
Pre-PCR Mutation-Enrichment Methods for Liquid Biopsy Applications


Abstract
:Simple Summary
Abstract
1. Introduction
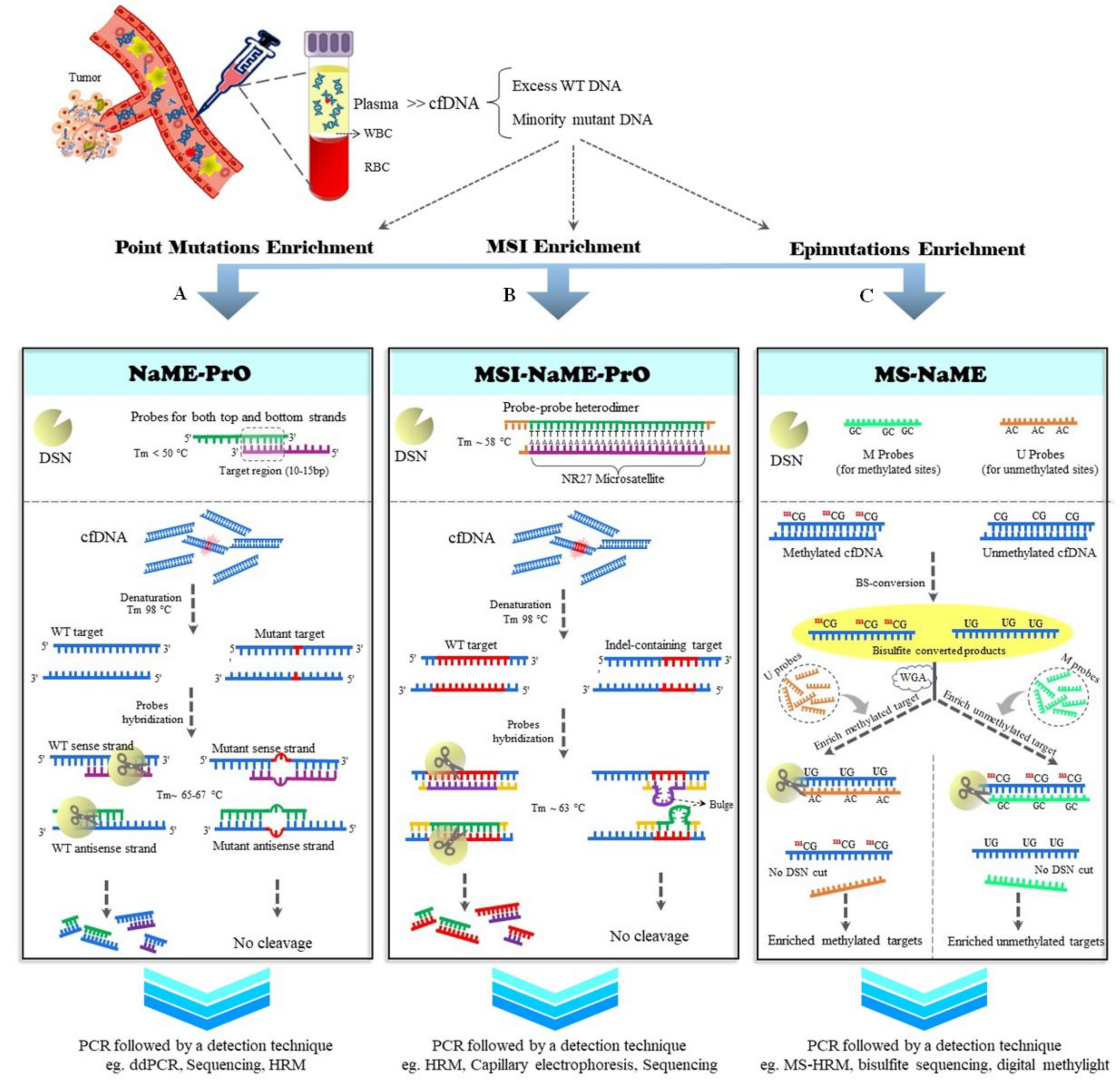
2. NaME-PrO
2.1. MSI-NaME-PrO
2.2. MS-NaME
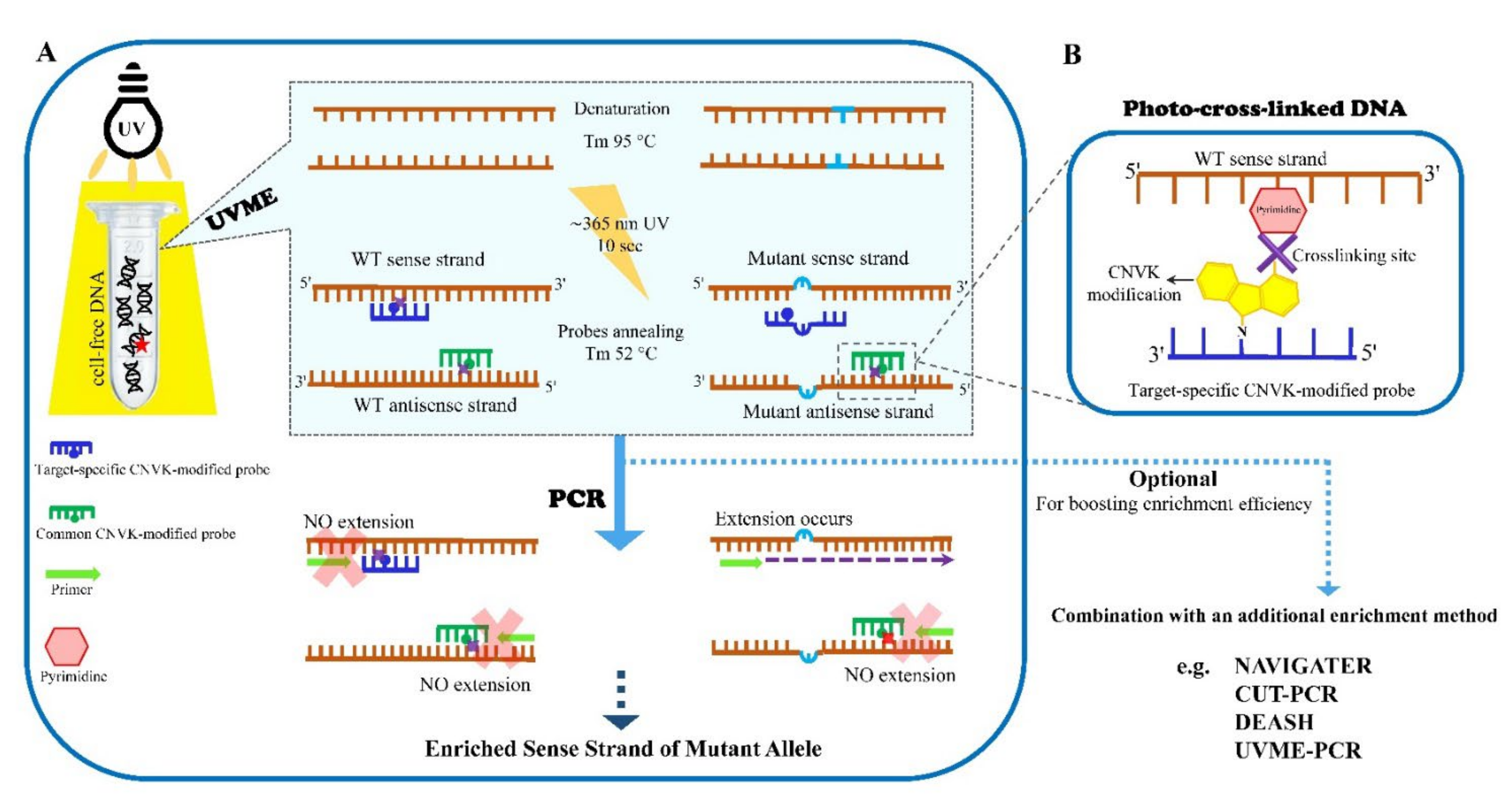
3. UVME
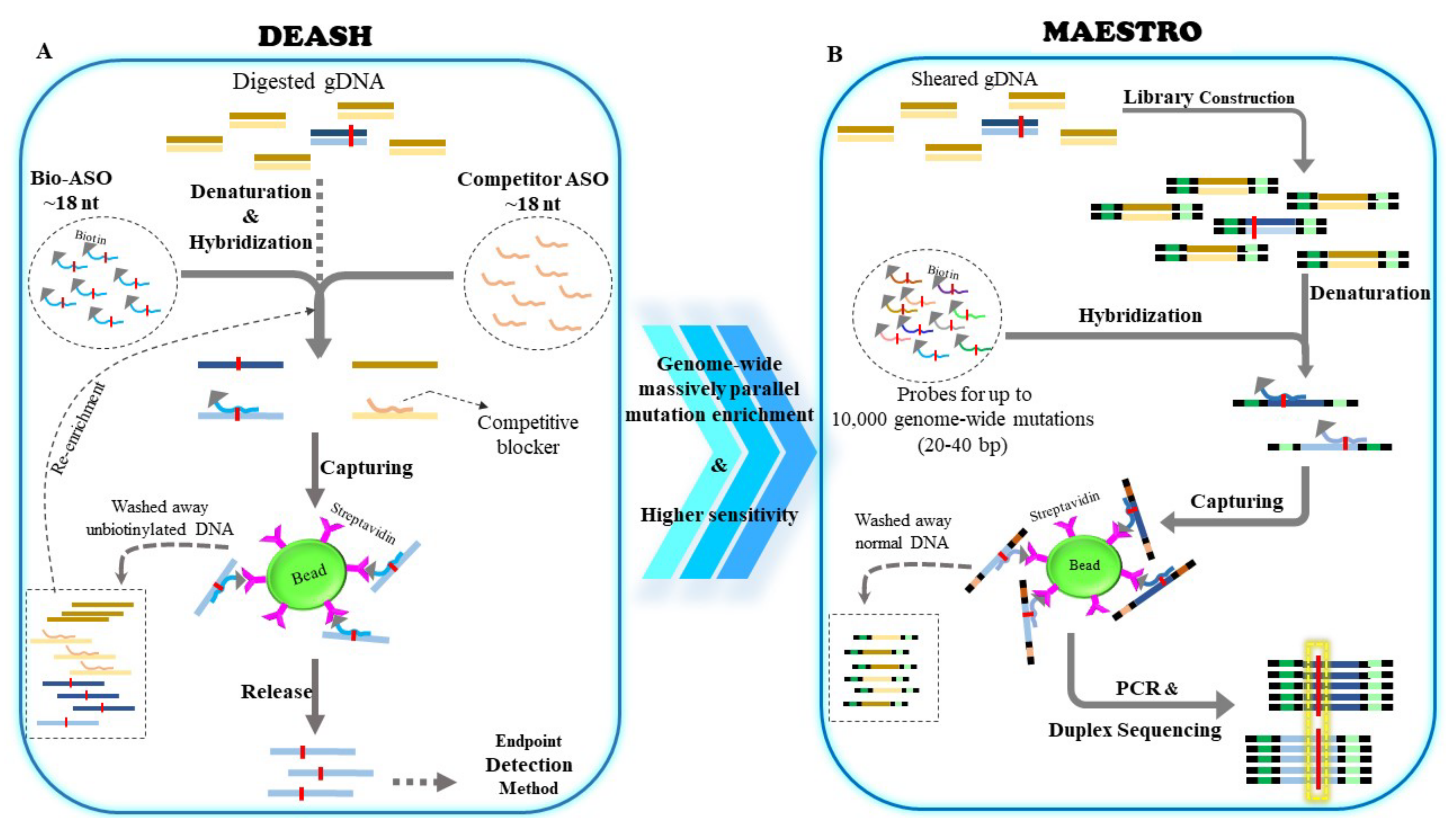
4. DEASH
5. Additional Methods for Mutation Enrichment Directly from Genomic DNA
5.1. Restriction-Enzyme-Mediated Enrichment
5.1.1. Restriction-Site Mutation (RSM) and Restriction-Fragment-Length Polymorphism (RFLP)-PCR
5.1.2. Random Mutation Capture (RMC)
5.2. CUT-PCR
5.3. NAVIGATER
5.4. PI Polyamides
6. Discussion and Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Giusti, I.; Bianchi, S.; Nottola, S.A.; Macchiarelli, G.; Dolo, V. Clinical Electron Microscopy in the Study of Human Ovarian Tissues. EuroMediterr. Biomed. J. 2019, 14, 145–151. [Google Scholar]
- Wan, J.C.; Massie, C.; Garcia-Corbacho, J.; Mouliere, F.; Brenton, J.D.; Caldas, C.; Pacey, S.; Baird, R.; Rosenfeld, N. Liquid biopsies come of age: Towards implementation of circulating tumour DNA. Nat. Rev. Cancer. 2017, 17, 223–238. [Google Scholar] [CrossRef] [PubMed]
- Alix-Panabieres, C.; Pantel, K. Clinical Applications of Circulating Tumor Cells and Circulating Tumor DNA as Liquid Biopsy. Cancer Discov. 2016, 6, 479–491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Murillas, I.; Schiavon, G.; Weigelt, B.; Ng, C.; Hrebien, S.; Cutts, R.J.; Cheang, M.; Osin, P.; Nerurkar, A.; Kozarewa, I.; et al. Mutation tracking in circulating tumor DNA predicts relapse in early breast cancer. Sci. Transl. Med. 2015, 7, 302ra133. [Google Scholar] [CrossRef]
- Newman, A.M.; Bratman, S.V.; To, J.; Wynne, J.F.; Eclov, N.C.; Modlin, L.A.; Liu, C.L.; Neal, J.W.; Wakelee, H.A.; Merritt, R.E.; et al. An ultrasensitive method for quantitating circulating tumor DNA with broad patient coverage. Nat. Med. 2014, 20, 548–554. [Google Scholar] [CrossRef]
- Parsons, H.A.; Rhoades, J.; Reed, S.C.; Gydush, G.; Ram, P.; Exman, P.; Xiong, K.; Lo, C.C.; Li, T.; Fleharty, M.; et al. Sensitive Detection of Minimal Residual Disease in Patients Treated for Early-Stage Breast Cancer. Clin. Cancer Res. 2020, 26, 2556–2564. [Google Scholar] [CrossRef] [Green Version]
- Reinert, T.; Henriksen, T.V.; Christensen, E.; Sharma, S.; Salari, R.; Sethi, H.; Knudsen, M.; Nordentoft, I.; Wu, H.T.; Tin, A.S.; et al. Analysis of Plasma Cell-Free DNA by Ultradeep Sequencing in Patients With Stages I to III Colorectal Cancer. JAMA Oncol. 2019, 5, 1124–1131. [Google Scholar] [CrossRef] [Green Version]
- Chabon, J.J.; Hamilton, E.G.; Kurtz, D.M.; Esfahani, M.S.; Moding, E.J.; Stehr, H.; Schroers-Martin, J.; Nabet, B.Y.; Chen, B.; Chaudhuri, A.A.; et al. Integrating genomic features for non-invasive early lung cancer detection. Nature 2020, 580, 245–251. [Google Scholar] [CrossRef]
- Cohen, J.D.; Li, L.; Wang, Y.; Thoburn, C.; Afsari, B.; Danilova, L.; Douville, C.; Javed, A.A.; Wong, F.; Mattox, A.; et al. Detection and localization of surgically resectable cancers with a multi-analyte blood test. Science 2018, 359, 926–930. [Google Scholar] [CrossRef] [Green Version]
- Liu, M.C.; Oxnard, G.R.; Klein, E.A.; Swanton, C.; Seiden, M.V.; Liu, M.C.; Oxnard, G.R.; Klein, E.A.; Smith, D.; Richards, D.; et al. Sensitive and specific multi-cancer detection and localization using methylation signatures in cell-free DNA. Ann. Oncol. 2020, 31, 745–759. [Google Scholar] [CrossRef]
- Phallen, J.; Sausen, M.; Adleff, V.; Leal, A.; Hruban, C.; White, J.; Anagnostou, V.; Fiksel, J.; Cristiano, S.; Papp, E.; et al. Direct detection of early-stage cancers using circulating tumor DNA. Sci. Transl. Med. 2017, 9, 403. [Google Scholar] [CrossRef] [Green Version]
