; Coghlan, V.M. A-Kinase-Anchoring Protein AKAP95 Is Targeted to the Nuclear Matrix and Associates with P68 RNA Helicase. J. Biol. Chem. 2001, 276, 17448–17454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, B.; Liu, C.; Shi, E.; Jin, Q.; Zhao, W.; Wang, J.; Ji, R. MiR-105-3p Acts as an Oncogene to Promote the Proliferation and Metastasis of Breast Cancer Cells by Targeting GOLIM4. BMC Cancer 2021, 21, 275. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Wang, J.; Zhou, R.; Yu, S.; Jiang, J.; Zhou, Q. Kinesin Family Member 23 Exerts a Protumor Function in Breast Cancer via Stimulation of the Wnt/β-Catenin Pathway. Toxicol. Appl. Pharmacol. 2022, 435, 115834. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Weng, W.; Zhang, Q.; Wu, Y.; Ni, S.; Tan, C.; Xu, M.; Sun, H.; Liu, C.; Wei, P.; et al. The LncRNA NEAT1 Activates Wnt/β-Catenin Signaling and Promotes Colorectal Cancer Progression via Interacting with DDX5. J. Hematol. Oncol. 2018, 11, 113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Liu, Y.; Wang, Q.; Song, S.; Feng, L.; Shi, C. The Alterations and Potential Roles of MCMs in Breast Cancer. J. Oncol. 2021, 2021, 7928937. [Google Scholar] [CrossRef] [PubMed]
- Mazurek, A.; Luo, W.; Krasnitz, A.; Hicks, J.; Scott Powers, R.; Stillman, B. DDX5 Regulates DNA Replication and Is Required for Cell Proliferation in a Subset of Breast Cancer Cells. Cancer Discov. 2012, 2, 812–825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, C.; Qin, X.; Wu, Z.; Shen, Q.; Yang, W.; Zhang, S.; Duan, J.; Liang, F.; Liu, C. Inhibitory Effect of MyoD on the Proliferation of Breast Cancer Cells. Oncol. Lett. 2016, 11, 3589–3596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giraud, G.; Terrone, S.; Bourgeois, C.F. Functions of DEAD Box RNA Helicases DDX5 and DDX17 in Chromatin Organization and Transcriptional Regulation. BMB Rep. 2018, 51, 613–622. [Google Scholar] [CrossRef] [Green Version]
- Du, J.X.; Luo, Y.H.; Zhang, S.J.; Wang, B.; Chen, C.; Zhu, G.Q.; Zhu, P.; Cai, C.Z.; Wan, J.L.; Cai, J.L.; et al. Splicing Factor SRSF1 Promotes Breast Cancer Progression via Oncogenic Splice Switching of PTPMT1. J. Exp. Clin. Cancer Res. 2021, 40, 171. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Niu, T.; Manley, J.L. The RNA Binding Protein RNPS1 Alleviates ASF/SF2 Depletion-Induced Genomic Instability. RNA 2007, 13, 2108–2115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, H.J.; Eom, H.-J.; Kim, H.; Myung, K.; Kwon, H.M.; Choi, J.H. Thrap3 Promotes R-Loop Resolution via Interaction with Methylated DDX5. Exp. Mol. Med. 2021, 53, 1602–1611. [Google Scholar] [CrossRef]
- Tabaglio, T.; Low, D.H.P.; Teo, W.K.L.; Goy, P.A.; Cywoniuk, P.; Wollmann, H.; Ho, J.; Tan, D.; Aw, J.; Pavesi, A.; et al. MBNL1 Alternative Splicing Isoforms Play Opposing Roles in Cancer. Life Sci. Alliance 2018, 1, e201800157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ray, D.; Yun, Y.C.; Idris, M.; Cheng, S.; Boot, A.; Huat Iain, T.B.; Rozen, S.G.; Tan, P.; Epstein, D.M. A Tumor-Associated Splice-Isoform of MAP2K7 Drives Dedifferentiation in MBNL1-Low Cancers via JNK Activation. Proc. Natl. Acad. Sci. USA 2020, 117, 16391–16400. [Google Scholar] [CrossRef] [PubMed]
