
p53 Isoforms as Cancer Biomarkers and Therapeutic Targets


Department of Cell and Molecular Biology, Uppsala University, SE-75124 Uppsala, Sweden
*
Author to whom correspondence should be addressed.
Cancers 2022, 14(13), 3145; https://doi.org/10.3390/cancers14133145
Submission received: 3 May 2022
/
Accepted: 22 June 2022
/
Published: 27 June 2022
(This article belongs to the Special Issue Targeting Therapies for the p53 Protein in Cancer Treatments)
Abstract
:Simple Summary
The well-known tumor suppressor protein p53 plays important roles in tumor prevention through transcriptional regulation of its target genes. Reactivation of p53 activity has been a potent strategy for cancer treatment. Accumulating evidences indicate that p53 isoforms truncated/modified in the N- or C-terminus can modulate the p53 pathway in a p53-dependent or p53-independent manner. It is thus imperative to characterize the roles of the p53 isoforms in cancer development. This review illustrates how p53 isoforms participate in tumor development and/or suppression. It also summarizes the knowledge about the p53 isoforms as promising cancer biomarkers and therapeutic targets.
Abstract
This review aims to summarize the implications of the major isoforms of the tumor suppressor protein p53 in aggressive cancer development. The current knowledge of p53 isoforms, their involvement in cell-signaling pathways, and their interactions with other cellular proteins or factors suggests the existence of an intricate molecular network that regulates their oncogenic function. Moreover, existing literature about the involvement of the p53 isoforms in various cancers leads to the proposition of therapeutic solutions by altering the cellular levels of the p53 isoforms. This review thus summarizes how the major p53 isoforms Δ40p53α/β/γ, Δ133p53α/β/γ, and Δ160p53α/β/γ might have clinical relevance in the diagnosis and effective treatments of cancer.
1. Introduction
p53 was discovered in 1979 as a 53 kD protein bound to simian virus (SV40) large T-antigen [1,2]. The p53 protein is a DNA sequence-specific transcription factor that suppresses tumor growth by regulating a large number of genes involved in cell cycle arrest, DNA repair, senescence, apoptosis, cell motility, adhesion, and migration [3,4,5]. p53 is the most frequently mutated gene in human cancers [6]. Loss or alteration of p53 functions for binding to p53 response elements located in its targeted genes leads to the development of cancer [7,8], thereby strengthening its significance in tumor suppression.
The p53 protein is a product of the TP53 gene that is highly conserved through evolution [9]. The human TP53 gene is located on the human chromosome 17p13.1 [10]. It is comprised of 11 exons, including 10 exons coding for the DNA sequence of the canonical or full-length p53 protein (FLp53, also termed p53α) and the first noncoding exon (exon 1) (Figure 1A) [11,12]. The TP53 gene expresses at least nine different mRNA transcripts encoding at least twelve different protein isoforms, namely p53α, p53β, p53γ, Δ40p53α, Δ40p53β, Δ40p53γ, Δ133p53α, Δ133p53β, Δ133p53γ, Δ160p53α, Δ160p53β, Δ160p53γ. These isoforms are the results of transcription initiation from different promotors, alternative splicing of the introns, and translation initiation at different codons [13,14].
It is interesting to note that although alternative splicing of p53 introns was first reported in the mid-1980s, the field of p53 isoforms did not emerge until quite recently [15,16]. In contrast, for proteins p63 and p73, both p53 homologues, the isoforms were identified almost immediately after their own discovery in the late 1990s [17,18,19]. However, the discovery of p63 and p73 isoforms accelerated the process of characterizing the first p53 isoform Δ40p53 [20]. Since then, studies on the p53 isoforms had been developing rapidly.
FLp53 consists of 393 amino acids and contains seven functional domains (Figure 1B) [14,21]. The two transactivation domains (TAD-1 and -2) are responsible for activating gene transcription [22]. The proline-rich domain (PRD) contributes to p53-mediated apoptosis [23]. The core DNA binding domain (DBD), as the name suggests, plays a key role in binding to specific DNA sequences [24]. The nuclear localization signaling region (NLS) is responsible for delivering p53 into the nucleus [25]. The oligomerization domain (OD) is involved in the formation of p53 tetramer [26]. Finally, the negative regulation domain (α) plays a crucial role in p53 post-translational modification that modulates the stability and activity of p53 [27,28].
The major p53 isoforms are truncated either in the N-terminus or in the C-terminus with some sequence alteration. The Δ40p53 isoform, without the first 40 amino acids, lacks the TAD-1 but still contains the TAD-2, which likely allows it to bind to both typical and atypical responsive elements [13,20]. The Δ133p53 and Δ160p53 isoforms without the first 133 and 160 amino acids, respectively, lack both transactivation domains TAD-1 and -2, and also a part of DBD supposedly modulating DNA binding property of p53 [13,21,22]. Other than these N-terminally truncated isoforms, p53 has two distinct C-terminally shortened variants (β and γ) [13,29]. In these isoforms, the OD is replaced by a stretch of 10 and 15 amino acids, with sequences DQTSFQKENC and MLLDLRWCYFLINSS respectively (Figure 1B) [29]. Given that the truncated p53 isoforms may have different structures from FL p53, it can be anticipated that they would display altered functions in relation to tumor suppression and can be even oncogenic [13,30,31].
Over the last decade, there has been a lot of evidence regarding the expression of p53 isoforms in various common cancers [13]. High p53β expression was positively associated with recurrence-free survival (RFS) and overall survival (OS) in patients with renal cell carcinoma [32]. Similarly, high levels of Δ133p53 expression have been correlated with better OS in patients with ovarian cancer [33,34]. In breast cancer, the most common type of female cancer, a low Δ40p53-to-FLp53 ratio is associated with increased disease-free survival (DFS) [35,36]. On the other hand, Δ40p53α overexpression exerts tumor suppressor activity by increasing the expression of p53 target genes in the most common type of primary liver cancer hepatocellular carcinoma [37]. Moreover, amyloid aggregation of Δ40p53 has been reported in the endometrial cancer cells [38]. The core domain of p53 also demonstrates aggregation propensity, particularly with the influence of various RNAs (mRNA and rRNA), which may have implications in cancer [39,40]. Treatment with the histone deacetylase inhibitor valproic acid resulted in downregulation of p53β/γ and upregulation of p53α in acute myeloid leukemia, suggesting that p53 isoforms have the potential to predict therapy response [41]. p53 isoforms, especially the N-terminally truncated ones Δ40p53, Δ133p53, and Δ160p53, play an important role in the development of cancers [13]. Altogether, p53 isoforms play critical roles in tumorigenesis and might be potential biomarkers and therapeutic targets.
2. Regulatory Mechanisms of p53 Isoform Expression
Current literature shows that the expression of the p53 isoforms is regulated by different mechanisms at different levels in the cells. The expression of the p53 isoforms is regulated at the transcriptional, post-transcriptional, translational, and post-translational levels. Moreover, different degradation mechanisms exist for different p53 isoforms, which also modulate their relative levels.
A major regulation occurs in the level of transcription by alternative promoter usage and alternative splicing [14], as elaborated below. TP53 gene has two different promoters, P1 and P2 (Figure 2A). FL p53 and Δ40p53 are encoded from the constitutive promoter P1 [42], whereas Δ133p53 and Δ160p53 mRNAs are transcribed from the promoter P2 [43,44]. 17β estradiol regulates p53 isoform expression through activating c-Myc that induces the P1 promoter of the TP53 gene in breast cancer cells [45]. Alternatively, p53 has been shown to increase the transcription of Δ133p53 by binding to the p53 response elements in the internal P2 promoter [44,46]. In addition, several p63/p73 isoforms such as p63β, ΔNp63α, ΔNp63β, and ΔNp73γ activate Δ133p53 expression by transactivating the promoter P2 [47].
Alternative splicing of the p53 mRNA plays a major role in the expression of the p53 isoforms. Differential splicing of intron 2 (i2) generates two different transcripts that encode the isoforms FLp53 and Δ40p533, respectively (Figure 1) [48]. A G-quadruplex structure in intron 3 (i3) regulates the splicing of i2 to generate the mRNAs for either FLp53 or the Δ40p53 isoform (Figure 2A) [49]. Alternatively, the mRNAs for the p53 α/β/γ are generated by alternative splicing of intron 9 (i9) (Figure 1A) under regulation of the serine/arginine-rich (SR) splicing factors (SRSFs) such as SRSF1 and SRSF3 (Figure 2A). Downregulation of SRSF1 stimulates transcription of the p53β and p53γ isoforms [50]. In contrast, SRSF3 downregulation only increases p53β [51]. Thus, alternative splicing mechanisms can be used to induce p53 isoform expression.
p53 isoform expression can also be regulated at translational and post-translational levels. The Δ40p53 isoform can be generated from the p53 transcript by alternative initiation of translation at the ATG40 codon. In that case, the stop codons retained in i2 of the p53 transcript can result in termination of translation followed by re-initiation at the AUG40 codon, thereby expressing the Δ40p53 isoform (Figure 1A and Figure 2A) [48]. FLp53 (p53α) and Δ40p53 expression are regulated by two distinct internal ribosome entry sites (IRESs) (Figure 2A) [52,53]. Multiple IRES-interacting trans-acting factors (ITAFs) have been found to regulate IRES-mediated translation of p53 mRNAs. These include polypyrimidine-tract-binding protein (PTB), PTB-associated splicing factor (PSF), Annexin A2, death-associated protein 5 (DAP5), translational control protein 80, and RNA helicase A [54,55,56,57,58,59]. Therefore, factors mediating expression of ITAFs also play a role in p53 isoform expression. The Δ133p53 and Δ160p53 isoforms are expressed from the same mRNA transcript. However, the Δ160p53 isoform is produced by alternative initiation of translation at ATG160 from the Δ133p53 transcript [43].
The stability of different p53 isoforms in the cells are also regulated by p53 degradation pathways. Under normal cellular conditions, p53 is regulated via a negative feedback loop involving its target gene E3 ubiquitin ligase mouse double minute 2 (MDM2). MDM2 binding to p53 TADs inhibits p53 activity by preventing it from regulating the target genes [60,61]. Alternatively, p53 can induce the expression of MDM2, thus forming a negative feedback loop [62]. Binding of MDM2 to p53 promotes its rapid degradation through ubiquitination using 26S proteasome pathway (Figure 2B, middle panel) [63,64]. The N- terminally truncated p53 isoforms lack the TADs partially or fully and thus may not be fully susceptible to MDM2 based ubiquitination. Earlier reports suggest that the Δ40p53 isoform can escape from MDM2-induced protein degradation [65]. However, another study reports that by retaining the DBD, and the C-terminal domain, the N-terminally truncated p53 isoforms can still bind to MDM2, which can be enough for p53 ubiquitination and proteasomal degradation [66]. It has also been demonstrated that Δ40p53 can protect p53 from MDM2-mediated degradation by interfering with p53-MDM2 binding (Figure 2B, middle panel) [65,67].
The p53β/γ isoforms tend not to be degraded by MDM2-mediated ubiquitination, although they contain TADs. The p53β/γ mRNAs are susceptible to nonsense-mediated decay (NMD) that can degrade mRNAs with premature translation termination codons (PTCs) (Figure 2B, left panel) resulting from alternative splicing of p53 i9 (Figure 2A) [68]. Thus, the expression of p53β and p53γ can be regulated at the post-transcriptional level.
A recent study suggests that the stability of the Δ133p53 isoform is not affected by a proteasome inhibitor, MG132 [69]. Δ133p53 can be degraded by the digestive organ expansion factor (Def) protein in a proteasome-independent degradation pathway [70]. Δ133p53α can also be degraded by autophagy during replicative senescence (Figure 2B, right panel) [71]. Downregulation of the chaperone-associated ubiquitin ligase STUB1 causes Δ133p53α autophagic degradation [71]. To the best of our knowledge, there have been no reports yet of the regulation of Δ160p53 degradation. Δ40p53 can also be generated post-translationally via a 20S proteasome-mediated cleavage of FLp53 [72]. Taken together, at least four levels of regulation govern the expression of p53 isoforms: transcriptional, post-transcriptional, translational, and post-translational.
3. Influence of the p53 Isoform Network on Biological Processes
The p53 network takes a central position in regulation of different cellular activities, but it is also regulated by external or internal factors. The p53 pathway is stimulated by various cellular stress factors such as DNA damage, cell contact, oncogene expression, oxidative stress, and endoplasmic reticulum (ER) stress (Figure 3). These in turn lead to multiple cellular effects including cell cycle arrest, senescence, cellular apoptosis, and DNA damage repair, resulting in tumor suppression [21]. p53, also called the ‘guardian of the genome’, acts as a transcription factor, regulating expressions over 3600 target genes [73,74]. Thus, the p53 pathway can play a critical role in tumor suppression by activating or repressing target genes [75].
Many studies have shown that p53 isoforms influence diverse biological activities of p53 by regulating p53 target genes. p53 isoforms can also interact with other specific proteins or factors to modulate the p53 pathway. For example, Δ133p53β can regulate apoptosis by inhibiting the activity of an anti-apoptotic protein, RhoB [76]. Netrin-1, an anti-apoptotic protein, can be positively regulated by Δ40p53 [77]. Similarly, overexpression of Δ133p53 has been shown to improve DNA double-strand break (DSB) repair by upregulating E2F1, a transcription factor that activates the DNA DSB repair factor RAD51 [78,79]. These studies demonstrated that p53 isoforms can mediate their network without relying on p53. Furthermore, several signaling pathways interact with the p53 pathway in a p53-isoforms-dependent manner (Figure 4) [75,80].
3.1. p53 Isoforms Regulating Cell Cycle Arrest and Senescence
Cellular senescence is usually used to describe an irreversible cell cycle arrest [81]. 14-3-3σ is a member of 14-3-3 family proteins that interact with p53 and affect its activity [82]. It has been shown that Δ40p53 can form a homotetramer complex and bind to the promoter of 14-3-3σ, activating the expression of 14-3-3σ and causing cell cycle arrest in response to ER stress [83]. Thus, the interaction between Δ40p53 and 14-3-3σ can play a role in cell cycle arrest. p53 can also regulate cell cycle arrest by activating its target gene p21 [84,85]. In addition, p53-induced production of microRNA-34a (miR-34a) leads to gene expression involved in cell cycle arrest and senescence in oncogenic cells [86,87]. Both p21 and miR-34a can act as the pro-senescence factors [30]. Ota et al. showed that Δ40p53 expression induced tumor cell senescence by upregulating p21 in hepatocellular carcinoma cells (Figure 3) [37]. In addition, overexpression of Δ133p53 induced cell cycle arrest and cellular senescence when exposed to oxidative stress by H2O2 in mouse embryonic fibroblasts [88]. Besides that, Δ133p53 expression inhibits senescence by repressing the expression of p21 and miR-34a (Figure 3) [69,89,90]. This is because Δ133p53 can prevent p53α from binding to the promoters of p21 and miR-34a [89]. Furthermore, p53β induces cellular senescence through forming complexes with p53α and upregulating p21 expression [69,91]. Thus, p53 isoforms modulate cell cycle arrest and senescence via inducing or suppressing p53 target genes.