- Shen, S.Y.; Singhania, R.; Fehringer, G.; Chakravarthy, A.; Roehrl, M.H.A.; Chadwick, D.; Zuzarte, P.C.; Borgida, A.; Wang, T.T.; Li, T.; et al. Sensitive tumour detection and classification using plasma cell-free DNA methylomes. Nature 2018, 563, 579–583. [Google Scholar] [CrossRef]
- Magbanua, M.J.M.; Swigart, L.B.; Wu, H.T.; Hirst, G.L.; Yau, C.; Wolf, D.M.; Tin, A.; Salari, R.; Shchegrova, S.; Pawar, H.; et al. Circulating tumor DNA in neoadjuvant-treated breast cancer reflects response and survival. Ann. Oncol. 2020, 32, 229–239. [Google Scholar] [CrossRef]
- Nabet, B.Y.; Esfahani, M.S.; Moding, E.J.; Hamilton, E.G.; Chabon, J.J.; Rizvi, H.; Steen, C.B.; Chaudhuri, A.A.; Liu, C.L.; Hui, A.B.; et al. Noninvasive Early Identification of Therapeutic Benefit from Immune Checkpoint Inhibition. Cell 2020, 183, 363–376.e313. [Google Scholar] [CrossRef]
- Diehl, F.; Schmidt, K.; Choti, M.A.; Romans, K.; Goodman, S.; Li, M.; Thornton, K.; Agrawal, N.; Sokoll, L.; Szabo, S.A. Circulating mutant DNA to assess tumor dynamics. Nat. Med. 2008, 14, 985–990. [Google Scholar] [CrossRef]
- Kuang, Y.; Rogers, A.; Yeap, B.Y.; Wang, L.; Makrigiorgos, M.; Vetrand, K.; Thiede, S.; Distel, R.J.; Jänne, P.A. Noninvasive detection of EGFR T790M in gefitinib or erlotinib resistant non–small cell lung cancer. Clin. Cancer Res. 2009, 15, 2630–2636. [Google Scholar] [CrossRef] [Green Version]
- Diaz, L.A., Jr.; Williams, R.T.; Wu, J.; Kinde, I.; Hecht, J.R.; Berlin, J.; Allen, B.; Bozic, I.; Reiter, J.G.; Nowak, M.A.; et al. The molecular evolution of acquired resistance to targeted EGFR blockade in colorectal cancers. Nature 2012, 486, 537–540. [Google Scholar] [CrossRef] [Green Version]
- Misale, S.; Yaeger, R.; Hobor, S.; Scala, E.; Janakiraman, M.; Liska, D.; Valtorta, E.; Schiavo, R.; Buscarino, M.; Siravegna, G.; et al. Emergence of KRAS mutations and acquired resistance to anti-EGFR therapy in colorectal cancer. Nature 2012, 486, 532–536. [Google Scholar] [CrossRef] [Green Version]
- Oxnard, G.R.; Paweletz, C.P.; Kuang, Y.; Mach, S.L.; O’connell, A.; Messineo, M.M.; Luke, J.J.; Butaney, M.; Kirschmeier, P.; Jackman, D.M.; et al. Noninvasive detection of response and resistance in EGFR-mutant lung cancer using quantitative next-generation genotyping of cell-free plasma DNA. Clin. Cancer Res. 2014, 20, 1698–1705. [Google Scholar] [CrossRef] [Green Version]
- Thress, K.S.; Paweletz, C.P.; Felip, E.; Cho, B.C.; Stetson, D.; Dougherty, B.; Lai, Z.; Markovets, A.; Vivancos, A.; Kuang, Y.; et al. Acquired EGFR C797S mutation mediates resistance to AZD9291 in non-small cell lung cancer harboring EGFR T790M. Nat. Med. 2015, 21, 560–562. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Milbury, C.A.; Li, C.; Makrigiorgos, G.M. Two-round coamplification at lower denaturation temperature–PCR (COLD-PCR)-based sanger sequencing identifies a novel spectrum of low-level mutations in lung adenocarcinoma. Hum. Mutat. 2009, 30, 1583–1590. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Wang, L.; Mamon, H.; Kulke, M.H.; Berbeco, R.; Makrigiorgos, G.M. Replacing PCR with COLD-PCR enriches variant DNA sequences and redefines the sensitivity of genetic testing. Nat. Med. 2008, 14, 579–584. [Google Scholar] [CrossRef]
- Milbury, C.A.; Li, J.; Liu, P.; Makrigiorgos, G.M. COLD-PCR: Improving the sensitivity of molecular diagnostics assays. Expert Rev. Mol. Diagn. 2011, 11, 159–169. [Google Scholar] [CrossRef] [Green Version]