- Laurent, F.X.; Sureau, A.; Klein, A.F.; Trouslard, F.; Gasnier, E.; Furling, D.; Marie, J. New Function for the RNA Helicase P68/DDX5 as a Modifier of MBNL1 Activity on Expanded CUG Repeats. Nucleic Acids Res. 2012, 40, 3159–3171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venables, J.P.; Brosseau, J.-P.; Gadea, G.; Klinck, R.; Prinos, P.; Beaulieu, J.-F.; Lapointe, E.; Durand, M.; Thibault, P.; Tremblay, K.; et al. RBFOX2 Is an Important Regulator of Mesenchymal Tissue-Specific Splicing in Both Normal and Cancer Tissues. Mol. Cell. Biol. 2013, 33, 396–405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Damianov, A.; Ying, Y.; Lin, C.H.; Lee, J.A.; Tran, D.; Vashisht, A.A.; Bahrami-Samani, E.; Xing, Y.; Martin, K.C.; Wohlschlegel, J.A.; et al. Rbfox Proteins Regulate Splicing as Part of a Large Multiprotein Complex LASR. Cell 2016, 165, 606–619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Lu, W.; Liu, S.; Wang, Y.; Li, S.; Xu, Y.; Xing, H.; Tang, K.; Tian, Z.; Rao, Q.; et al. ZFP36L2, a Novel AML1 Target Gene, Induces AML Cells Apoptosis and Inhibits Cell Proliferation. Leuk. Res. 2018, 68, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Suk, F.M.; Chang, C.C.; Lin, R.J.; Lin, S.Y.; Liu, S.C.; Jau, C.F.; Liang, Y.C. ZFP36L1 and ZFP36L2 Inhibit Cell Proliferation in a Cyclin D-Dependent and P53-Independent Manner. Sci. Rep. 2018, 8, 2742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Cairns, M.; Rose, B.; O’Brien, C.; Shannon, K.; Clark, J.; Gamble, J.; Tran, N. Alterations in MiRNA Processing and Expression in Pleomorphic Adenomas of the Salivary Gland. Int. J. Cancer 2009, 124, 2855–2863. [Google Scholar] [CrossRef] [PubMed]
- Yonemori, K.; Seki, N.; Kurahara, H.; Osako, Y.; Idichi, T.; Arai, T.; Koshizuka, K.; Kita, Y.; Maemura, K.; Natsugoe, S. ZFP36L2 Promotes Cancer Cell Aggressiveness and Is Regulated by Antitumor MicroRNA-375 in Pancreatic Ductal Adenocarcinoma. Cancer Sci. 2017, 108, 124–135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dun, M.D.; Mannan, A.; Rigby, C.J.; Butler, S.; Toop, H.D.; Beck, D.; Connerty, P.; Sillar, J.; Kahl, R.G.S.; Duchatel, R.J.; et al. Shwachman-Bodian-Diamond Syndrome (SBDS) Protein Is a Direct Inhibitor of Protein Phosphatase 2A (PP2A) Activity and Overexpressed in Acute Myeloid Leukaemia. Leukemia 2020, 34, 3393–3397. [Google Scholar] [CrossRef] [Green Version]
- Zan, J.; Xu, R.; Tang, X.; Lu, M.; Xie, S.; Cai, J.; Huang, Z.; Zhang, J. RNA Helicase DDX5 Suppresses IFN-I Antiviral Innate Immune Response by Interacting with PP2A-Cβ to Deactivate IRF3. Exp. Cell Res. 2020, 396, 112332. [Google Scholar] [CrossRef] [PubMed]
- Lian, Y.F.; Huang, Y.L.; Wang, J.L.; Deng, M.H.; Xia, T.L.; Zeng, M.S.; Chen, M.S.; Wang, H.B.; Huang, Y.H. Anillin Is Required for Tumor Growth and Regulated by MiR-15a/MiR-16-1 in HBV-Related Hepatocellular Carcinoma. Aging (Albany NY) 2018, 10, 1884–1901. [Google Scholar] [CrossRef]
- Beezhold, K.J.; Castranova, V.; Chen, F. Microprocessor of MicroRNAs: Regulation and Potential for Therapeutic Intervention. Mol. Cancer 2010, 9, 134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hulleman, E.; Quarto, M.; Vernell, R.; Masserdotti, G.; Colli, E.; Kros, J.M.; Levi, D.; Gaetani, P.; Tunici, P.; Finocchiaro, G.; et al. A Role for the Transcription Factor HEY1 in Glioblastoma. J. Cell. Mol. Med. 2009, 13, 136–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, J.X.; Sun, C.Y.; Tian, S.; Yu, C.; Chen, M.Y.; Zhang, H. Tumor suppressor Fbxw7 antagonizes WNT signaling by targeting β-catenin for degradation in pancreatic cancer. Tumour Biol. 2016, 37, 13893–13902. [Google Scholar] [CrossRef]