3.2. p53 Isoforms—The Role in Apoptosis
The Bcl-2 protein family, which includes Bcl-2, Bcl-xL, Bax, NOXA, and PUMA, is important for cell apoptosis [92]. p53 is known to induce apoptosis by inhibiting the expression of the anti-apoptotic proteins such as Bcl-2 and Bcl-xL and activating pro-apoptotic proteins such as Bax, NOXA, and PUMA [92,93,94]. In addition, p53-induced protein with a death domain (PIDD) can promote p53-mediated apoptosis [95]. Several p53 isoforms have been linked to the regulation of apoptosis. For instance, p53β can form a complex with p53α and boost p53 transcriptional activity on the Bax promoter, thereby enhancing apoptosis (Figure 3) [91]. Alternatively, Δ133p53/Δ113p53 has been shown to inhibit p53-mediated apoptosis through activating Bcl-xL in zebrafish (Figure 3) [96]. In response to low or moderate stress, Δ133p53 and Δ123p53 (Δ133p53 mouse ortholog) can form a complex with p53α and switch the p53 binding sites within the Bcl-2 promoter from repressive to activating p53 response elements, thereby resulting in Bcl-2 induction and anti-apoptosis (Figure 3) [97]. These studies suggest that Δ133p53 prefers to be an antagonist of p53-mediated apoptosis. In contrast, when the expression of Δ40p53 is lower or equal to that of p53α, it induces the expression of Bax and PIDD, resulting in apoptosis (Figure 3) [98,99]. Together, these results suggest that the p53 isoforms influence apoptosis primarily through interactions with p53α.
3.3. p53 Isoforms and DNA DSB Repair
DNA DSBs are one of the most harmful types of DNA damage [100]. Correct DNA DSB repair is important for prohibiting tumorigenesis [100]. It is known that p53 is activated in response to DNA damage [101], which, in turn, inhibits DNA DSB repair [102] by downregulating DNA DSB repair genes including RAD51, RAD52, LIG4, WRN, and XRCC4 [103]. Interestingly, the p53 isoform Δ133p53 plays a significant role in promoting DNA DSB repair [104]. Δ133p53/Δ113p53 (zebrafish ortholog of Δ133p53) can switch p53 from repression to activation DNA DSB repair via upregulating the expression of RAD51, RAD52, and LIG4 (Figure 3) [103,105,106]. In addition, upon γ-irradiation, Δ133p53 can form a complex with p73 and can stimulate the expression of genes RAD51, RAD52, and LIG4 with higher efficiency [107]. Thus, with a prominent role in DNA DSB repair, Δ133p53 can be strongly associated with tumorigenesis.
3.4. p53 Isoforms and NF-κB Signaling
Nuclear factor-κB (NF-κB) has both positive and negative effects on tumor progression [108]. The interaction of two critical signaling pathways, NF-κB and p53, can regulate several important cellular processes involved in tumorigenesis, including senescence, apoptosis, inflammation, and DNA damage repair [109]. Using a luciferase reporter assay, it has been shown that Δ133p53 can induce NF-κB activity upon infection by Helicobacter pylori, a strong risk factor for gastric cancer [110]. This, in turn, induces the expression of the NF-κB target genes including anti-apoptotic protein Bcl-2 and pro-inflammatory cytokines interleukin 6 (IL-6) and IL-8 (Figure 4) [110]. However, NF-κB is blocked in p53-null cells [110,111]. Thus, Δ133p53 activates NF-κB activity in a p53-dependent manner, which would inhibit apoptosis and promote inflammation during H. pylori infection (Figure 4). Another study reported that Δ133p53 mRNA level was downregulated upon treatment with NF-κB inhibitor pyrrolidine dithiocarbamate in MKN45 gastric cancer cells [112]. Thus, Δ133p53 and NF-κB expression levels are positively correlated, and downregulation of Δ133p53 may disrupt NF-κB signaling, which is significant for tumor development.
3.5. p53 Isoforms, JAK- STAT and RhoA-ROCK Signaling
Janus kinase (JAK)-signal transducer and activator of transcription (STAT), commonly called JAK-STAT signaling, is closely associated with cancer progression [113]. The JAK kinases activate the STAT family proteins through phosphorylation of tyrosine and serine residues [114,115]. STAT3, a member of the STAT family, is a transcription factor that can bind to various gene promoters [116]. These genes can be involved in various cellular processes such as anti-apoptosis and cell cycle progression (Figure 4) [114,116]. Of interest, STAT3 is necessary for the activity of NF-κB [117]. Both STAT3 and NF-κB induce the expression of the genes involved in anti-apoptosis, such as Bcl-2 and Bcl-xL, and cell cycle progression, such as c-Myc [116,118]. Thus, the activation of JAK-STAT and NF-κB signaling pathways contributes to cancer progression. Furthermore, IL-6 is considered as an activator of JAK-STAT signaling (Figure 4) [113]. As mentioned above, Δ133p53 induces the expression of NF-κB target genes including IL-6 [110]. Thus, Δ133p53 may activate the JAK-STAT signaling pathway by interacting with NF-κB signaling.
RhoA-ROCK signaling pathway consists of a small GTPase protein RhoA and its effector Rho-kinase (ROCK) [119]. RhoA cyclically exchanges between an active GTP-bound state and an inactive GDP-bound state (Figure 4) [120]. Interestingly, RhoA can drive STAT3 activation [121], suggesting an interplay between the RhoA with STAT. Furthermore, JAK activates ROCK; in turn, ROCK induces STAT3 phosphorylation (Figure 4) [122,123]. A study has shown that the increased expression of Δ133p53 activated the RhoA-ROCK signaling pathway probably by activating IL-6 and, further, by inducing the cooperation between JAK-STAT and RhoA-ROCK pathways [123]. Altogether, Δ133p53 expression can regulate the coordinated action of the NF-κB, JAK-STAT, and RhoA-ROCK signaling pathways (Figure 4).
3.6. p53 Isoforms and IFN Signaling
Interferons (IFNs) are pleiotropic cytokines that have important functions/properties including antiviral, antiproliferative response, immunoregulation, and antitumor [124,125]. There are three types of IFNs in humans: type I (IFN-α/β), type II (IFN-γ), and type III (IFN-λs) [124,126,127]. Several studies have shown that p53 isoforms are involved in IFN signaling [128,129,130,131]. Both p53β and p53γ isoforms were shown to be involved in the induced production of IFN-α and IFN-β, which have a critical role in the antiviral response [131,132]. In turn, IFN-β can induce both FLp53 (p53α) and p53β/γ isoforms in human peripheral blood mononuclear cells, potentially altering the expression of p53 target genes involved in cell cycle arrest or apoptosis [133]. Type II IFN IFN-γ is associated with antiviral, antiproliferative, immunomodulatory, and antitumor responses [124,134,135]. In estrogen-receptor-positive breast cancer with mutant p53, Δ133p53 can activate the expression of IFN-γ signaling genes [129]. Additionally, overexpression of Δ133p53β increased the expression of genes involved in the IFN-γ signaling pathway in prostate cancer [128]. Thus, Δ133p53 isoforms could activate IFN-γ signaling in different cancer types. Moreover, IFN-α/β/γ can induce the JAK-STAT signaling pathway that plays an important role in tumor progression (Figure 4) [126,130,136]. Collectively, these studies suggest that p53 isoforms that interact with the IFN signaling pathway have an important influence on antiviral immune responses and the risk of developing cancer.
Except for wild-type p53, Δ133p53 is the most well studied p53 isoform. In comparison, little is known about Δ160p53 and p53γ activity in modulating the p53 signaling pathway. As a result, much research remains to be done in the field of the p53 isoform network. Collectively, the biological activities of p53 signaling and several other signaling pathways can be affected by p53 isoforms.
4. p53 Isoforms as Promising Cancer Biomarkers
Many clinical studies have attempted to evaluate the clinical relevance of the p53 isoforms in various types of human cancers, both at the mRNA and the protein levels [13]. Endogenous p53 isoforms are generally found to be overexpressed in tumors when compared to nontumor cell lines, making p53 isoforms promising cancer biomarkers. In human cells, p53 isoforms express tissue specifically at both the mRNA and protein levels [21,91]. It is therefore important to quantify the expression levels of p53 isoforms in cells and tissues to be able to use those as cancer biomarkers.
To detect the p53 isoforms, several p53 isoform-specific antibodies that recognize different epitopes have been developed. However, these antibiotics lack specificity due to the similar epitopes in other p53 isoforms [78]. Due to unavailability of the antibodies specific to each p53 isoform, the expression of different p53 isoforms is mostly quantified at the mRNA level using RNA-seq [137,138]. The abundance of TP53 transcripts is quantified in many clinical studies using short-read RNA sequencing, which cannot distinguish 5′ variants of the α, β, and γ transcripts [139,140]. Recently, several studies have developed new approaches to quantitate distinct transcripts, such as RNAscope and multiplex probe-based long amplicon droplet digital PCR [128,140,141]. However, the correlation between the amount of the mRNAs and the abundance of their corresponding proteins can be weak, even for general proteins [142,143]. Furthermore, RNA-seq analysis has a disadvantage in detecting less-abundant p53 isoform transcripts [138]. To detect each C-terminal p53 isoform, a new technique combining molecularly imprinted polymers (MIPs) and LC-MS/MS-based targeted proteomics has recently been developed [144]. Thus, the development of novel and highly specific anti-p53 isoform approaches is very desirable.
Deregulation of p53 isoforms can either promote or inhibit tumor progression in a variety of cancers. Following the literature, different p53 isoform expression profiles have been correlated with different cancer types (Table 1 and Table 2). So far, only a few studies have shown that p53 isoforms expression can slow down tumor progression (Table 1). For example, high p53β expression is associated with a good prognosis in clear-cell renal cell carcinoma [32]. Similarly, high Δ40p53 expression has been inferred to be a prognostic marker for recurrence-free survival (RFS) in mucinous ovarian cancer [145]. High Δ40p53α expression has been reported to inhibit tumor cell growth and, at the same time, increase cellular senescence in hepatocellular carcinoma [37]. Moreover, high ∆133p53 mRNA expression levels are found to be a predictor of improved overall survival (OS) in patients diagnosed with high-grade serous ovarian carcinoma [34]. The p53 isoform expression profile in myeloma has recently been identified [146]. In these patients with high-risk cytogenetics, high expression of the short p53 isoforms Δ133p53 and Δ160p53 has been reported to better OS [146]. Furthermore, in brain cancer patients, ∆133p53 expression helped to reduce the side effects of radiation, such as astrocyte senescence and astrocyte-mediated neuro-inflammation [79]. These studies, thus demonstrate that high levels of p53 isoform expression can be used as a survival-associated biomarker for several types of cancer.
The p53 isoforms are often reported to promote tumor progression in a variety of cancers (Table 1). The Δ40p53 isoform has been identified as the major component of p53 amyloid aggregates in endometrial carcinoma cancer. It plays an important role in modifying p53 aggregation properties, which is associated with tumor progression [38]. The increased expression of Δ133p53β has been predicted to have poorer outcomes in glioblastoma, melanoma, breast, and prostate cancer patients [128,141,147,148]. Additionally, elevated Δ133p53 mRNA levels were thought to be a potential marker for lung and colorectal cancer malignancy [123,149]. A recent study also revealed that overexpression of Δ160p53 can stimulate the proliferation and migration of melanoma cells [150]. Besides, the N-terminally shortened p53 isoforms, the C-terminal truncated variants p53β and -γ can also aid in cancer prognosis. In uterine serous carcinoma, the high expression of p53γ has been associated with reduced progression-free survival (PFS) [151]. Moreover, high levels of p53β and -γ expression had a negative effect on the survival of myeloma patients [146]. These findings suggest that high p53 isoform expression could be a potential biomarker for cancer survival outcome.
Other than high or low expressions, proper Δ40p53-to-p53α ratio can be important for suppressing cancer [67]. In breast cancer, a high Δ40p53-to-p53α ratio is associated with poorer outcomes (Table 2) [152]. Patients with cholangiocarcinoma who have a high ratio of Δ133p53-to-p53α have a poor prognosis (Table 2) [153]. Furthermore, a high ∆133p53-to-p53β ratio increases cancer aggressiveness in colorectal cancer (Table 2) [69]. In acute myeloid leukemia, a high degree of p53β/γ-to-p53α predicts a better prognosis (Table 2) [154]. These studies suggest that the ratio of p53 isoforms can also be a potential biomarker in cancer and can be involved in clinical cancer therapy.
Mutations in the TP53 gene are seen to occur in ~50–60% of human cancers [7]. The majority of these missense mutations in the DBD cause p53 to lose its tumor-suppressive function and acquire novel oncogenic functions [7,155,156]. There are a few reports which suggest that p53 isoforms are beneficial for inhibiting the oncogenic activities of the p53 mutants. For example, Δ133p53 isoform can interact with the mutant FLp53 and have a beneficial effect in advanced serous carcinomas [157]. In addition, Δ133p53 RNA can upregulate the expression of IFN-γ signaling genes in mutant TP53 oestrogen receptor positive breast cancer, which is associated with a better patient outcome [129]. Furthermore, when compared to patients with wild-type p53 breast cancer, patients with mutant p53 breast cancer who expressed the p53γ isoform had a lower risk of cancer recurrence and a better OS [158]. In contrast, high levels of ∆160p53 expression contribute to the proliferation of cancer cells with p53 mutations [159]. Based on current evidence, it can be stated that the p53 isoforms can be prognostic biomarkers in human cancer.