- Murphy, D.M.; Bejar, R.; Stevenson, K.; Neuberg, D.; Shi, Y.; Cubrich, C.; Richardson, K.; Eastlake, P.; Garcia-Manero, G.; Kantarjian, H.; et al. NRAS mutations with low allele burden have independent prognostic significance for patients with lower risk myelodysplastic syndromes. Leukemia 2013, 27, 2077–2081. [Google Scholar] [CrossRef] [Green Version]
- Girotti, M.R.; Gremel, G.; Lee, R.; Galvani, E.; Rothwell, D.; Viros, A.; Mandal, A.K.; Lim, K.H.; Saturno, G.; Furney, S.J.; et al. Application of Sequencing, Liquid Biopsies, and Patient-Derived Xenografts for Personalized Medicine in Melanoma. Cancer Discov. 2016, 6, 286–299. [Google Scholar] [CrossRef] [Green Version]
- Schwaederle, M.; Husain, H.; Fanta, P.T.; Piccioni, D.E.; Kesari, S.; Schwab, R.B.; Patel, S.P.; Harismendy, O.; Ikeda, M.; Parker, B.A.; et al. Use of Liquid Biopsies in Clinical Oncology: Pilot Experience in 168 Patients. Clin. Cancer Res. 2016, 22, 5497–5505. [Google Scholar] [CrossRef] [Green Version]
- Thierry, A.R.; Mouliere, F.; El Messaoudi, S.; Mollevi, C.; Lopez-Crapez, E.; Rolet, F.; Gillet, B.; Gongora, C.; Dechelotte, P.; Robert, B.; et al. Clinical validation of the detection of KRAS and BRAF mutations from circulating tumor DNA. Nat. Med. 2014, 20, 430–435. [Google Scholar] [CrossRef]
- Dawson, S.J.; Tsui, D.W.; Murtaza, M.; Biggs, H.; Rueda, O.M.; Chin, S.F.; Dunning, M.J.; Gale, D.; Forshew, T.; Mahler-Araujo, B.; et al. Analysis of circulating tumor DNA to monitor metastatic breast cancer. N. Engl. J. Med. 2013, 368, 1199–1209. [Google Scholar] [CrossRef] [Green Version]
- Fernandez-Garcia, D.; Hills, A.; Page, K.; Hastings, R.K.; Toghill, B.; Goddard, K.S.; Ion, C.; Ogle, O.; Boydell, A.R.; Gleason, K. Plasma cell-free DNA (cfDNA) as a predictive and prognostic marker in patients with metastatic breast cancer. Breast Cancer Res. 2019, 21, 149. [Google Scholar] [CrossRef]
- Panagopoulou, M.; Karaglani, M.; Balgkouranidou, I.; Biziota, E.; Koukaki, T.; Karamitrousis, E.; Nena, E.; Tsamardinos, I.; Kolios, G.; Lianidou, E. Circulating cell-free DNA in breast cancer: Size profiling, levels, and methylation patterns lead to prognostic and predictive classifiers. Oncogene 2019, 38, 3387–3401. [Google Scholar] [CrossRef]
- Leon, S.A.; Shapiro, B.; Sklaroff, D.M.; Yaros, M.J. Free DNA in the serum of cancer patients and the effect of therapy. Cancer Res. 1977, 37, 646–650. [Google Scholar] [PubMed]
- Vandeputte, C.; Kehagias, P.; El Housni, H.; Ameye, L.; Laes, J.F.; Desmedt, C.; Sotiriou, C.; Deleporte, A.; Puleo, F.; Geboes, K.; et al. Circulating tumor DNA in early response assessment and monitoring of advanced colorectal cancer treated with a multi-kinase inhibitor. Oncotarget 2018, 9, 17756–17769. [Google Scholar] [CrossRef] [PubMed]
- Bronkhorst, A.J.; Ungerer, V.; Holdenrieder, S. The emerging role of cell-free DNA as a molecular marker for cancer management. Biomol. Detect. Quantif. 2019, 17, 100087. [Google Scholar] [CrossRef] [PubMed]
- Lo, Y.M.; Leung, S.F.; Chan, L.Y.; Chan, A.T.; Lo, K.W.; Johnson, P.J.; Huang, D.P. Kinetics of plasma Epstein-Barr virus DNA during radiation therapy for nasopharyngeal carcinoma. Cancer Res. 2000, 60, 2351–2355. [Google Scholar]
- Kassis, A.I.; Wen, P.Y.; Van Den Abbeele, A.D.; Baranowska-Kortylewicz, J.; Makrigiorgos, G.M.; Metz, K.R.; Matalka, K.Z.; Cook, C.U.; Sahu, S.K.; Black, P.M.; et al. 5-[125I]iodo-2′-deoxyuridine in the radiotherapy of brain tumors in rats. J. Nucl. Med. 1998, 39, 1148–1154. [Google Scholar]
- Makrigiorgos, G.M.; Berman, R.M.; Baranowska-Kortylewicz, J.; Bump, E.; Humm, J.L.; Adelstein, S.J.; Kassis, A.I. DNA damage produced in V79 cells by DNA-incorporated iodine-123: A comparison with iodine-125. Radiat. Res. 1992, 129, 309–314. [Google Scholar] [CrossRef]