- Wlodarchak, N.; Xing, Y. PP2A as a master regulator of the cell cycle. Crit. Rev. Biochem. Mol. Biol. 2016, 51, 162–184. [Google Scholar] [CrossRef] [PubMed]
- Ji, R.; Ji, Y.; Ma, L.; Ge, S.; Chen, J.; Wu, S.; Huang, T.; Sheng, Y.; Wang, L.; Yi, N.; et al. Keratin 17 upregulation promotes cell metastasis and angiogenesis in colonadenocarcinoma. Bioengineered 2021, 12, 12598–12611. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Pan, H.; He, Y.; Hu, C.; Gu, Y. Functional roles of the SHCBP1 and KIF23 interaction in modulating the cell-cycle and cisplatin resistance of head and neck squamous cell carcinoma. Head Neck 2022, 44, 591–605. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Cancer | Role 1 | Mechanism | Ref. |
|---|---|---|---|
| Breast | + | downregulates KLF4 expression; modulates E-cadherin via HIF-1α; activates ERα | [20,21,22] |
| Colorectal | dual | +: upregulated KRAS leads to HIF-1α/Yap1 or β-catenin/ZEB1 pathway; −: DDX3X/Snail/E-cadherin axis | [23,24,25] |
| Lung | dual | +: activates Wnt/β-catenin pathway −: enhances p53-activated p21 transcription; prevents E-cadherin degradation by MDM2 transcription | [26,27,28,29] |
| Hepatocellular carcinoma | − | overexpressed in HCC cells but blocks cell cycle progression via regulation of cyclin D1 and p21; expression of miRNA; Rotterlin upregulation of DDX3X | [30,31,32,33,34] |
| Prostate cancer and Ewing sarcoma | + | inhibition of DDX3X leads to a decrease in cellular proliferation | [35,36,37] |
| Oral squamous cell carcinoma | dual | +: high DDX3X expression is associated with cancer progression in smokers; promotes AREG translation −: low DDX3X expression is associated with poor prognosis in non-smoker patients | [38,39,40] |
| Head and neck squamous cell carcinoma | + | forms the CBC/DDX3X/eIF3 complex to promotes ATF4 translation | [41] |
| Glioblastoma | + | high DDX3X level is linked with poor prognosis | [42,43] |
| Medulloblastoma | + | mutated DDX3X activates WNT/β-catenin signaling and drives stress granules formation | [44,45,46] |
| Gallbladder carcinoma and | + | high DDX3X level is linked with poor prognosis | [47] |
| Pancreatic ductal adenocarcinoma | + | high DDX3X level is linked with poor prognosis; promotes p62 accumulation | [48,49] |
| Chronic myeloid leukemia | + | mutated DDX3X is associated with poor prognosis | [49] |
| Melanoma | − | promotes MITF translation | [50,51] |
| NK/T-cell lymphoma | − | DDX3X mutant alters NF-κB and MAPK pathways and increases STAT3/p42/p44 phosphorylation | [52,53] |
| Cancer | Role 1 | Mechanism | Ref. |
|---|---|---|---|
| Colorectal | + | DDX5 overexpression promotes cancer by AKT/mTOR signaling or association with AldoA | [62,63,64,65,66] |
| Breast | + | upregulates a subset of miRNAs; highly correlated with Ki67; involved in β-catenin/Wnt pathway | [67,68,69] |
| Leukemia | + | DDX5 depletion selectively induces stress in AML cells; positive regulator of NOTCH1 signaling; DDX5 inhibition reduces tumor proliferation | [70,71,72] |
| Non-small-cell lung/small cell lung | + | induces β-catenin to promote cell proliferation | [73,74] |
| Osteosarcoma | + | lncRNA DLEU1/miR-671-5p/DDX5 interaction to promote cancer progression | [75] |
| Prostate | + | lncRNA CCAT1/DDX5/miR-28-5p interaction to promote cancer progression; DDX5-ETV4 fusion protein | [76,77] |
| Gastric | + | high DDX5 expression activates mTOR/SK61 pathway to induce cancer progression; lnc MIAT interaction | [78,79] |