5. p53 Isoforms as Potential Therapeutic Target in Cancer
The therapeutic values of p53 isoforms are yet to be investigated. The p53 isoforms have good potential as therapeutic targets in cancer due to their various regulatory expression mechanisms, complex interaction network, and different expression levels in tumor versus nontumor cells. Because relative expressions of the p53 isoforms are relevant in various cancers, the factors affecting the ratio of the p53 isoforms can also be therapeutic targets. For example, ITAFs, which regulate the IRESs responsible for the relative expression of FLp53 and ∆40p53 [52], can be targets for cancer treatment [160]. However, more research on ITAFs will be required to identify the specific targets for the first and the second IRES in order to use those for cancer therapy.
The aberrant alternative splicing of p53 mRNA has shown to be a hallmark of cancer [161,162]. Recently, three major therapeutic approaches, namely, small molecule inhibitors, splice-switching antisense oligonucleotides (SSOs), and clustered regularly interspaced short palindromic repeats (CRISPR)-Cas system, have been developed to target the splicing factors, providing a new strategy for cancer treatment [161,163]. Small molecules that affect the phosphorylation of serine/arginine-rich (SR) splicing factors (SRSFs) seem to have the potential to modify isoform production and provide therapeutic benefits [164,165]. Caffeine has been shown as a small molecule that downregulates the expression of the splicing factor SRSF3, altering splicing pattern of the gene TP53 and also other SRSF3 target genes [51,166]. An appropriate dose of caffeine decreases p53α expression while increasing p53β expression, which improves cellular senescence. Thus, caffeine can be used to modulate the p53β-to-p53α ratio, a ratio that is important for tumor treatment [51,69,166]. The SR proteins, which regulate alternative splicing of the p53 mRNA, are regulated by SR-protein-specific kinases such as Cdc2-like kinases (Clks), which phosphorylate SR proteins [167,168,169]. Thus, small molecules which can inhibit Clks may contribute to cancer treatment. For example, the Clks inhibitor TG003 can induce the expression of p53β and -γ in the MCF7 breast cancer cell line, which promotes cell apoptosis [50]. Other Clks inhibitors, such as KH-CB19 and GPS167, have also been shown to inhibit the phosphorylation of splicing factors [170,171] and thus can have therapeutic value. However, targeting Clks with small molecules is still facing clinical implementation challenges [163].
SSOs are short (15 to 30 nucleotide), chemically-modified RNA molecules which compete with splicing factors for binding to pre-mRNAs [161,163,172]. Though it is not yet approved for clinical trials, research is ongoing in cancer model systems to implement this strategy in cancer treatment [163]. In addition, the CRISPR-Cas gene-editing system has also been developed for alternative splicing of target mRNAs [163,173]. There is no doubt that splicing modulation is an important strategy for cancer treatment. However, more research is needed in order to implement that as an effective and safe therapeutic tool.
The mechanisms of p53 isoform degradation and aggregation can play an important role in regulating the expression of p53 isoforms. Gudikote et al. showed that upregulating p53β and -γ by inhibition of NMD can restore p53 activity in p53-deficient tumors caused by MDM2 overexpression or by inactivating mutations downstream of exon 9 [68]. There are mainly two reasons to consider NMD inhibition as a therapeutic strategy for p53 reactivation.
First, while p53β and -γ are significantly less susceptible to MDM2-mediated degradation, they are highly susceptible to NMD. Second, they intrinsically promote p53 transcriptional activity [68,91,174]. Furthermore, a peptide covering residues 107–129 of wild-type p53 could inhibit the aggregation of destabilized Δ133p53β and restore it to the wild-type p53 conformation, simultaneously restoring p53’s tumor suppressor activity [175]. Thus, the available p53-reactivation strategies could provide effective cancer therapies, highlighting the attractiveness of p53 isoforms as a therapeutic target. In terms of autophagic degradation of ∆133p53α, knockdown of autophagy-related proteins and a regulator protein STUB1, respectively, can decrease and increase the degradation process [71]. This strategy opens up a new avenue for regulating the abundance of ∆133p53α via autophagic degradation. Overall, modulating the stability of p53 isoforms may be a useful tool for cancer treatment.
Recently, Sun et al. reported that ∆40p53 expression is positively correlated with netrin-1, a cancer biomarker and therapeutic target protein that can inhibit apoptosis in several aggressive cancers [77,176]. Netrin-1 inhibition has been shown to be a highly promising strategy in human cancers with high levels of ∆40p53 expression [77]. Similarly, patients whose cancers have high Δ133p53 expression may benefit from JAK-STAT signaling inhibitors [123,177,178]. Thus, indirect modulation of p53 isoform expression by specific factors has the potential to be used as a therapeutic strategy in tumors expressing p53 isoforms.
6. Conclusions
The tumor suppressor protein p53 has been well studied in terms of its role in cancer development and progression. The TP53 gene does not only express just one protein p53; at least twelve different p53 isoforms are produced by alternative promoter usage, alternative splicing, and alternative translation initiation events. Current studies have revealed that p53 isoform expression is regulated at several levels, including transcription, post-transcription, translation, and post-translation. The distinct expression profiles of p53 isoforms in tumor and normal cells, as well as the deregulation of p53 isoforms as an emerging contributor to cancer development, make p53 isoforms biomarkers in cancer. Alternative-splicing-mediated transcription and IRES-mediated translation are important tools for regulating the expression of p53 isoforms, which would likely provide attractive strategies for cancer therapeutics. Furthermore, the intricate p53 isoform network modulates many biological processes, cooperation with other proteins, etc. Thus, a thorough understanding of the p53-isoform-mediated biological processes can open up new avenues for cancer therapeutics.
Author Contributions
Conceptualization and writing, L.Z. and S.S.; funding acquisition, S.S. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Swedish Research Council, grant numbers 2016-06264, 2018-05946 and 2018-05498; Knut and Alice Wallenberg Foundation, Grant number KAW 2017.0055, Carl Trygger Foundation, Grant numbers CTS 18:338 and CTS 19:806, and Wenner-Gren Foundation, Grant number UPD2017:0238 to S.S.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Linzer, D.I.; Levine, A.J. Characterization of a 54K dalton cellular SV40 tumor antigen present in SV40-transformed cells and uninfected embryonal carcinoma cells. Cell 1979, 17, 43–52. [Google Scholar] [CrossRef]
- Lane, D.P.; Crawford, L.V. T antigen is bound to a host protein in SV40-transformed cells. Nature 1979, 278, 261–263. [Google Scholar] [CrossRef] [PubMed]
- Wei, C.L.; Wu, Q.; Vega, V.B.; Chiu, K.P.; Ng, P.; Zhang, T.; Shahab, A.; Yong, H.C.; Fu, Y.; Weng, Z.; et al. A global map of p53 transcription-factor binding sites in the human genome. Cell 2006, 124, 207–219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vogelstein, B.; Lane, D.; Levine, A.J. Surfing the p53 network. Nature 2000, 408, 307–310. [Google Scholar] [CrossRef]
- Hernández Borrero, L.J.; El-Deiry, W.S. Tumor suppressor p53: Biology, signaling pathways, and therapeutic targeting. Biochimica et biophysica acta. Rev. Cancer 2021, 1876, 188556. [Google Scholar]
- Hollstein, M.; Sidransky, D.; Vogelstein, B.; Harris, C.C. p53 mutations in human cancers. Science 1991, 253, 49–53. [Google Scholar] [CrossRef] [Green Version]
- Baugh, E.H.; Ke, H.; Levine, A.J.; Bonneau, R.A.; Chan, C.S. Why are there hotspot mutations in the TP53 gene in human cancers? Cell Death Differ. 2018, 25, 154–160. [Google Scholar] [CrossRef]
- Kearns, S.; Lurz, R.; Orlova, E.V.; Okorokov, A.L. Two p53 tetramers bind one consensus DNA response element. Nucleic Acids Res. 2016, 44, 6185–6199. [Google Scholar] [CrossRef] [Green Version]
- Belyi, V.A.; Ak, P.; Markert, E.; Wang, H.; Hu, W.; Puzio-Kuter, A.; Levine, A.J. The origins and evolution of the p53 family of genes. Cold Spring Harb. Perspect. Biol. 2010, 2, a001198. [Google Scholar] [CrossRef] [Green Version]
- McBride, O.W.; Merry, D.; Givol, D. The gene for human p53 cellular tumor antigen is located on chromosome 17 short arm (17p13). Proc. Natl. Acad. Sci. USA 1986, 83, 130–134. [Google Scholar] [CrossRef] [Green Version]
- Surget, S.; Khoury, M.P.; Bourdon, J.C. Uncovering the role of p53 splice variants in human malignancy: A clinical perspective. OncoTargets Ther. 2013, 7, 57–68. [Google Scholar]
- Hainaut, P.; Pfeifer, G.P. Somatic TP53 Mutations in the Era of Genome Sequencing. Cold Spring Harb. Perspect. Med. 2016, 6, a026179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vieler, M.; Sanyal, S. p53 Isoforms and Their Implications in Cancer. Cancers 2018, 10, 288. [Google Scholar] [CrossRef] [Green Version]
- Joruiz, S.M.; Bourdon, J.C. p53 Isoforms: Key Regulators of the Cell Fate Decision. Cold Spring Harb. Perspect. Med. 2016, 6, a026039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matlashewski, G.; Pim, D.; Banks, L.; Crawford, L. Alternative splicing of human p53 transcripts. Oncogene Res. 1987, 1, 77–85. [Google Scholar]
- Arai, N.; Nomura, D.; Yokota, K.; Wolf, D.; Brill, E.; Shohat, O.; Rotter, V. Immunologically distinct p53 molecules generated by alternative splicing. Mol. Cell. Biol. 1986, 6, 3232–3239. [Google Scholar]
- De Laurenzi, V.; Costanzo, A.; Barcaroli, D.; Terrinoni, A.; Falco, M.; Annicchiarico-Petruzzelli, M.; Levrero, M.; Melino, G. Two new p73 splice variants, gamma and delta, with different transcriptional activity. J. Exp. Med. 1998, 188, 1763–1768. [Google Scholar] [CrossRef] [Green Version]
- Yang, A.; Kaghad, M.; Wang, Y.; Gillett, E.; Fleming, M.D.; Dötsch, V.; Andrews, N.C.; Caput, D.; McKeon, F. p63, a p53 homolog at 3q27-29, encodes multiple products with transactivating, death-inducing, and dominant-negative activities. Mol. Cell 1998, 2, 305–316. [Google Scholar] [CrossRef]
- Kaghad, M.; Bonnet, H.; Yang, A.; Creancier, L.; Biscan, J.C.; Valent, A.; Minty, A.; Chalon, P.; Lelias, J.M.; Dumont, X.; et al. Monoallelically expressed gene related to p53 at 1p36, a region frequently deleted in neuroblastoma and other human cancers. Cell 1997, 90, 809–819. [Google Scholar] [CrossRef] [Green Version]
- Courtois, S.; Verhaegh, G.; North, S.; Luciani, M.G.; Lassus, P.; Hibner, U.; Oren, M.; Hainaut, P. DeltaN-p53, a natural isoform of p53 lacking the first transactivation domain, counteracts growth suppression by wild-type p53. Oncogene 2002, 21, 6722–6728. [Google Scholar] [CrossRef] [Green Version]
- Hayman, L.; Chaudhry, W.R.; Revin, V.V.; Zhelev, N.; Bourdon, J.C. What is the potential of p53 isoforms as a predictive biomarker in the treatment of cancer? Expert Rev. Mol. Diagn. 2019, 19, 149–159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, J.; Kim, D.H.; Lee, S.W.; Choi, K.Y.; Sung, Y.C. Transactivation ability of p53 transcriptional activation domain is directly related to the binding affinity to TATA-binding protein. J. Biol. Chem. 1995, 270, 25014–25019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, J.; Zhang, S.; Jiang, J.; Chen, X. Definition of the p53 functional domains necessary for inducing apoptosis. J. Biol. Chem. 2000, 275, 39927–39934. [Google Scholar] [CrossRef] [Green Version]
- Pavletich, N.P.; Chambers, K.A.; Pabo, C.O. The DNA-binding domain of p53 contains the four conserved regions and the major mutation hot spots. Genes Dev. 1993, 7, 2556–2564. [Google Scholar] [CrossRef] [Green Version]
- Shaulsky, G.; Goldfinger, N.; Ben-Ze’ev, A.; Rotter, V. Nuclear accumulation of p53 protein is mediated by several nuclear localization signals and plays a role in tumorigenesis. Mol. Cell. Biol. 1990, 10, 6565–6577. [Google Scholar] [PubMed]