- Makrigiorgos, G.; Adelstein, S.J.; Kassis, A.I. Auger electron emitters: Insights gained from in vitro experiments. Radiat. Environ. Biophys. 1990, 29, 75–91. [Google Scholar] [CrossRef]
- Makrigiorgos, G.M.; Adelstein, S.J.; Kassis, A.I. Cellular radiation dosimetry and its implications for estimation of radiation risks. Illustrative results with technetium 99m-labeled microspheres and macroaggregates. JAMA 1990, 264, 592–595. [Google Scholar] [CrossRef]
- Makrigiorgos, G.M.; Ito, S.; Baranowska-Kortylewicz, J.; Vinter, D.W.; Iqbal, A.; Van Den Abbeele, A.D.; Adelstein, S.J.; Kassis, A.I. Inhomogeneous deposition of radiopharmaceuticals at the cellular level: Experimental evidence and dosimetric implications. J. Nucl. Med. 1990, 31, 1358–1363. [Google Scholar]
- Kinde, I.; Wu, J.; Papadopoulos, N.; Kinzler, K.W.; Vogelstein, B. Detection and quantification of rare mutations with massively parallel sequencing. Proc. Natl. Acad. Sci. USA 2011, 108, 9530–9535. [Google Scholar] [CrossRef] [Green Version]
- Milbury, C.A.; Correll, M.; Quackenbush, J.; Rubio, R.; Makrigiorgos, G.M. COLD-PCR enrichment of rare cancer mutations prior to targeted amplicon resequencing. Clin. Chem. 2012, 58, 580–589. [Google Scholar] [CrossRef] [Green Version]
- Newman, A.M.; Lovejoy, A.F.; Klass, D.M.; Kurtz, D.M.; Chabon, J.J.; Scherer, F.; Stehr, H.; Liu, C.L.; Bratman, S.V.; Say, C.; et al. Integrated digital error suppression for improved detection of circulating tumor DNA. Nat. Biotechnol. 2016, 34, 547–555. [Google Scholar] [CrossRef]
- Schmitt, M.W.; Kennedy, S.R.; Salk, J.J.; Fox, E.J.; Hiatt, J.B.; Loeb, L.A. Detection of ultra-rare mutations by next-generation sequencing. Proc. Natl. Acad. Sci. USA 2012, 109, 14508–14513. [Google Scholar] [CrossRef] [Green Version]
- Forshew, T.; Murtaza, M.; Parkinson, C.; Gale, D.; Tsui, D.W.; Kaper, F.; Dawson, S.J.; Piskorz, A.M.; Jimenez-Linan, M.; Bentley, D.; et al. Noninvasive identification and monitoring of cancer mutations by targeted deep sequencing of plasma DNA. Sci. Transl. Med. 2012, 4, 136ra168. [Google Scholar] [CrossRef]
- Narayan, A.; Carriero, N.J.; Gettinger, S.N.; Kluytenaar, J.; Kozak, K.R.; Yock, T.I.; Muscato, N.E.; Ugarelli, P.; Decker, R.H.; Patel, A.A. Ultrasensitive measurement of hotspot mutations in tumor DNA in blood using error-suppressed multiplexed deep sequencing. Cancer Res. 2012, 72, 3492–3498. [Google Scholar] [CrossRef] [Green Version]
- Lou, D.I.; Hussmann, J.A.; Mcbee, R.M.; Acevedo, A.; Andino, R.; Press, W.H.; Sawyer, S.L. High-throughput DNA sequencing errors are reduced by orders of magnitude using circle sequencing. Proc. Natl. Acad. Sci. USA 2013, 110, 19872–19877. [Google Scholar] [CrossRef] [Green Version]
- Gregory, M.T.; Bertout, J.A.; Ericson, N.G.; Taylor, S.D.; Mukherjee, R.; Robins, H.S.; Drescher, C.W.; Bielas, J.H. Targeted single molecule mutation detection with massively parallel sequencing. Nucleic Acids Res. 2016, 44, e22. [Google Scholar] [CrossRef] [Green Version]
- Milbury, C.A.; Li, J.; Makrigiorgos, G.M. PCR-based methods for the enrichment of minority alleles and mutations. Clin. Chem. 2009, 55, 632–640. [Google Scholar] [CrossRef] [Green Version]
- Galbiati, S.; Brisci, A.; Lalatta, F.; Seia, M.; Makrigiorgos, G.M.; Ferrari, M.; Cremonesi, L. Full COLD-PCR protocol for noninvasive prenatal diagnosis of genetic diseases. Clin. Chem. 2011, 57, 136–138. [Google Scholar] [CrossRef] [Green Version]