| Glioblastoma | + | NF-κB p50 subunit activation; lncRNA LINC01116 interaction; hyperactivation of ERK and downregulation of DUSP5 | [80,81,82] |
| Cervical | + | stimulation of the expression of TGF-β1 in CaSki cells | [83] |
| Endometrial | + | HDGF/DDX5 interaction to induce β-catenin | [84] |
| Squamous cell carcinoma (HNSCC) | + | high DDX5 expression is associated with cancer progression | [85] |
| Squamous cell carcinoma (ESCC) | + | decreased DDX5 expression is associated with inhibition of cancer progression | [86] |
| Human hepatocellular carcinoma (HCC) associated with HBV | − | inhibits cancer progression by interacting with p62/SQSTM1, by regulation of miRNAs and by associating with lncRNA HOTAIR | [87,88,89,90,91] |
| Interactor | Function | Tumor-Specific Interactome | Role 1 | Mechanism | Roles of DDX3X/DDX5 in the Same Tumor 1 | Ref. |
|---|---|---|---|---|---|---|
| RPA2 | ss DNA-binding protein | Breast Cancer | + | NF-κB activation | +/+ | [92,93] |
| CTCF | Transcriptional coactivator | Breast Cancer | − | Regulation of transcription | +/+ | [94,95] |
| DHX9 | RNA/DNA helicase | Breast Cancer | + | Upregulation of lncRNA | +/+ | [96,97] |
| CUL1 | Ubiquitination cofactor | Breast Cancer | + | Positive regulation of proliferation | +/+ | [98] |
| CUL5 | Ubiquitination cofactor | Breast Cancer | − | Negative regulation of proliferation | +/+ | [99] |
| YBX1 | Transcription factor | Breast Cancer | + | Interaction with lncRNA AC073352.1 | +/+ | [100] |
| RPS6KB2 | Protein kinase | Breast Cancer | + | Activation of estrogen receptor-alpha | +/+ | [101,102] |
| AGR2 | Disulfide isomerase | Breast Cancer | + | Aberrant protein maturation in the ER | +/+ | [103,104] |
| YY1 | Transcription factor | Cervical cancer | Dual | Gene-specific recruitment of transcriptional activators or repressors | ?/+ | [105] |
| CHD4 | Chromatin remodeling | Endometrial cancer | − | Activation of the TGF-β pathway | ?/+ | [106] |
| TAF15 | mRNA metabolism | Gastric cancer | + | Modulation of stress response | −/+ | [107] |
| MATR3 | DNA/RNA-binding protein | Lung cancer | + | Undetermined | Dual/+ | [108,109,110] |
| DYRK2 | Protein kinase | Glioblastoma | − | Negative regulation of cell migration | +/+ | [111] |
| PINK1 | Mitochondrial protein kinase | Glioblastoma | − | Negative regulation of oxidative stress | +/+ | [112] |
| SYNCRIP | RNA-binding protein | Leukemia | + | Activation of oncogenes | +/+ | [113] |
| ITGA4 | Integrin | Leukemia | + | Upregulation of BCL-2/negative regulation of apoptosis | +/+ | [114] |
| MED12 | Protein kinase | Prostate cancer | + | Activation of Wnt/β-catenin and TGF-β signaling | +/+ | [115] |
| CNEPA | Kinetochore subunit | Liver cancer | + | Aberrant chromosomal segregation | ?/− | [116] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Secchi, M.; Lodola, C.; Garbelli, A.; Bione, S.; Maga, G. DEAD-Box RNA Helicases DDX3X and DDX5 as Oncogenes or Oncosuppressors: A Network Perspective. Cancers 2022, 14, 3820. https://doi.org/10.3390/cancers14153820
Secchi M, Lodola C, Garbelli A, Bione S, Maga G. DEAD-Box RNA Helicases DDX3X and DDX5 as Oncogenes or Oncosuppressors: A Network Perspective. Cancers. 2022; 14(15):3820. https://doi.org/10.3390/cancers14153820
Chicago/Turabian StyleSecchi, Massimiliano, Camilla Lodola, Anna Garbelli, Silvia Bione, and Giovanni Maga. 2022. "DEAD-Box RNA Helicases DDX3X and DDX5 as Oncogenes or Oncosuppressors: A Network Perspective" Cancers 14, no. 15: 3820. https://doi.org/10.3390/cancers14153820