- Friedman, P.N.; Chen, X.; Bargonetti, J.; Prives, C. The p53 protein is an unusually shaped tetramer that binds directly to DNA. Proc. Natl. Acad. Sci. USA 1993, 90, 3319–3323. [Google Scholar] [CrossRef] [Green Version]
- Meek, D.W.; Anderson, C.W. Posttranslational modification of p53: Cooperative integrators of function. Cold Spring Harb. Perspect. Biol. 2009, 1, a000950. [Google Scholar] [CrossRef] [Green Version]
- Feng, L.; Lin, T.; Uranishi, H.; Gu, W.; Xu, Y. Functional analysis of the roles of posttranslational modifications at the p53 C terminus in regulating p53 stability and activity. Mol. Cell. Biol. 2005, 25, 5389–5395. [Google Scholar] [CrossRef] [Green Version]
- Marcel, V.; Dichtel-Danjoy, M.L.; Sagne, C.; Hafsi, H.; Ma, D.; Ortiz-Cuaran, S.; Olivier, M.; Hall, J.; Mollereau, B.; Hainaut, P.; et al. Biological functions of p53 isoforms through evolution: Lessons from animal and cellular models. Cell Death Differ. 2011, 18, 1815–1824. [Google Scholar] [CrossRef] [Green Version]
- Olivares-Illana, V.; Fåhraeus, R. p53 isoforms gain functions. Oncogene 2010, 29, 5113–5119. [Google Scholar] [CrossRef] [Green Version]
- Anbarasan, T.; Bourdon, J.C. The Emerging Landscape of p53 Isoforms in Physiology, Cancer and Degenerative Diseases. Int. J. Mol. Sci. 2019, 20, 6257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Zhao, Y.; Sun, P.; Zhao, M.; Su, Z.; Jin, X.; Song, W. p53β: A new prognostic marker for patients with clear-cell renal cell carcinoma from 5.3 years of median follow-up. Carcinogenesis 2018, 39, 368–374. [Google Scholar] [CrossRef]
- Chambers, S.K.; Martinez, J.D. The significance of p53 isoform expression in serous ovarian cancer. Future Oncol. 2012, 8, 683–686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bischof, K.; Knappskog, S.; Hjelle, S.M.; Stefansson, I.; Woie, K.; Salvesen, H.B.; Gjertsen, B.T.; Bjorge, L. Influence of p53 Isoform Expression on Survival in High-Grade Serous Ovarian Cancers. Sci. Rep. 2019, 9, 5244. [Google Scholar] [CrossRef] [Green Version]
- Morten, B.C.; Wong-Brown, M.W.; Scott, R.J.; Avery-Kiejda, K.A. The presence of the intron 3 16 bp duplication polymorphism of p53 (rs17878362) in breast cancer is associated with a low Δ40p53:p53 ratio and better outcome. Carcinogenesis 2016, 37, 81–86. [Google Scholar] [CrossRef]
- Zhang, X.; Groen, K.; Morten, B.C.; Steffens Reinhardt, L.; Campbell, H.G.; Braithwaite, A.W.; Bourdon, J.C.; Avery-Kiejda, K.A. Effect of p53 and its N-terminally truncated isoform, Δ40p53, on breast cancer migration and invasion. Mol. Oncol. 2021, 16, 447–465. [Google Scholar] [CrossRef]
- Ota, A.; Nakao, H.; Sawada, Y.; Karnan, S.; Wahiduzzaman, M.; Inoue, T.; Kobayashi, Y.; Yamamoto, T.; Ishii, N.; Ohashi, T.; et al. Δ40p53α suppresses tumor cell proliferation and induces cellular senescence in hepatocellular carcinoma cells. J. Cell Sci. 2017, 130, 614–625. [Google Scholar] [PubMed] [Green Version]
- Dos Santos, N.M.; de Oliveira, G.A.P.; Rocha, M.R.; Pedrote, M.M.; da Silva Ferretti, G.D.; Rangel, L.P.; Morgado-Diaz, J.A.; Silva, J.L.; Gimba, E.R.P. Loss of the p53 transactivation domain results in high amyloid aggregation of the Δ40p53 isoform in endometrial carcinoma cells. J. Biol. Chem. 2019, 294, 9430–9439. [Google Scholar] [CrossRef] [Green Version]
- Silva, J.L.; Vieira, T.C.; Cordeiro, Y.; de Oliveira, G.A.P. Nucleic acid actions on abnormal protein aggregation, phase transitions and phase separation. Curr. Opin. Struct. Biol. 2022, 73, 102346. [Google Scholar] [CrossRef]
- Kovachev, P.S.; Banerjee, D.; Rangel, L.P.; Eriksson, J.; Pedrote, M.M.; Martins-Dinis, M.; Edwards, K.; Cordeiro, Y.; Silva, J.L.; Sanyal, S. Distinct modulatory role of RNA in the aggregation of the tumor suppressor protein p53 core domain. J. Biol. Chem. 2017, 292, 9345–9357. [Google Scholar] [CrossRef] [Green Version]
- Haaland, I.; Hjelle, S.M.; Reikvam, H.; Sulen, A.; Ryningen, A.; McCormack, E.; Bruserud, Ø.; Gjertsen, B.T. p53 Protein Isoform Profiles in AML: Correlation with Distinct Differentiation Stages and Response to Epigenetic Differentiation Therapy. Cells 2021, 10, 833. [Google Scholar] [CrossRef] [PubMed]
- Khoury, M.P.; Bourdon, J.C. The isoforms of the p53 protein. Cold Spring Harb. Perspect. Biol. 2010, 2, a000927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marcel, V.; Perrier, S.; Aoubala, M.; Ageorges, S.; Groves, M.J.; Diot, A.; Fernandes, K.; Tauro, S.; Bourdon, J.C. Δ160p53 is a novel N-terminal p53 isoform encoded by Δ133p53 transcript. FEBS Lett. 2010, 584, 4463–4468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marcel, V.; Vijayakumar, V.; Fernández-Cuesta, L.; Hafsi, H.; Sagne, C.; Hautefeuille, A.; Olivier, M.; Hainaut, P. p53 regulates the transcription of its Delta133p53 isoform through specific response elements contained within the TP53 P2 internal promoter. Oncogene 2010, 29, 2691–2700. [Google Scholar] [CrossRef] [Green Version]
- Hurd, C.; Dinda, S.; Khattree, N.; Moudgil, V.K. Estrogen-dependent and independent activation of the P1 promoter of the p53 gene in transiently transfected breast cancer cells. Oncogene 1999, 18, 1067–1072. [Google Scholar] [CrossRef] [Green Version]
- Aoubala, M.; Murray-Zmijewski, F.; Khoury, M.P.; Fernandes, K.; Perrier, S.; Bernard, H.; Prats, A.C.; Lane, D.P.; Bourdon, J.C. p53 directly transactivates Δ133p53α, regulating cell fate outcome in response to DNA damage. Cell Death Differ. 2011, 18, 248–258. [Google Scholar] [CrossRef] [Green Version]
- Marcel, V.; Petit, I.; Murray-Zmijewski, F.; de Rugy, T.G.; Fernandes, K.; Meuray, V.; Diot, A.; Lane, D.P.; Aberdam, D.; Bourdon, J.C. Diverse p63 and p73 isoforms regulate Δ133p53 expression through modulation of the internal TP53 promoter activity. Cell Death Differ. 2012, 19, 816–826. [Google Scholar] [CrossRef]
- Ghosh, A.; Stewart, D.; Matlashewski, G. Regulation of human p53 activity and cell localization by alternative splicing. Mol. Cell. Biol. 2004, 24, 7987–7997. [Google Scholar] [CrossRef] [Green Version]
- Marcel, V.; Tran, P.L.; Sagne, C.; Martel-Planche, G.; Vaslin, L.; Teulade-Fichou, M.P.; Hall, J.; Mergny, J.L.; Hainaut, P.; Van Dyck, E. G-quadruplex structures in TP53 intron 3: Role in alternative splicing and in production of p53 mRNA isoforms. Carcinogenesis 2011, 32, 271–278. [Google Scholar] [CrossRef] [Green Version]
- Marcel, V.; Fernandes, K.; Terrier, O.; Lane, D.P.; Bourdon, J.C. Modulation of p53β and p53γ expression by regulating the alternative splicing of TP53 gene modifies cellular response. Cell Death Differ. 2014, 21, 1377–1387. [Google Scholar] [CrossRef] [Green Version]
- Tang, Y.; Horikawa, I.; Ajiro, M.; Robles, A.I.; Fujita, K.; Mondal, A.M.; Stauffer, J.K.; Zheng, Z.M.; Harris, C.C. Downregulation of splicing factor SRSF3 induces p53β, an alternatively spliced isoform of p53 that promotes cellular senescence. Oncogene 2013, 32, 2792–2798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ray, P.S.; Grover, R.; Das, S. Two internal ribosome entry sites mediate the translation of p53 isoforms. EMBO Rep. 2006, 7, 404–410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharathchandra, A.; Katoch, A.; Das, S. IRES mediated translational regulation of p53 isoforms. Wiley interdisciplinary reviews. RNA 2014, 5, 131–139. [Google Scholar] [PubMed]
- Grover, R.; Ray, P.S.; Das, S. Polypyrimidine tract binding protein regulates IRES-mediated translation of p53 isoforms. Cell Cycle 2008, 7, 2189–2198. [Google Scholar] [CrossRef] [Green Version]
- Sharathchandra, A.; Lal, R.; Khan, D.; Das, S. Annexin A2 and PSF proteins interact with p53 IRES and regulate translation of p53 mRNA. RNA Biol. 2012, 9, 1429–1439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weingarten-Gabbay, S.; Khan, D.; Liberman, N.; Yoffe, Y.; Bialik, S.; Das, S.; Oren, M.; Kimchi, A. The translation initiation factor DAP5 promotes IRES-driven translation of p53 mRNA. Oncogene 2014, 33, 611–618. [Google Scholar] [CrossRef] [PubMed]
- Halaby, M.J.; Li, Y.; Harris, B.R.; Jiang, S.; Miskimins, W.K.; Cleary, M.P.; Yang, D.Q. Translational Control Protein 80 Stimulates IRES-Mediated Translation of p53 mRNA in Response to DNA Damage. BioMed Res. Int. 2015, 2015, 708158. [Google Scholar] [CrossRef] [Green Version]
- Halaby, M.J.; Harris, B.R.; Miskimins, W.K.; Cleary, M.P.; Yang, D.Q. Deregulation of Internal Ribosome Entry Site-Mediated p53 Translation in Cancer Cells with Defective p53 Response to DNA Damage. Mol. Cell. Biol. 2015, 35, 4006–4017. [Google Scholar] [CrossRef] [Green Version]
- Ji, B.; Harris, B.R.; Liu, Y.; Deng, Y.; Gradilone, S.A.; Cleary, M.P.; Liu, J.; Yang, D.Q. Targeting IRES-Mediated p53 Synthesis for Cancer Diagnosis and Therapeutics. Int. J. Mol. Sci. 2017, 18, 93. [Google Scholar] [CrossRef] [Green Version]
- Picksley, S.M.; Lane, D.P. The p53-mdm2 autoregulatory feedback loop: A paradigm for the regulation of growth control by p53? BioEssays News Rev. Mol. Cell. Dev. Biol. 1993, 15, 689–690. [Google Scholar] [CrossRef]
- Chen, J.; Lin, J.; Levine, A.J. Regulation of transcription functions of the p53 tumor suppressor by the mdm-2 oncogene. Mol. Med. 1995, 1, 142–152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michael, D.; Oren, M. The p53-Mdm2 module and the ubiquitin system. Semin. Cancer Biol. 2003, 13, 49–58. [Google Scholar] [CrossRef]
- Haupt, Y.; Maya, R.; Kazaz, A.; Oren, M. Mdm2 promotes the rapid degradation of p53. Nature 1997, 387, 296–299. [Google Scholar] [CrossRef] [PubMed]
- Shirangi, T.R.; Zaika, A.; Moll, U.M. Nuclear degradation of p53 occurs during down-regulation of the p53 response after DNA damage. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2002, 16, 420–422. [Google Scholar] [CrossRef]
- Hafsi, H.; Santos-Silva, D.; Courtois-Cox, S.; Hainaut, P. Effects of Δ40p53, an isoform of p53 lacking the N-terminus, on transactivation capacity of the tumor suppressor protein p53. BMC Cancer 2013, 13, 134. [Google Scholar] [CrossRef] [Green Version]
- Ma, J.; Martin, J.D.; Zhang, H.; Auger, K.R.; Ho, T.F.; Kirkpatrick, R.B.; Grooms, M.H.; Johanson, K.O.; Tummino, P.J.; Copeland, R.A.; et al. A second p53 binding site in the central domain of Mdm2 is essential for p53 ubiquitination. Biochemistry 2006, 45, 9238–9245. [Google Scholar] [CrossRef]
- Reinhardt, L.S.; Zhang, X.; Wawruszak, A.; Groen, K.; De Iuliis, G.N.; Avery-Kiejda, K.A. Good Cop, Bad Cop: Defining the Roles of Δ40p53 in Cancer and Aging. Cancers 2020, 12, 1659. [Google Scholar] [CrossRef]
- Gudikote, J.P.; Cascone, T.; Poteete, A.; Sitthideatphaiboon, P.; Wu, Q.; Morikawa, N.; Zhang, F.; Peng, S.; Tong, P.; Li, L.; et al. Inhibition of nonsense-mediated decay rescues p53β/γ isoform expression and activates the p53 pathway in MDM2-overexpressing and select p53-mutant cancers. J. Biol. Chem. 2021, 297, 101163. [Google Scholar] [CrossRef]
- Fujita, K.; Mondal, A.M.; Horikawa, I.; Nguyen, G.H.; Kumamoto, K.; Sohn, J.J.; Bowman, E.D.; Mathe, E.A.; Schetter, A.J.; Pine, S.R.; et al. p53 isoforms Delta133p53 and p53beta are endogenous regulators of replicative cellular senescence. Nat. Cell Biol. 2009, 11, 1135–1142. [Google Scholar] [CrossRef]
- Tao, T.; Shi, H.; Guan, Y.; Huang, D.; Chen, Y.; Lane, D.P.; Chen, J.; Peng, J. Def defines a conserved nucleolar pathway that leads p53 to proteasome-independent degradation. Cell Res. 2013, 23, 620–634. [Google Scholar] [CrossRef] [Green Version]
- Horikawa, I.; Fujita, K.; Jenkins, L.M.; Hiyoshi, Y.; Mondal, A.M.; Vojtesek, B.; Lane, D.P.; Appella, E.; Harris, C.C. Autophagic degradation of the inhibitory p53 isoform Δ133p53α as a regulatory mechanism for p53-mediated senescence. Nat. Commun. 2014, 5, 4706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Solomon, H.; Bräuning, B.; Fainer, I.; Ben-Nissan, G.; Rabani, S.; Goldfinger, N.; Moscovitz, O.; Shakked, Z.; Rotter, V.; Sharon, M. Post-translational regulation of p53 function through 20S proteasome-mediated cleavage. Cell Death Differ. 2017, 24, 2187–2198. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; He, Y.; Dubois, W.; Wu, X.; Shi, J.; Huang, J. Distinct regulatory mechanisms and functions for p53-activated and p53-repressed DNA damage response genes in embryonic stem cells. Mol. Cell 2012, 46, 30–42. [Google Scholar] [CrossRef] [Green Version]