- Gill, P.; Ghaemi, A. Nucleic acid isothermal amplification technologies: A review. Nucleosides Nucleotides Nucleic Acids 2008, 27, 224–243. [Google Scholar] [CrossRef]
- Taly, V.; Huggett, J. Digital PCR, a technique for the future. Biomol. Detect. Quantif. 2016, 10, 1. [Google Scholar] [CrossRef] [Green Version]
- Amicarelli, G.; Shehi, E.; Makrigiorgos, G.M.; Adlerstein, D. FLAG assay as a novel method for real-time signal generation during PCR: Application to detection and genotyping of KRAS codon 12 mutations. Nucleic Acids Res. 2007, 35, e131. [Google Scholar] [CrossRef]
- Wittwer, C.T.; Reed, G.H.; Gundry, C.N.; Vandersteen, J.G.; Pryor, R.J. High-resolution genotyping by amplicon melting analysis using LCGreen. Clin. Chem. 2003, 49, 853–860. [Google Scholar] [CrossRef] [Green Version]
- Song, C.; Liu, Y.; Fontana, R.; Makrigiorgos, A.; Mamon, H.; Kulke, M.H.; Makrigiorgos, G.M. Elimination of unaltered DNA in mixed clinical samples via nuclease-assisted minor-allele enrichment. Nucleic Acids Res. 2016, 44, e146. [Google Scholar] [CrossRef] [Green Version]
- Shagin, D.A.; Rebrikov, D.V.; Kozhemyako, V.B.; Altshuler, I.M.; Shcheglov, A.S.; Zhulidov, P.A.; Bogdanova, E.A.; Staroverov, D.B.; Rasskazov, V.A.; Lukyanov, S. A novel method for SNP detection using a new duplex-specific nuclease from crab hepatopancreas. Genome Res. 2002, 12, 1935–1942. [Google Scholar] [CrossRef] [Green Version]
- Snyder, T.M.; Khush, K.K.; Valantine, H.A.; Quake, S.R. Universal noninvasive detection of solid organ transplant rejection. Proc. Natl. Acad. Sci. USA 2011, 108, 6229–6234. [Google Scholar] [CrossRef] [Green Version]
- Yu, F.; Leong, K.W.; Makrigiorgos, G.M. Nuclease-Assisted, Multiplexed Minor-Allele Enrichment: Application in Liquid Biopsy of Cancer. Methods Mol. Biol. 2022, 2394, 433–451. [Google Scholar]
- Ladas, I.; Fitarelli-Kiehl, M.; Song, C.; Adalsteinsson, V.A.; Parsons, H.A.; Lin, N.U.; Wagle, N.; Makrigiorgos, G.M. Multiplexed Elimination of Wild-Type DNA and High-Resolution Melting Prior to Targeted Resequencing of Liquid Biopsies. Clin. Chem. 2017, 63, 1605–1613. [Google Scholar] [CrossRef] [Green Version]
- Dobrovic, A. DNA Breathing Enables Closed-Tube Mutant Allele Enrichment for Circulating Tumor DNA Analysis. Clin. Chem. 2017, 63, e1–e3. [Google Scholar] [CrossRef]
- Markou, A.; Tzanikou, E.; Ladas, I.; Makrigiorgos, G.M.; Lianidou, E. Nuclease-Assisted Minor Allele Enrichment Using Overlapping Probes-Assisted Amplification-Refractory Mutation System: An Approach for the Improvement of Amplification-Refractory Mutation System-Polymerase Chain Reaction Specificity in Liquid Biopsies. Anal. Chem. 2019, 91, 13105–13111. [Google Scholar] [CrossRef]
- Mollon, L.E.; Anderson, E.J.; Dean, J.L.; Warholak, T.L.; Aizer, A.; Platt, E.A.; Tang, D.H.; Davis, L.E. A Systematic Literature Review of the Prognostic and Predictive Value of PIK3CA Mutations in HR+/HER2− Metastatic Breast Cancer. Clin. Breast Cancer. 2020, 20, e232–e243. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Shi, Y.-L.; Zhou, K.; Wang, L.-L.; Yan, Z.-X.; Liu, Y.-L.; Xu, L.-L.; Zhao, S.-W.; Chu, H.-L.; Shi, T.-T. PIK3CA mutations confer resistance to first-line chemotherapy in colorectal cancer. Cell Death Dis. 2018, 9, 739. [Google Scholar] [CrossRef] [PubMed]
- Stergiopoulou, D.; Markou, A.; Tzanikou, E.; Ladas, I.; Makrigiorgos, G.M.; Georgoulias, V.; Lianidou, E. ESR1 NAPA Assay: Development and Analytical Validation of a Highly Sensitive and Specific Blood-Based Assay for the Detection of ESR1 Mutations in Liquid Biopsies. Cancers 2021, 13, 556. [Google Scholar] [CrossRef] [PubMed]