- Fischer, M. Census and evaluation of p53 target genes. Oncogene 2017, 36, 3943–3956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujita, K. p53 Isoforms in Cellular Senescence- and Ageing-Associated Biological and Physiological Functions. Int. J. Mol. Sci. 2019, 20, 6023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arsic, N.; Ho-Pun-Cheung, A.; Lopez-Crapez, E.; Assenat, E.; Jarlier, M.; Anguille, C.; Colard, M.; Pezet, M.; Roux, P.; Gadea, G. The p53 isoform delta133p53ß regulates cancer cell apoptosis in a RhoB-dependent manner. PLoS ONE 2017, 12, e0172125. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Manceau, A.; Frydman, L.; Cappuccio, L.; Neves, D.; Basso, V.; Wang, H.; Fombonne, J.; Maisse, C.; Mehlen, P.; et al. Δ40p53 isoform up-regulates netrin-1/UNC5B expression and potentiates netrin-1 pro-oncogenic activity. Proc. Natl. Acad. Sci. USA. 2021, 118, e2103319118. [Google Scholar] [CrossRef]
- von Muhlinen, N.; Horikawa, I.; Alam, F.; Isogaya, K.; Lissa, D.; Vojtesek, B.; Lane, D.P.; Harris, C.C. p53 isoforms regulate premature aging in human cells. Oncogene 2018, 37, 2379–2393. [Google Scholar] [CrossRef]
- Turnquist, C.; Beck, J.A.; Horikawa, I.; Obiorah, I.E.; Von Muhlinen, N.; Vojtesek, B.; Lane, D.P.; Grunseich, C.; Chahine, J.J.; Ames, H.M.; et al. Radiation-induced astrocyte senescence is rescued by Δ133p53. Neuro-Oncol. 2019, 21, 474–485. [Google Scholar] [CrossRef] [Green Version]
- Mehta, S.; Campbell, H.; Drummond, C.J.; Li, K.; Murray, K.; Slatter, T.; Bourdon, J.C.; Braithwaite, A.W. Adaptive homeostasis and the p53 isoform network. EMBO Rep. 2021, 22, e53085. [Google Scholar] [CrossRef]
- Gorgoulis, V.; Adams, P.D.; Alimonti, A.; Bennett, D.C.; Bischof, O.; Bishop, C.; Campisi, J.; Collado, M.; Evangelou, K.; Ferbeyre, G.; et al. Cellular Senescence: Defining a Path Forward. Cell 2019, 179, 813–827. [Google Scholar] [CrossRef] [PubMed]
- Falcicchio, M.; Ward, J.A.; Macip, S.; Doveston, R.G. Regulation of p53 by the 14-3-3 protein interaction network: New opportunities for drug discovery in cancer. Cell Death Discov. 2020, 6, 126. [Google Scholar] [CrossRef] [PubMed]
- Bourougaa, K.; Naski, N.; Boularan, C.; Mlynarczyk, C.; Candeias, M.M.; Marullo, S.; Fåhraeus, R. Endoplasmic reticulum stress induces G2 cell-cycle arrest via mRNA translation of the p53 isoform p53/47. Mol. Cell 2010, 38, 78–88. [Google Scholar] [CrossRef] [PubMed]
- Niculescu, A.B.; 3rd Chen, X.; Smeets, M.; Hengst, L.; Prives, C.; Reed, S.I. Effects of p21(Cip1/Waf1) at both the G1/S and the G2/M cell cycle transitions: pRb is a critical determinant in blocking DNA replication and in preventing endoreduplication. Mol. Cell. Biol. 1998, 18, 629–643. [Google Scholar] [CrossRef] [Green Version]
- Barr, A.R.; Cooper, S.; Heldt, F.S.; Butera, F.; Stoy, H.; Mansfeld, J.; Novák, B.; Bakal, C. DNA damage during S-phase mediates the proliferation-quiescence decision in the subsequent G1 via p21 expression. Nat. Commun. 2017, 8, 14728. [Google Scholar] [CrossRef] [Green Version]
- Chen, F.; Hu, S.J. Effect of microRNA-34a in cell cycle, differentiation, and apoptosis: A review. J. Biochem. Mol. Toxicol. 2012, 26, 79–86. [Google Scholar] [CrossRef]
- Blagosklonny, M.V. Cell cycle arrest is not senescence. Aging 2011, 3, 94–101. [Google Scholar] [CrossRef] [Green Version]
- Gambino, V.; De Michele, G.; Venezia, O.; Migliaccio, P.; Dall’Olio, V.; Bernard, L.; Minardi, S.P.; Della Fazia, M.A.; Bartoli, D.; Servillo, G.; et al. Oxidative stress activates a specific p53 transcriptional response that regulates cellular senescence and aging. Aging Cell 2013, 12, 435–445. [Google Scholar] [CrossRef] [Green Version]
- Horikawa, I.; Park, K.Y.; Isogaya, K.; Hiyoshi, Y.; Li, H.; Anami, K.; Robles, A.I.; Mondal, A.M.; Fujita, K.; Serrano, M.; et al. Δ133p53 represses p53-inducible senescence genes and enhances the generation of human induced pluripotent stem cells. Cell Death Differ. 2017, 24, 1017–1028. [Google Scholar] [CrossRef]
- Mondal, A.M.; Horikawa, I.; Pine, S.R.; Fujita, K.; Morgan, K.M.; Vera, E.; Mazur, S.J.; Appella, E.; Vojtesek, B.; Blasco, M.A.; et al. p53 isoforms regulate aging- and tumor-associated replicative senescence in T lymphocytes. J. Clin. Investig. 2013, 123, 5247–5257. [Google Scholar] [CrossRef] [Green Version]
- Bourdon, J.C.; Fernandes, K.; Murray-Zmijewski, F.; Liu, G.; Diot, A.; Xirodimas, D.P.; Saville, M.K.; Lane, D.P. p53 isoforms can regulate p53 transcriptional activity. Genes Dev. 2005, 19, 2122–2137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adams, C.M.; Clark-Garvey, S.; Porcu, P.; Eischen, C.M. Targeting the Bcl-2 Family in B Cell Lymphoma. Front. Oncol. 2018, 8, 636. [Google Scholar] [CrossRef] [PubMed]
- Antonsson, B.; Conti, F.; Ciavatta, A.; Montessuit, S.; Lewis, S.; Martinou, I.; Bernasconi, L.; Bernard, A.; Mermod, J.J.; Mazzei, G.; et al. Inhibition of Bax channel-forming activity by Bcl-2. Science 1997, 277, 370–372. [Google Scholar] [CrossRef] [PubMed]
- Schott, A.F.; Apel, I.J.; Nuñez, G.; Clarke, M.F. Bcl-XL protects cancer cells from p53-mediated apoptosis. Oncogene 1995, 11, 1389–1394. [Google Scholar]
- Berube, C.; Boucher, L.M.; Ma, W.; Wakeham, A.; Salmena, L.; Hakem, R.; Yeh, W.C.; Mak, T.W.; Benchimol, S. Apoptosis caused by p53-induced protein with death domain (PIDD) depends on the death adapter protein RAIDD. Proc. Natl. Acad. Sci. USA 2005, 102, 14314–14320. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Ng, S.M.; Chang, C.; Zhang, Z.; Bourdon, J.C.; Lane, D.P.; Peng, J. p53 isoform delta113p53 is a p53 target gene that antagonizes p53 apoptotic activity via BclxL activation in zebrafish. Genes Dev. 2009, 23, 278–290. [Google Scholar] [CrossRef] [Green Version]
- Gong, L.; Pan, X.; Abali, G.K.; Little, J.B.; Yuan, Z.M. Functional interplay between p53 and Δ133p53 in adaptive stress response. Cell Death Differ. 2020, 27, 1618–1632. [Google Scholar] [CrossRef]
- Yin, Y.; Stephen, C.W.; Luciani, M.G.; Fåhraeus, R. p53 Stability and activity is regulated by Mdm2-mediated induction of alternative p53 translation products. Nat. Cell Biol. 2002, 4, 462–467. [Google Scholar] [CrossRef]
- Takahashi, R.; Markovic, S.N.; Scrable, H.J. Dominant effects of Δ40p53 on p53 function and melanoma cell fate. J. Investig. Dermatol. 2014, 134, 791–800. [Google Scholar] [CrossRef] [Green Version]
- da Silva, M.S. DNA Double-Strand Breaks: A Double-Edged Sword for Trypanosomatids. Front. Cell Dev. Biol. 2021, 9, 669041. [Google Scholar] [CrossRef]
- Lakin, N.D.; Jackson, S.P. Regulation of p53 in response to DNA damage. Oncogene 1999, 18, 7644–7655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keimling, M.; Wiesmüller, L. DNA double-strand break repair activities in mammary epithelial cells—Influence of endogenous p53 variants. Carcinogenesis 2009, 30, 1260–1268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, L.; Gong, H.; Pan, X.; Chang, C.; Ou, Z.; Ye, S.; Yin, L.; Yang, L.; Tao, T.; Zhang, Z.; et al. p53 isoform Δ113p53/Δ133p53 promotes DNA double-strand break repair to protect cell from death and senescence in response to DNA damage. Cell Res. 2015, 25, 351–369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.X.; Pan, W.Y.; Chen, J. p53 and its isoforms in DNA double-stranded break repair. J. Zhejiang Univ. Sci. B 2019, 20, 457–466. [Google Scholar] [CrossRef]
- Gong, L.; Chen, J. Δ113p53/Δ133p53 converts P53 from a repressor to a promoter of DNA double-stand break repair. Mol. Cell. Oncol. 2016, 3, e1033587. [Google Scholar] [CrossRef] [Green Version]
- Gong, L.; Pan, X.; Chen, H.; Rao, L.; Zeng, Y.; Hang, H.; Peng, J.; Xiao, L.; Chen, J. p53 isoform Δ133p53 promotes efficiency of induced pluripotent stem cells and ensures genomic integrity during reprogramming. Sci. Rep. 2016, 6, 37281. [Google Scholar] [CrossRef] [Green Version]
- Gong, H.; Zhang, Y.; Jiang, K.; Ye, S.; Chen, S.; Zhang, Q.; Peng, J.; Chen, J. p73 coordinates with Δ133p53 to promote DNA double-strand break repair. Cell Death Differ. 2018, 25, 1063–1079. [Google Scholar] [CrossRef]
- Ben-Neriah, Y.; Karin, M. Inflammation meets cancer, with NF-κB as the matchmaker. Nat. Immunol. 2011, 12, 715–723. [Google Scholar] [CrossRef]
- Carrà, G.; Lingua, M.F.; Maffeo, B.; Taulli, R.; Morotti, A. P53 vs NF-κB: The role of nuclear factor-kappa B in the regulation of p53 activity and vice versa. Cell. Mol. Life Sci. CMLS 2020, 77, 4449–4458. [Google Scholar] [CrossRef]
- Wei, J.; Noto, J.; Zaika, E.; Romero-Gallo, J.; Correa, P.; El-Rifai, W.; Peek, R.M.; Zaika, A. Pathogenic bacterium Helicobacter pylori alters the expression profile of p53 protein isoforms and p53 response to cellular stresses. Proc. Natl. Acad. Sci. USA 2012, 109, E2543–E2550. [Google Scholar] [CrossRef] [Green Version]
- Zaika, A.; Wei, J.; Noto, J.; Peek, R., Jr. Regulation of the p53 by Helicobacter pylori. Oncotarget 2012, 3, 1057–1058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.M.; Sang, X.G.; Wang, Y.Z.; Cui, C.; Zhang, L.; Ji, W.S. Role of Δ133p53 isoform in NF-κB inhibitor PDTC-mediated growth inhibition of MKN45 gastric cancer cells. World J. Gastroenterol. 2017, 23, 2716–2722. [Google Scholar] [CrossRef] [PubMed]
- Brooks, A.J.; Putoczki, T. JAK-STAT Signalling Pathway in Cancer. Cancers 2020, 12, 1971. [Google Scholar] [CrossRef] [PubMed]
- Hirano, T.; Ishihara, K.; Hibi, M. Roles of STAT3 in mediating the cell growth, differentiation and survival signals relayed through the IL-6 family of cytokine receptors. Oncogene 2000, 19, 2548–2556. [Google Scholar] [CrossRef] [PubMed]
- Wen, Z.; Zhong, Z.; Darnell, J.E., Jr. Maximal activation of transcription by Stat1 and Stat3 requires both tyrosine and serine phosphorylation. Cell 1995, 82, 241–250. [Google Scholar] [CrossRef] [Green Version]
- Grivennikov, S.I.; Karin, M. Dangerous liaisons: STAT3 and NF-kappaB collaboration and crosstalk in cancer. Cytokine Growth Factor Rev. 2010, 21, 11–19. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.; Herrmann, A.; Deng, J.H.; Kujawski, M.; Niu, G.; Li, Z.; Forman, S.; Jove, R.; Pardoll, D.M.; Yu, H. Persistently activated Stat3 maintains constitutive NF-kappaB activity in tumors. Cancer Cell 2009, 15, 283–293. [Google Scholar] [CrossRef] [Green Version]
- Bollrath, J.; Phesse, T.J.; von Burstin, V.A.; Putoczki, T.; Bennecke, M.; Bateman, T.; Nebelsiek, T.; Lundgren-May, T.; Canli, O.; Schwitalla, S.; et al. gp130-mediated Stat3 activation in enterocytes regulates cell survival and cell-cycle progression during colitis-associated tumorigenesis. Cancer Cell 2009, 15, 91–102. [Google Scholar] [CrossRef] [Green Version]
- Zeng, C.; Zeng, B.; Dong, C.; Liu, J.; Xing, F. Rho-ROCK signaling mediates entotic cell death in tumor. Cell Death Discov. 2020, 6, 4. [Google Scholar] [CrossRef] [Green Version]
- Citalán-Madrid, A.F.; García-Ponce, A.; Vargas-Robles, H.; Betanzos, A.; Schnoor, M. Small GTPases of the Ras superfamily regulate intestinal epithelial homeostasis and barrier function via common and unique mechanisms. Tissue Barriers 2013, 1, e26938. [Google Scholar] [CrossRef] [Green Version]
- Corry, J.; Mott, H.R.; Owen, D. Activation of STAT transcription factors by the Rho-family GTPases. Biochem. Soc. Trans. 2020, 48, 2213–2227. [Google Scholar] [CrossRef] [PubMed]
- Sanz-Moreno, V.; Gaggioli, C.; Yeo, M.; Albrengues, J.; Wallberg, F.; Viros, A.; Hooper, S.; Mitter, R.; Féral, C.C.; Cook, M.; et al. ROCK and JAK1 signaling cooperate to control actomyosin contractility in tumor cells and stroma. Cancer Cell 2011, 20, 229–245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campbell, H.; Fleming, N.; Roth, I.; Mehta, S.; Wiles, A.; Williams, G.; Vennin, C.; Arsic, N.; Parkin, A.; Pajic, M.; et al. ∆133p53 isoform promotes tumour invasion and metastasis via interleukin-6 activation of JAK-STAT and RhoA-ROCK signalling. Nat. Commun. 2018, 9, 254. [Google Scholar] [CrossRef] [Green Version]
- Castro, F.; Cardoso, A.P.; Gonçalves, R.M.; Serre, K.; Oliveira, M.J. Interferon-Gamma at the Crossroads of Tumor Immune Surveillance or Evasion. Front. Immunol. 2018, 9, 847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rudick, R.A.; Ransohoff, R.M. Biologic effects of interferons: Relevance to multiple sclerosis. Mult. Scler. 1995, 1 (Suppl. 1), S12–S16. [Google Scholar]
- Ivashkiv, L.B.; Donlin, L.T. Regulation of type I interferon responses. Nature reviews. Immunology 2014, 14, 36–49. [Google Scholar]
- Wack, A.; Terczyńska-Dyla, E.; Hartmann, R. Guarding the frontiers: The biology of type III interferons. Nat. Immunol. 2015, 16, 802–809. [Google Scholar] [CrossRef]