- Keraite, I.; Alvarez-Garcia, V.; Garcia-Murillas, I.; Beaney, M.; Turner, N.C.; Bartos, C.; Oikonomidou, O.; Kersaudy-Kerhoas, M.; Leslie, N.R. PIK3CA mutation enrichment and quantitation from blood and tissue. Sci. Rep. 2020, 10, 17082. [Google Scholar] [CrossRef]
- Cortes-Ciriano, I.; Lee, S.; Park, W.-Y.; Kim, T.-M.; Park, P.J. A molecular portrait of microsatellite instability across multiple cancers. Nat. Commun. 2017, 8, 15180. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, H.; Watanabe, Y.; Maehata, T.; Imai, K.; Itoh, F. Microsatellite instability in cancer: A novel landscape for diagnostic and therapeutic approach. Arch. Toxicol. 2020, 94, 3349–3357. [Google Scholar] [CrossRef]
- Ladas, I.; Yu, F.; Leong, K.W.; Fitarelli-Kiehl, M.; Song, C.; Ashtaputre, R.; Kulke, M.; Mamon, H.; Makrigiorgos, G.M. Enhanced detection of microsatellite instability using pre-PCR elimination of wild-type DNA homo-polymers in tissue and liquid biopsies. Nucleic Acids Res. 2018, 46, e74. [Google Scholar] [CrossRef] [Green Version]
- Baudrin, L.G.; Deleuze, J.F.; How-Kit, A. Molecular and Computational Methods for the Detection of Microsatellite Instability in Cancer. Front. Oncol. 2018, 8, 621. [Google Scholar] [CrossRef]
- Lehmann-Werman, R.; Neiman, D.; Zemmour, H.; Moss, J.; Magenheim, J.; Vaknin-Dembinsky, A.; Rubertsson, S.; Nellgård, B.; Blennow, K.; Zetterberg, H. Identification of tissue-specific cell death using methylation patterns of circulating DNA. Proc. Natl. Acad. Sci. USA 2016, 113, E1826–E1834. [Google Scholar] [CrossRef] [Green Version]
- Moran, S.; Martínez-Cardús, A.; Sayols, S.; Musulén, E.; Balañá, C.; Estival-Gonzalez, A.; Moutinho, C.; Heyn, H.; Diaz-Lagares, A.; De Moura, M.C. Epigenetic profiling to classify cancer of unknown primary: A multicentre, retrospective analysis. Lancet Oncol. 2016, 17, 1386–1395. [Google Scholar] [CrossRef]
- Sun, K.; Jiang, P.; Chan, K.A.; Wong, J.; Cheng, Y.K.; Liang, R.H.; Chan, W.-K.; Ma, E.S.; Chan, S.L.; Cheng, S.H. Plasma DNA tissue mapping by genome-wide methylation sequencing for noninvasive prenatal, cancer, and transplantation assessments. Proc. Natl. Acad. Sci. USA 2015, 112, E5503–E5512. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Song, C.; Ladas, I.; Fitarelli-Kiehl, M.; Makrigiorgos, G.M. Methylation-sensitive enrichment of minor DNA alleles using a double-strand DNA-specific nuclease. Nucleic Acids Res. 2017, 45, e39. [Google Scholar] [CrossRef] [Green Version]
- Hayatsu, H.; Wataya, Y.; Kai, K.; Iida, S. Reaction of sodium bisulfite with uracil, cytosine, and their derivatives. Biochemistry 1970, 9, 2858–2865. [Google Scholar] [CrossRef]
- Leong, K.W.; Yu, F.; Makrigiorgos, G.M. Mutation enrichment in human DNA samples via UV-mediated cross-linking. Nucleic Acids Res. 2022, 50, e32. [Google Scholar] [CrossRef]
- Fujimoto, K.; Yamada, A.; Yoshimura, Y.; Tsukaguchi, T.; Sakamoto, T. Details of the Ultrafast DNA Photo-Cross-Linking Reaction of 3-Cyanovinylcarbazole Nucleoside: Cis-Trans Isomeric Effect and the Application for SNP-Based Genotyping. J. Am. Chem. Soc. 2013, 135, 16161–16167. [Google Scholar] [CrossRef]
- Yoshimura, Y.; Fujimoto, K. Ultrafast reversible photo-cross-linking reaction: Toward in situ DNA manipulation. Org. Lett. 2008, 10, 3227–3230. [Google Scholar] [CrossRef]
- Jeffreys, A.J.; May, C.A. DNA enrichment by allele-specific hybridization (DEASH): A novel method for haplotyping and for detecting low-frequency base substitutional variants and recombinant DNA molecules. Genome Res. 2003, 13, 2316–2324. [Google Scholar] [CrossRef] [Green Version]