- Kazantseva, M.; Mehta, S.; Eiholzer, R.A.; Gimenez, G.; Bowie, S.; Campbell, H.; Reily-Bell, A.L.; Roth, I.; Ray, S.; Drummond, C.J.; et al. The Δ133p53β isoform promotes an immunosuppressive environment leading to aggressive prostate cancer. Cell Death Dis. 2019, 10, 631. [Google Scholar] [CrossRef]
- Mehta, S.Y.; Morten, B.C.; Antony, J.; Henderson, L.; Lasham, A.; Campbell, H.; Cunliffe, H.; Horsfield, J.A.; Reddel, R.R.; Avery-Kiejda, K.A.; et al. Regulation of the interferon-gamma (IFN-γ) pathway by p63 and Δ133p53 isoform in different breast cancer subtypes. Oncotarget 2018, 9, 29146–29161. [Google Scholar] [CrossRef] [Green Version]
- Regis, G.; Conti, L.; Boselli, D.; Novelli, F. IFNgammaR2 trafficking tunes IFNgamma-STAT1 signaling in T lymphocytes. Trends Immunol. 2006, 27, 96–101. [Google Scholar] [CrossRef]
- Dubois, J.; Traversier, A.; Julien, T.; Padey, B.; Lina, B.; Bourdon, J.C.; Marcel, V.; Boivin, G.; Rosa-Calatrava, M.; Terrier, O. The Nonstructural NS1 Protein of Influenza Viruses Modulates TP53 Splicing through Host Factor CPSF4. J. Virol. 2019, 93, e02168-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suarez, B.; Prats-Mari, L.; Unfried, J.P.; Fortes, P. LncRNAs in the Type I Interferon Antiviral Response. Int. J. Mol. Sci. 2020, 21, 6447. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Sriram, S. Identification and characterization of the interferon-beta-mediated p53 signal pathway in human peripheral blood mononuclear cells. Immunology 2009, 128, e905–e918. [Google Scholar] [CrossRef]
- Schroder, K.; Hertzog, P.J.; Ravasi, T.; Hume, D.A. Interferon-gamma: An overview of signals, mechanisms and functions. J. Leukoc. Biol. 2004, 75, 163–189. [Google Scholar] [CrossRef] [PubMed]
- Saha, B.; Jyothi Prasanna, S.; Chandrasekar, B.; Nandi, D. Gene modulation and immunoregulatory roles of interferon gamma. Cytokine 2010, 50, 1–14. [Google Scholar] [CrossRef]
- Levy, D.E.; Darnell, J.E., Jr. Stats: Transcriptional control and biological impact. Nature reviews. Mol. Cell Biol. 2002, 3, 651–662. [Google Scholar]
- Bourdon, J.C. p53 and its isoforms in cancer. Br. J. Cancer 2007, 97, 277–282. [Google Scholar] [CrossRef]
- Mehta, S.; Tsai, P.; Lasham, A.; Campbell, H.; Reddel, R.; Braithwaite, A.; Print, C. A Study of TP53 RNA Splicing Illustrates Pitfalls of RNA-seq Methodology. Cancer Res. 2016, 76, 7151–7159. [Google Scholar] [CrossRef] [Green Version]
- Joruiz, S.M.; Beck, J.A.; Horikawa, I.; Harris, C.C. The Δ133p53 Isoforms, Tuners of the p53 Pathway. Cancers 2020, 12, 3422. [Google Scholar] [CrossRef]
- Lasham, A.; Tsai, P.; Fitzgerald, S.J.; Mehta, S.Y.; Knowlton, N.S.; Braithwaite, A.W.; Print, C.G. Accessing a New Dimension in TP53 Biology: Multiplex Long Amplicon Digital PCR to Specifically Detect and Quantitate Individual TP53 Transcripts. Cancers 2020, 12, 769. [Google Scholar] [CrossRef] [Green Version]
- Kazantseva, M.; Eiholzer, R.A.; Mehta, S.; Taha, A.; Bowie, S.; Roth, I.; Zhou, J.; Joruiz, S.M.; Royds, J.A.; Hung, N.A.; et al. Elevation of the TP53 isoform Δ133p53β in glioblastomas: An alternative to mutant p53 in promoting tumor development. J. Pathol. 2018, 246, 77–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kosti, I.; Jain, N.; Aran, D.; Butte, A.J.; Sirota, M. Cross-tissue Analysis of Gene and Protein Expression in Normal and Cancer Tissues. Sci. Rep. 2016, 6, 24799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vogel, C.; Marcotte, E.M. Insights into the regulation of protein abundance from proteomic and transcriptomic analyses. Nature reviews. Genetics 2012, 13, 227–232. [Google Scholar]
- Jiang, W.; Liu, L.; Chen, Y. Simultaneous Detection of Human C-Terminal p53 Isoforms by Single Template Molecularly Imprinted Polymers (MIPs) Coupled with Liquid Chromatography-Tandem Mass Spectrometry (LC-MS/MS)-Based Targeted Proteomics. Anal. Chem. 2018, 90, 3058–3066. [Google Scholar] [CrossRef]
- Hofstetter, G.; Berger, A.; Berger, R.; Zorić, A.; Braicu, E.I.; Reimer, D.; Fiegl, H.; Marth, C.; Zeimet, A.G.; Ulmer, H.; et al. The N-terminally truncated p53 isoform Δ40p53 influences prognosis in mucinous ovarian cancer. Int. J. Gynecol. Cancer Off. J. Int. Gynecol. Cancer Soc. 2012, 22, 372–379. [Google Scholar] [CrossRef]
- Rojas, E.A.; Corchete, L.A.; De Ramón, C.; Krzeminski, P.; Quwaider, D.; García-Sanz, R.; Martínez-López, J.; Oriol, A.; Rosiñol, L.; Bladé, J.; et al. Expression of p53 protein isoforms predicts survival in patients with multiple myeloma. Am. J. Hematol. 2022, 97, 700–710. [Google Scholar] [CrossRef] [PubMed]
- Gadea, G.; Arsic, N.; Fernandes, K.; Diot, A.; Joruiz, S.M.; Abdallah, S.; Meuray, V.; Vinot, S.; Anguille, C.; Remenyi, J.; et al. TP53 drives invasion through expression of its Δ133p53β variant. eLife 2016, 5, e14734. [Google Scholar] [CrossRef]
- Ozretić, P.; Hanžić, N.; Proust, B.; Sabol, M.; Trnski, D.; Radić, M.; Musani, V.; Ciribilli, Y.; Milas, I.; Puljiz, Z.; et al. Expression profiles of p53/p73, NME and GLI families in metastatic melanoma tissue and cell lines. Sci. Rep. 2019, 9, 12470. [Google Scholar] [CrossRef]
- Fragou, A.; Tzimagiorgis, G.; Karageorgopoulos, C.; Barbetakis, N.; Lazopoulos, A.; Papaioannou, M.; Haitoglou, C.; Kouidou, S. Increased Δ133p53 mRNA in lung carcinoma corresponds with reduction of p21 expression. Mol. Med. Rep. 2017, 15, 1455–1460. [Google Scholar] [CrossRef] [Green Version]
- Tadijan, A.; Precazzini, F.; Hanžić, N.; Radić, M.; Gavioli, N.; Vlašić, I.; Ozretić, P.; Pinto, L.; Škreblin, L.; Barban, G.; et al. Altered Expression of Shorter p53 Family Isoforms Can Impact Melanoma Aggressiveness. Cancers 2021, 13, 5231. [Google Scholar] [CrossRef]
- Bischof, K.; Knappskog, S.; Stefansson, I.; McCormack, E.M.; Trovik, J.; Werner, H.M.J.; Woie, K.; Gjertsen, B.T.; Bjorge, L. High expression of the p53 isoform γ is associated with reduced progression-free survival in uterine serous carcinoma. BMC Cancer 2018, 18, 684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Avery-Kiejda, K.A.; Morten, B.; Wong-Brown, M.W.; Mathe, A.; Scott, R.J. The relative mRNA expression of p53 isoforms in breast cancer is associated with clinical features and outcome. Carcinogenesis 2014, 35, 586–596. [Google Scholar] [CrossRef] [PubMed]
- Nutthasirikul, N.; Limpaiboon, T.; Leelayuwat, C.; Patrakitkomjorn, S.; Jearanaikoon, P. Ratio disruption of the ∆133p53 and TAp53 isoform equilibrium correlates with poor clinical outcome in intrahepatic cholangiocarcinoma. Int. J. Oncol. 2013, 42, 1181–1188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ånensen, N.; Hjelle, S.M.; Van Belle, W.; Haaland, I.; Silden, E.; Bourdon, J.C.; Hovland, R.; Taskén, K.; Knappskog, S.; Lønning, P.E.; et al. Correlation analysis of p53 protein isoforms with NPM1/FLT3 mutations and therapy response in acute myeloid leukemia. Oncogene 2012, 31, 1533–1545. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.P.; Lozano, G. Mutant p53 partners in crime. Cell Death Differ. 2018, 25, 161–168. [Google Scholar] [CrossRef]
- Mantovani, F.; Collavin, L.; Del Sal, G. Mutant p53 as a guardian of the cancer cell. Cell Death Differ. 2019, 26, 199–212. [Google Scholar] [CrossRef]
- Hofstetter, G.; Berger, A.; Schuster, E.; Wolf, A.; Hager, G.; Vergote, I.; Cadron, I.; Sehouli, J.; Braicu, E.I.; Mahner, S.; et al. Δ133p53 is an independent prognostic marker in p53 mutant advanced serous ovarian cancer. Br. J. Cancer 2011, 105, 1593–1599. [Google Scholar] [CrossRef] [Green Version]
- Bourdon, J.C.; Khoury, M.P.; Diot, A.; Baker, L.; Fernandes, K.; Aoubala, M.; Quinlan, P.; Purdie, C.A.; Jordan, L.B.; Prats, A.C.; et al. p53 mutant breast cancer patients expressing p53γ have as good a prognosis as wild-type p53 breast cancer patients. Breast Cancer Res. BCR 2011, 13, R7. [Google Scholar] [CrossRef]
- Candeias, M.M.; Hagiwara, M.; Matsuda, M. Cancer-specific mutations in p53 induce the translation of Δ160p53 promoting tumorigenesis. EMBO Rep. 2016, 17, 1542–1551. [Google Scholar] [CrossRef]
- Godet, A.C.; David, F.; Hantelys, F.; Tatin, F.; Lacazette, E.; Garmy-Susini, B.; Prats, A.C. IRES Trans-Acting Factors, Key Actors of the Stress Response. Int. J. Mol. Sci. 2019, 20, 924. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Qian, J.; Gu, C.; Yang, Y. Alternative splicing and cancer: A systematic review. Signal Transduct. Target. Ther. 2021, 6, 78. [Google Scholar] [CrossRef] [PubMed]
- Ladomery, M. Aberrant alternative splicing is another hallmark of cancer. Int. J. Cell Biol. 2013, 2013, 463786. [Google Scholar] [CrossRef] [PubMed]
- Jbara, A.; Siegfried, Z.; Karni, R. Splice-switching as cancer therapy. Curr. Opin. Pharmacol. 2021, 59, 140–148. [Google Scholar] [CrossRef] [PubMed]
- Ohe, K.; Hagiwara, M. Modulation of alternative splicing with chemical compounds in new therapeutics for human diseases. ACS Chem. Biol. 2015, 10, 914–924. [Google Scholar] [CrossRef]
- Chang, J.G.; Yang, D.M.; Chang, W.H.; Chow, L.P.; Chan, W.L.; Lin, H.H.; Huang, H.D.; Chang, Y.S.; Hung, C.H.; Yang, W.K. Small molecule amiloride modulates oncogenic RNA alternative splicing to devitalize human cancer cells. PLoS ONE 2011, 6, e18643. [Google Scholar] [CrossRef] [Green Version]
- Lu, G.Y.; Huang, S.M.; Liu, S.T.; Liu, P.Y.; Chou, W.Y.; Lin, W.S. Caffeine induces tumor cytotoxicity via the regulation of alternative splicing in subsets of cancer-associated genes. Int. J. Biochem. Cell Biol. 2014, 47, 83–92. [Google Scholar] [CrossRef]
- Ghosh, G.; Adams, J.A. Phosphorylation mechanism and structure of serine-arginine protein kinases. FEBS J. 2011, 278, 587–597. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Z.; Fu, X.D. Regulation of splicing by SR proteins and SR protein-specific kinases. Chromosoma 2013, 122, 191–207. [Google Scholar] [CrossRef]
- Jeong, S. SR Proteins: Binders, Regulators, and Connectors of RNA. Mol. Cells 2017, 40, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Fedorov, O.; Huber, K.; Eisenreich, A.; Filippakopoulos, P.; King, O.; Bullock, A.N.; Szklarczyk, D.; Jensen, L.J.; Fabbro, D.; Trappe, J.; et al. Specific CLK inhibitors from a novel chemotype for regulation of alternative splicing. Chem. Biol. 2011, 18, 67–76. [Google Scholar] [CrossRef] [Green Version]
- Sohail, M.; Shkreta, L.; Toutant, J.; Rabea, S.; Babeu, J.P.; Huard, C.; Coulombe-Huntington, J.; Delannoy, A.; Placet, M.; Geha, S.; et al. A novel class of inhibitors that target SRSF10 and promote p53-mediated cytotoxicity on human colorectal cancer cells. NAR Cancer 2021, 3, zcab019. [Google Scholar] [CrossRef] [PubMed]
- Bauman, J.; Jearawiriyapaisarn, N.; Kole, R. Therapeutic potential of splice-switching oligonucleotides. Oligonucleotides 2009, 19, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Fu, Y.; Thakur, C.; Bi, Z.; Wadgaonkar, P.; Qiu, Y.; Xu, L.; Rice, M.; Zhang, W.; Almutairy, B.; et al. CRISPR-Cas9 gene editing causes alternative splicing of the targeting mRNA. Biochem. Biophys. Res. Commun. 2020, 528, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Cowen, L.E.; Tang, Y. Identification of nonsense-mediated mRNA decay pathway as a critical regulator of p53 isoform β. Sci. Rep. 2017, 7, 17535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lei, J.; Qi, R.; Tang, Y.; Wang, W.; Wei, G.; Nussinov, R.; Ma, B. Conformational stability and dynamics of the cancer-associated isoform Δ133p53β are modulated by p53 peptides and p53-specific DNA. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2019, 33, 4225–4235. [Google Scholar] [CrossRef] [PubMed]
- Paradisi, A.; Creveaux, M.; Gibert, B.; Devailly, G.; Redoulez, E.; Neves, D.; Cleyssac, E.; Treilleux, I.; Klein, C.; Niederfellner, G.; et al. Combining chemotherapeutic agents and netrin-1 interference potentiates cancer cell death. EMBO Mol. Med. 2013, 5, 1821–1834. [Google Scholar] [CrossRef] [Green Version]
- Kazantseva, M.; Mehta, S.; Eiholzer, R.A.; Hung, N.; Wiles, A.; Slatter, T.L.; Braithwaite, A.W. A mouse model of the Δ133p53 isoform: Roles in cancer progression and inflammation. Mamm. Genome Off. J. Int. Mamm. Genome Soc. 2018, 29, 831–842. [Google Scholar] [CrossRef]
- Bose, S.; Banerjee, S.; Mondal, A.; Chakraborty, U.; Pumarol, J.; Croley, C.R.; Bishayee, A. Targeting the JAK/STAT Signaling Pathway Using Phytocompounds for Cancer Prevention and Therapy. Cells 2020, 9, 1451. [Google Scholar] [CrossRef]
Figure 1.
Constitution of the human TP53 gene and p53 isoforms. (A) Schematic representation of different elements of the TP53 gene. The TP53 gene is composed of 11 exons marked with numbers 1–11. It has two promoter regions P1 and P2, which produce transcripts of different length for expression of different p53 isoforms. The RNA transcripts for FL53 and its isoforms are also generated by alternative splicing of the introns (i2 and i9), and alternative initiation of translation. The stop codons (T) present in i2 and exon 11 are indicated with T. (B) Schematic representation of the p53 isoforms. p53α (FLp53) has seven structural components: two transactivation domains (TAD-1 and -2), a proline-rich domain (PRD), a DNA binding domain (DBD), a nuclear localization signaling region (NLS), oligomerization domain (OD), and a negative regulation domain (α). Δ40p53, Δ133p53, and Δ160p53 isoforms have a deletion of 40, 133, and 160 amino acids from the N-terminus, respectively. Their start codons are indicated. The β and γ isoforms lack the OD and the α domains in the C-terminus, and instead have extensions DQTSFQKENC and MLLDLRWCYFLINSS, respectively.
Figure 1.
Constitution of the human TP53 gene and p53 isoforms. (A) Schematic representation of different elements of the TP53 gene. The TP53 gene is composed of 11 exons marked with numbers 1–11. It has two promoter regions P1 and P2, which produce transcripts of different length for expression of different p53 isoforms. The RNA transcripts for FL53 and its isoforms are also generated by alternative splicing of the introns (i2 and i9), and alternative initiation of translation. The stop codons (T) present in i2 and exon 11 are indicated with T. (B) Schematic representation of the p53 isoforms. p53α (FLp53) has seven structural components: two transactivation domains (TAD-1 and -2), a proline-rich domain (PRD), a DNA binding domain (DBD), a nuclear localization signaling region (NLS), oligomerization domain (OD), and a negative regulation domain (α). Δ40p53, Δ133p53, and Δ160p53 isoforms have a deletion of 40, 133, and 160 amino acids from the N-terminus, respectively. Their start codons are indicated. The β and γ isoforms lack the OD and the α domains in the C-terminus, and instead have extensions DQTSFQKENC and MLLDLRWCYFLINSS, respectively.

Figure 2.
Expression of the p53 isoforms is regulated by different mechanisms. (A) Regulation at the levels of transcription and translation. At the transcriptional level, alternative promoter usage (P1 and P2) lead to the production of mRNAs for different p53 isoforms. Moreover, alternative splicing (Λ) of the introns i2 and i9, guided by G-quadruplex structures and serine/arginine-rich (SR) splicing factors (SRSFs), respectively, govern expression of the p53 N- and C- terminal variant isoforms. In addition, two distinct internal ribosome entry sites (IRESs) under action of the IRES-interacting trans-acting factors (ITAFs) can modulate the expression of p53 and Δ40p53 at the level of translation. (B) The degradation mechanisms of p53 and its isoforms. Nonsense-mediated decay (NMD) degrades the p53β and p53γ mRNAs post-transcriptionally. The MDM2-mediated ubiquitination-26S proteasome pathway degrades wild-type p53. Δ40p53 forms a heterotetramer with p53, which prohibits MDM2-mediated degradation. Proteasome-independent degradation pathways, such as the digestive organ expansion factor (Def) protein-mediated pathway and autophagy, can degrade Δ133p53α. STUB1, a chaperone-associated ubiquitin ligase, can protect Δ133p53α from autophagic degradation.
Figure 2.
Expression of the p53 isoforms is regulated by different mechanisms. (A) Regulation at the levels of transcription and translation. At the transcriptional level, alternative promoter usage (P1 and P2) lead to the production of mRNAs for different p53 isoforms. Moreover, alternative splicing (Λ) of the introns i2 and i9, guided by G-quadruplex structures and serine/arginine-rich (SR) splicing factors (SRSFs), respectively, govern expression of the p53 N- and C- terminal variant isoforms. In addition, two distinct internal ribosome entry sites (IRESs) under action of the IRES-interacting trans-acting factors (ITAFs) can modulate the expression of p53 and Δ40p53 at the level of translation. (B) The degradation mechanisms of p53 and its isoforms. Nonsense-mediated decay (NMD) degrades the p53β and p53γ mRNAs post-transcriptionally. The MDM2-mediated ubiquitination-26S proteasome pathway degrades wild-type p53. Δ40p53 forms a heterotetramer with p53, which prohibits MDM2-mediated degradation. Proteasome-independent degradation pathways, such as the digestive organ expansion factor (Def) protein-mediated pathway and autophagy, can degrade Δ133p53α. STUB1, a chaperone-associated ubiquitin ligase, can protect Δ133p53α from autophagic degradation.

Figure 3.
p53 isoforms modulate the p53-mediated cellular activities. Different cellular stresses such as ER stress, cell contact, DNA damage, oncogene expression, and oxidative stress activate the p53 signaling pathway. The TP53 gene produces multiple isoforms: p53α, p53β, p53γ, Δ40p53α/β/γ, Δ133p53α/β/γ, and Δ160p53α/β/γ. Isoforms Δ40p53, Δ133p53, and Δ160p53 have been reported to affect biological activities such as cell cycle arrest, senescence, apoptosis, and DNA DAB repair by inducing (green line) or repressing (red line) p53 target gene expression. Furthermore, p53 isoforms can regulate the expression of p53 target genes either directly (solid red and green line) or indirectly (dash red and green line). 14-3-3σ is a target gene of p53, and it induces cell cycle arrest. p53 can induce senescence by transactivating its target genes p21 and miR-34a. Activating p53 target genes such as Bcl-2 and Bcl-xL inhibits apoptosis, while p53 can induce apoptosis by activating pro-apoptotic proteins such as Bax and PIDD. Activating several p53 target genes such as RAD51, RAD52, and LIG4 contributes to DNA DSB repair.
Figure 3.
p53 isoforms modulate the p53-mediated cellular activities. Different cellular stresses such as ER stress, cell contact, DNA damage, oncogene expression, and oxidative stress activate the p53 signaling pathway. The TP53 gene produces multiple isoforms: p53α, p53β, p53γ, Δ40p53α/β/γ, Δ133p53α/β/γ, and Δ160p53α/β/γ. Isoforms Δ40p53, Δ133p53, and Δ160p53 have been reported to affect biological activities such as cell cycle arrest, senescence, apoptosis, and DNA DAB repair by inducing (green line) or repressing (red line) p53 target gene expression. Furthermore, p53 isoforms can regulate the expression of p53 target genes either directly (solid red and green line) or indirectly (dash red and green line). 14-3-3σ is a target gene of p53, and it induces cell cycle arrest. p53 can induce senescence by transactivating its target genes p21 and miR-34a. Activating p53 target genes such as Bcl-2 and Bcl-xL inhibits apoptosis, while p53 can induce apoptosis by activating pro-apoptotic proteins such as Bax and PIDD. Activating several p53 target genes such as RAD51, RAD52, and LIG4 contributes to DNA DSB repair.

Figure 4.
Mechanisms of Δ133p53-induced tumor development via several signaling pathways, including NF-κB, JAK-STAT, RhoA-ROCK, and IFN. In cancer cells, Δ133p53 expression induces the expression of NF-κB target genes, including IL-6, a key factor for activating phosphorylation of STAT. In addition, type I and II IFNs IFN-α, IFN-β, and IFN-γ can also activate the JAK-STAT signaling. STAT can stimulate the transcription of its target genes including Bcl-xL, Bcl-2, and c-Myc. STAT can also activate NF-κB. The activated NF-κB can also induce the transcription of various target genes, such as IL-6, IL-8, Bcl-xL, Bcl-2, and c-Myc. The RhoA-ROCK signaling pathway can interact with the JAK-STAT signaling pathway, resulting in increased phosphorylation of STAT. These pathways regulate genes involved in several processes, such as pro-inflammation, anti-apoptosis, and cell cycle progression, all of which aid in tumor development.
Figure 4.
Mechanisms of Δ133p53-induced tumor development via several signaling pathways, including NF-κB, JAK-STAT, RhoA-ROCK, and IFN. In cancer cells, Δ133p53 expression induces the expression of NF-κB target genes, including IL-6, a key factor for activating phosphorylation of STAT. In addition, type I and II IFNs IFN-α, IFN-β, and IFN-γ can also activate the JAK-STAT signaling. STAT can stimulate the transcription of its target genes including Bcl-xL, Bcl-2, and c-Myc. STAT can also activate NF-κB. The activated NF-κB can also induce the transcription of various target genes, such as IL-6, IL-8, Bcl-xL, Bcl-2, and c-Myc. The RhoA-ROCK signaling pathway can interact with the JAK-STAT signaling pathway, resulting in increased phosphorylation of STAT. These pathways regulate genes involved in several processes, such as pro-inflammation, anti-apoptosis, and cell cycle progression, all of which aid in tumor development.