- Gydush, G.; Nguyen, E.; Bae, J.H.; Blewett, T.; Rhoades, J.; Reed, S.C.; Shea, D.; Xiong, K.; Liu, R.; Yu, F. Massively parallel enrichment of low-frequency alleles enables duplex sequencing at low depth. Nat. Biomed. Eng. 2022, 6, 257–266. [Google Scholar] [CrossRef]
- Pingoud, A.; Fuxreiter, M.; Pingoud, V.; Wende, W. Type II restriction endonucleases: Structure and mechanism. Cell. Mol. Life Sci. 2005, 62, 685–707. [Google Scholar] [CrossRef]
- Di Felice, F.; Micheli, G.; Camilloni, G. Restriction enzymes and their use in molecular biology: An overview. J. Biosci. 2019, 44, 38. [Google Scholar] [CrossRef]
- Parry, J.; Shamsher, M.; Skibinski, D. Restriction site mutation analysis, a proposed methodology for the detection and study of DNA base changes following mutagen exposure. Mutagenesis 1990, 5, 209–212. [Google Scholar] [CrossRef]
- Jenkins, G. REVIEW The restriction site mutation (RSM) method: Clinical applications. Mutagenesis 2004, 19, 3–11. [Google Scholar] [CrossRef] [Green Version]
- Sandy, M.S.; Chiocca, S.M.; Cerutti, P.A. Genotypic analysis of mutations in Taq I restriction recognition sites by restriction fragment length polymorphism/polymerase chain reaction. Proc. Natl. Acad. Sci. USA 1992, 89, 890–894. [Google Scholar] [CrossRef] [Green Version]
- Felley-Bosco, E.; Pourzand, C.; Zijlstra, J.; Amstad, P.; Cerutti, P. A genotypic mutation system measuring mutations in restriction recognition sequences. Nucleic Acids Res. 1991, 19, 2913–2919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bielas, J.H.; Loeb, L.A. Quantification of random genomic mutations. Nat. Methods 2005, 2, 285–290. [Google Scholar] [CrossRef] [PubMed]
- Wright, J.H.; Modjeski, K.L.; Bielas, J.H.; Preston, B.D.; Fausto, N.; Loeb, L.A.; Campbell, J.S. A random mutation capture assay to detect genomic point mutations in mouse tissue. Nucleic Acids Res. 2011, 39, e73. [Google Scholar] [CrossRef] [PubMed]
- Loeb, L.A.; Loeb, K.R.; Anderson, J.P. Multiple mutations and cancer. Proc. Natl. Acad. Sci. USA 2003, 100, 776–781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bielas, J.H.; Loeb, K.R.; Rubin, B.P.; True, L.D.; Loeb, L.A. Human cancers express a mutator phenotype. Proc. Natl. Acad. Sci. USA 2006, 103, 18238–18242. [Google Scholar] [CrossRef] [Green Version]
- Aalipour, A.; Dudley, J.C.; Park, S.-M.; Murty, S.; Chabon, J.J.; Boyle, E.A.; Diehn, M.; Gambhir, S.S. Deactivated CRISPR associated protein 9 for minor-allele enrichment in cell-free DNA. Clin. Chem. 2018, 64, 307–316. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Qiu, T.; Mauk, M.G.; Fan, Y.; Jiang, Y.; Ying, J.; Zhou, Q.; Qiao, Y.; Bau, H.H.; Song, J. CRISPR Cas9-Mediated Selective Isothermal Amplification for Sensitive Detection of Rare Mutant Alleles. Clin. Chem. 2021, 67, 1569–1571. [Google Scholar] [CrossRef]
- Gu, W.; Crawford, E.D.; O’donovan, B.; Wilson, M.R.; Chow, E.D.; Retallack, H.; Derisi, J.L. Depletion of Abundant Sequences by Hybridization (DASH): Using Cas9 to remove unwanted high-abundance species in sequencing libraries and molecular counting applications. Genome Biol. 2016, 17, 41. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.H.; Yu, J.; Hwang, G.H.; Kim, S.; Kim, H.; Ye, S.; Kim, K.; Park, J.; Park, D.; Cho, Y.-K. CUT-PCR: CRISPR-mediated, ultrasensitive detection of target DNA using PCR. Oncogene 2017, 36, 6823–6829. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Jiang, J.; Li, X.; Li, K.; He, R.; Li, J.; Duan, L.; Luo, W.; Hu, Z.; Luo, D. Improved EGFR mutation detection sensitivity after enrichment by Cas9/sgRNA digestion and PCR amplification. Acta Biochim. Biophys. Sin. 2020, 52, 1316–1324. [Google Scholar] [CrossRef]
- Song, J.; Hegge, J.W.; Mauk, M.G.; Chen, J.; Till, J.E.; Bhagwat, N.; Azink, L.T.; Peng, J.; Sen, M.; Mays, J. Highly specific enrichment of rare nucleic acid fractions using Thermus thermophilus argonaute with applications in cancer diagnostics. Nucleic Acids Res. 2020, 48, e19. [Google Scholar] [CrossRef]
- Wang, F.; Wang, L.; Briggs, C.; Sicinska, E.; Gaston, S.M.; Mamon, H.; Kulke, M.H.; Zamponi, R.; Loda, M.; Maher, E.; et al. DNA Degradation Test Predicts Success in Whole-Genome Amplification from Diverse Clinical Samples. J. Mol. Diagn. 2007, 9, 441–451. [Google Scholar] [CrossRef] [Green Version]
- Kitagawa, Y.; Okumura, K.; Watanabe, T.; Tsukamoto, K.; Kitano, S.; Nankinzan, R.; Suzuki, T.; Hara, T.; Soda, H.; Denda, T. Enrichment technique to allow early detection and monitor emergence of KRAS mutation in response to treatment. Sci. Rep. 2019, 9, 11346. [Google Scholar] [CrossRef]
- Kaur, M.; Makrigiorgos, G.M. Novel amplification of DNA in a hairpin structure: Towards a radical elimination of PCR errors from amplified DNA. Nucl. Acids. Res. 2003, 31, e26. [Google Scholar] [CrossRef] [Green Version]
- Keohavong, P.; Thilly, W.G. Fidelity of DNA polymerases in DNA amplification. Proc. Natl. Acad. Sci. USA 1989, 86, 9253–9257. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Method | Operation | Target Motif Requirement | Mutation Type | LOD (%MAF) | Multiplexity | Adjustment for Liquid Biopsy |
|---|---|---|---|---|---|---|
| NaME-PrO 1 | Specific probes and duplex-specific nuclease | NO | Point Mutations | 10−2 | Moderate | YES |
| MSI-NaME-PrO 2 | Specific probes and duplex-specific nuclease | NO | Microsatellite instability | 10−2 | Low | YES |
| NaMSIE 3 | LNA probes and duplex-specific nuclease | NO | Microsatellite instability | 5 × 10−1 | Low | NO |
| MS-NaME 4 | Specific probes and duplex-specific nuclease | NO | Epimutations | 10−2 | Moderate | YES |
| Pre-PCR-UVME 5 | Specific probes and UV irradiation | NO | Point Mutations | 10−2 | High | YES |
| DEASH 6 | Biotinylated allele-specific oligonucleotides and magnetic bead capture | NO | Point Mutations | 10−3–10−5 | Low | NO |
| MAESTRO 7 | NGS library construction, biotinylated allele-specific oligonucleotides, magnetic bead capture, and duplex sequencing | NO | Point Mutations | 10−3 | High | YES |
| RSM 8 | Restriction endonuclease | YES | Point Mutations | 10−1–10−4 | Low | NO |
| RFLP-PCR 9 | Restriction endonuclease | YES | Point Mutations | 10−5 | Low | NO |
| RMC 10 | Magnetic bead capture and restriction endonuclease | YES | Point Mutations | 10−1–10−6 | Low | NO |
| CUT-PCR 11 | CRISPR-Cas9 and guide RNA | YES | Point Mutations | 10−2 | Low | YES |
| NAVIGATER 12 | TtAgo endonuclease and guide DNA | NO | Point Mutations | 10−1–10−2 | Moderate | YES |
| PI Polyamides 13 | Biotinylated PI polyamides and magnetic bead capture | YES | Point Mutations | 10−1 | Low | YES |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Darbeheshti, F.; Yu, F.; Makrigiorgos, G.M. Pre-PCR Mutation-Enrichment Methods for Liquid Biopsy Applications. Cancers 2022, 14, 3143. https://doi.org/10.3390/cancers14133143
Darbeheshti F, Yu F, Makrigiorgos GM. Pre-PCR Mutation-Enrichment Methods for Liquid Biopsy Applications. Cancers. 2022; 14(13):3143. https://doi.org/10.3390/cancers14133143
Chicago/Turabian StyleDarbeheshti, Farzaneh, Fangyan Yu, and G. Mike Makrigiorgos. 2022. "Pre-PCR Mutation-Enrichment Methods for Liquid Biopsy Applications" Cancers 14, no. 13: 3143. https://doi.org/10.3390/cancers14133143