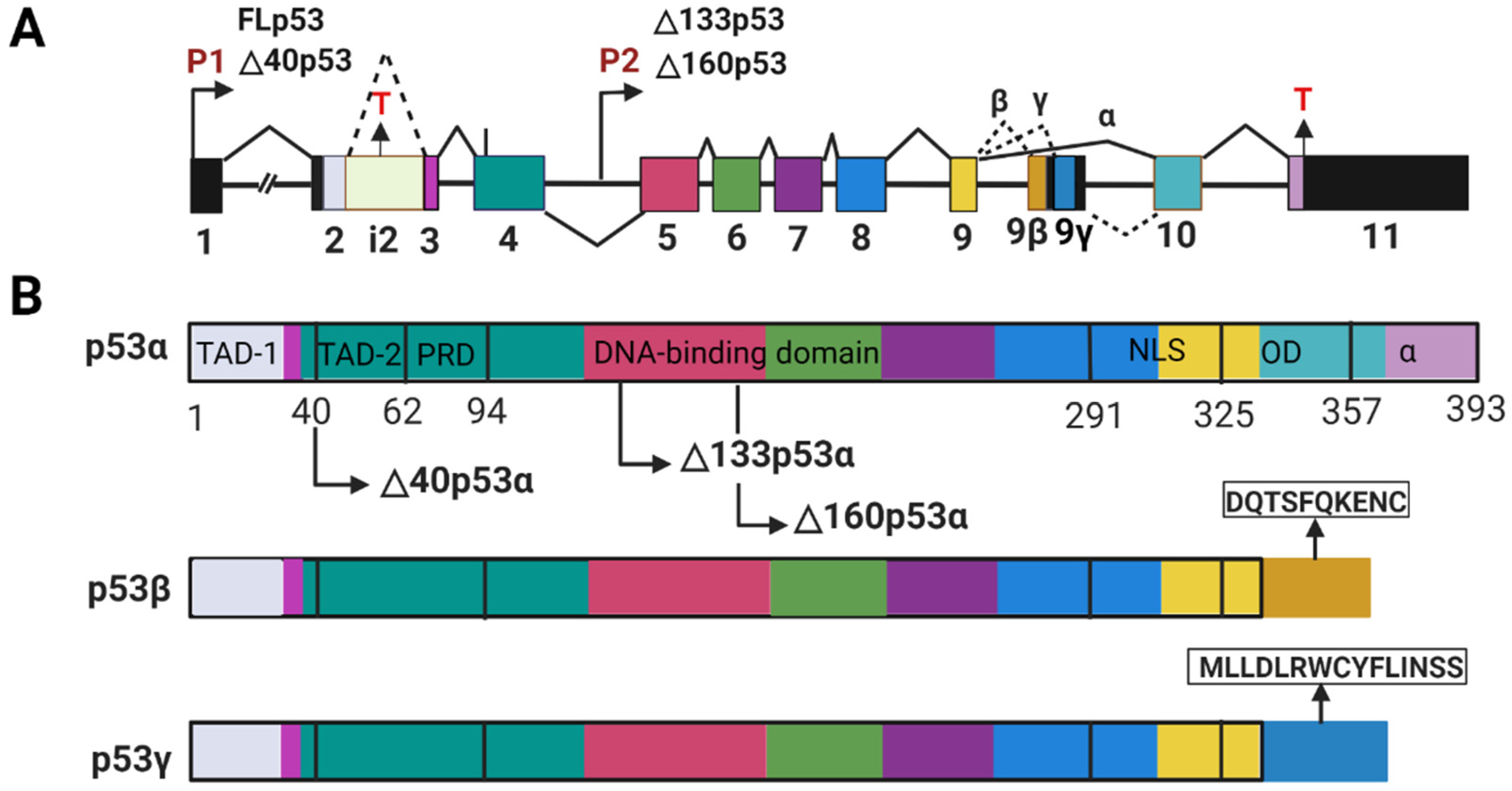{kind=link}
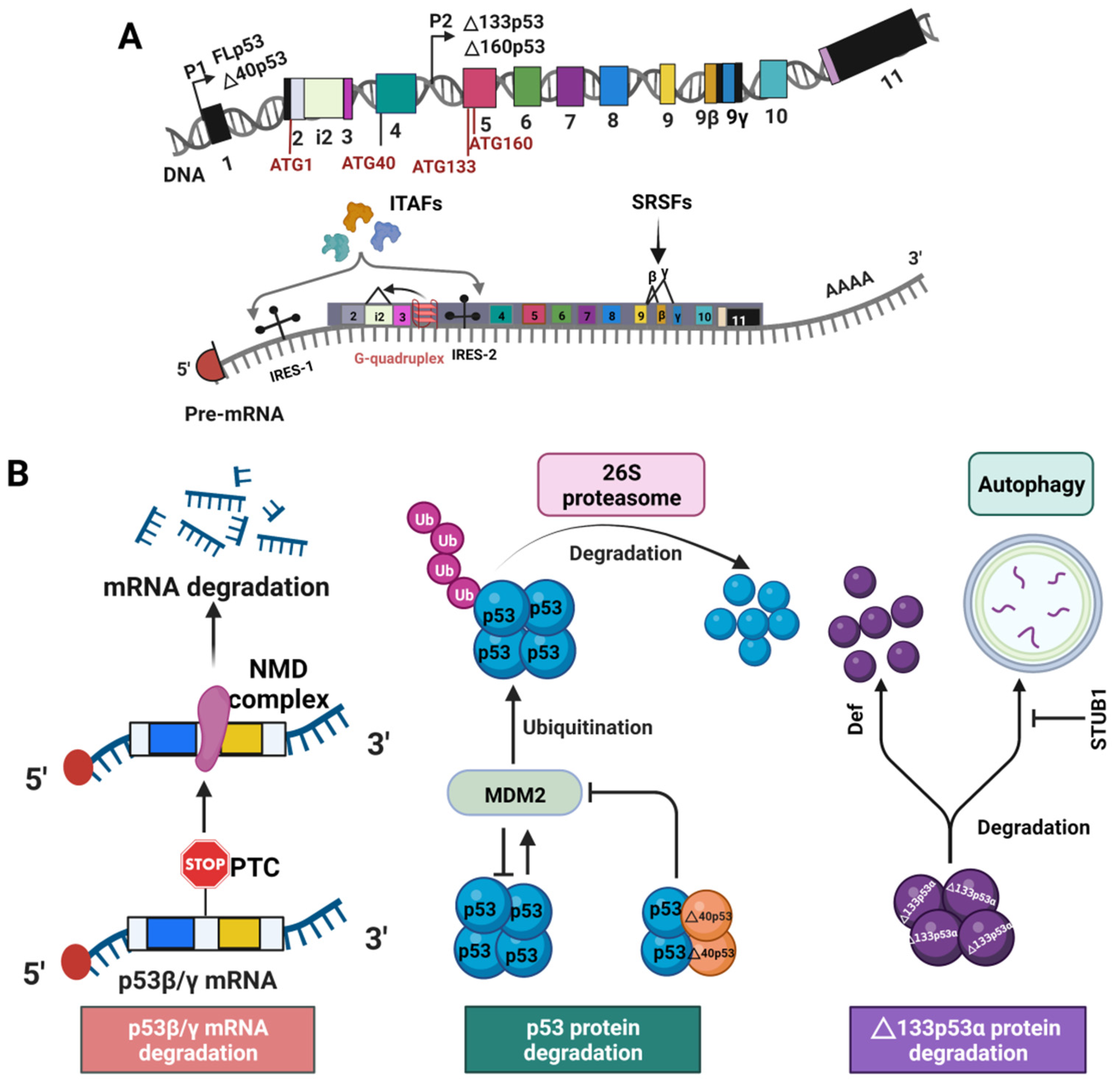{kind=link}
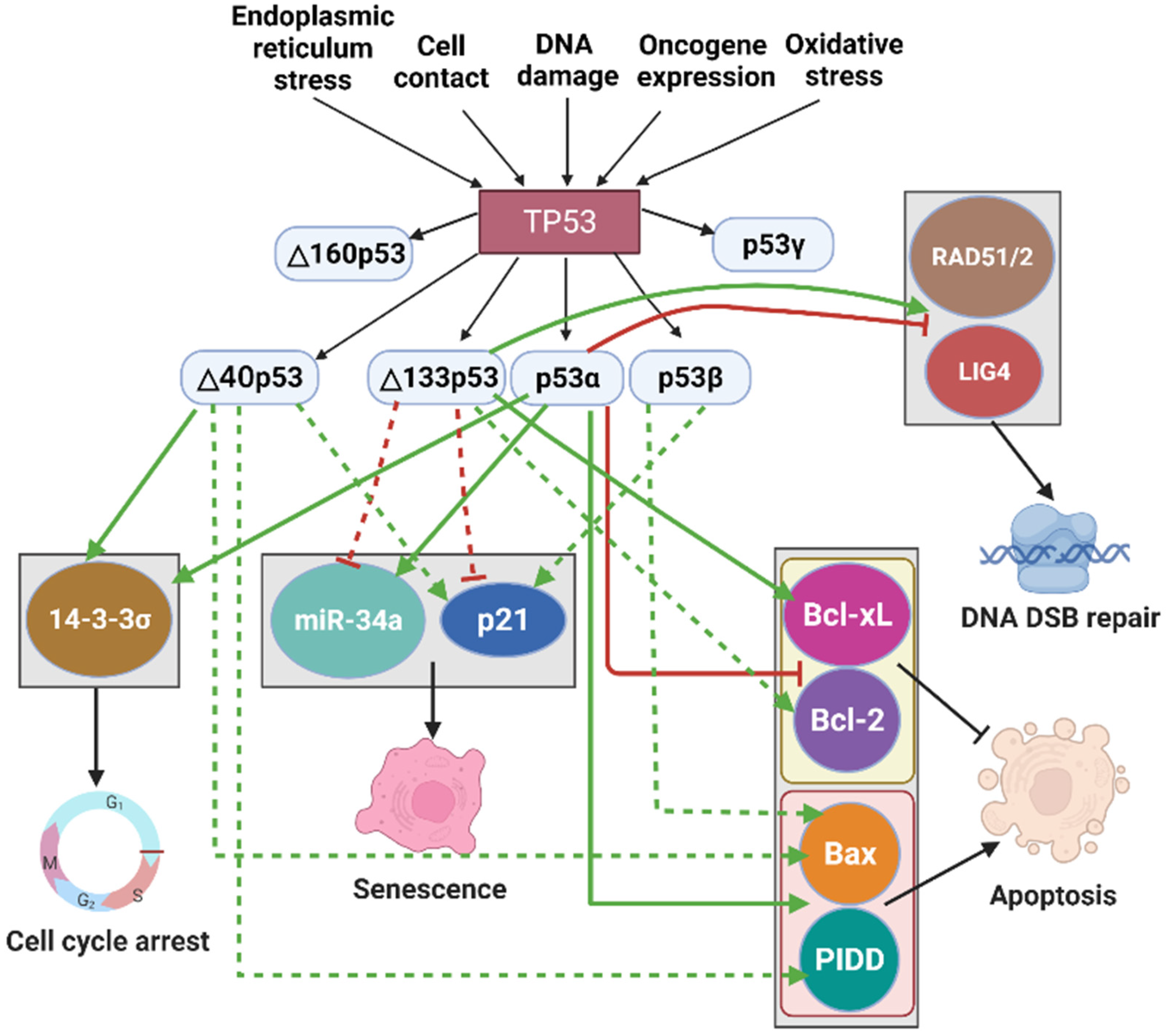{kind=link}
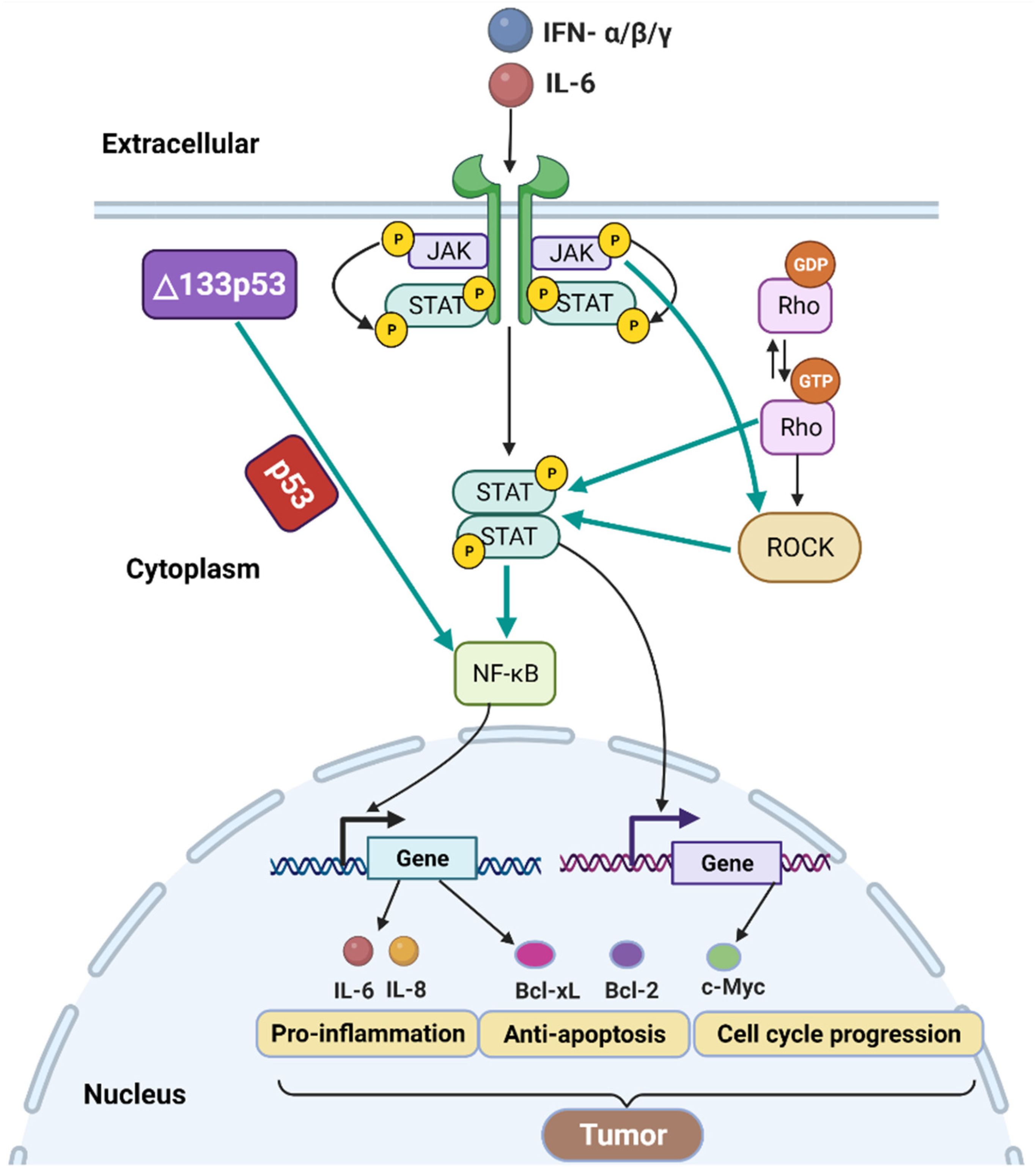{kind=link}
Table 1.
The effects of increased expression of p53 isoforms on human cancer progression.
| Tumor Types | Name of Proteins | Molecule Detected | Tumor Progression * | Reference |
|---|---|---|---|---|
| High-grade serous ovarian carcinoma | ∆133p53 | mRNA | ↓ | [34] |
| Mucinous ovarian cancer | ∆40p53 | mRNA and protein | ↓ | [145] |
| Hepatocellular carcinoma | ∆40p53 | protein | ↓ | [37] |
| Renal cell carcinoma | p53β | mRNA and protein | ↓ | [32] |
| Myeloma | ∆133p53/∆160p53 | mRNA and protein | ↓ | [146] |
| p53β/γ | mRNA and protein | ↑ | [146] | |
| Breast cancer | ∆133p53β | mRNA and protein | ↑ | [147] |
| Melanoma | ∆160p53 | mRNA and protein | ↑ | [150] |
| ∆133p53β | mRNA and protein | ↑ | [148] | |
| Colorectal cancer | ∆133p53 | mRNA | ↑ | [123] |
| Prostate cancer | ∆133p53β | mRNA | ↑ | [128] |
| Glioblastoma | ∆133p53β | mRNA | ↑ | [141] |
| Lung cancer | ∆133p53 | mRNA | ↑ | [149] |
| Endometrial carcinoma | ∆40p53 | mRNA and protein | ↑ | [38] |
| Uterine serous carcinoma | p53γ | mRNA | ↑ | [151] |
* ↑ to accelerate tumor progression, ↓ to slow down tumor progression.
Table 2.
The effects of high ratio of p53 isoforms to p53α/β on human cancer progression.
| Tumor Types | NAME of PROTEINS | Molecule Detected | Tumor Progression * | Reference |
|---|---|---|---|---|
| Breast cancer | ∆40p53:p53α | mRNA | ↑ | [152] |
| Colorectal cancer | ∆133p53:p53β | mRNA | ↑ | [69] |
| Cholangiocarcinoma | ∆133p53:p53α | mRNA | ↑ | [153] |
| Acute myeloid leukemia | p53β/γ:p53α | protein | ↓ | [154] |
* ↑ to accelerate tumor progression, ↓ to slow down tumor progression.
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Zhao, L.; Sanyal, S. p53 Isoforms as Cancer Biomarkers and Therapeutic Targets. Cancers 2022, 14, 3145. https://doi.org/10.3390/cancers14133145
AMA Style
Zhao L, Sanyal S. p53 Isoforms as Cancer Biomarkers and Therapeutic Targets. Cancers. 2022; 14(13):3145. https://doi.org/10.3390/cancers14133145
Chicago/Turabian StyleZhao, Liuqun, and Suparna Sanyal. 2022. "p53 Isoforms as Cancer Biomarkers and Therapeutic Targets" Cancers 14, no. 13: 3145. https://doi.org/10.3390/cancers14133145
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.