
In Vitro and In Vivo Characterization of MCT1 Inhibitor AZD3965 Confirms Preclinical Safety Compatible with Breast Cancer Treatment

 , , and
, , and 
Abstract
:Simple Summary
Abstract
1. Introduction
2. Results
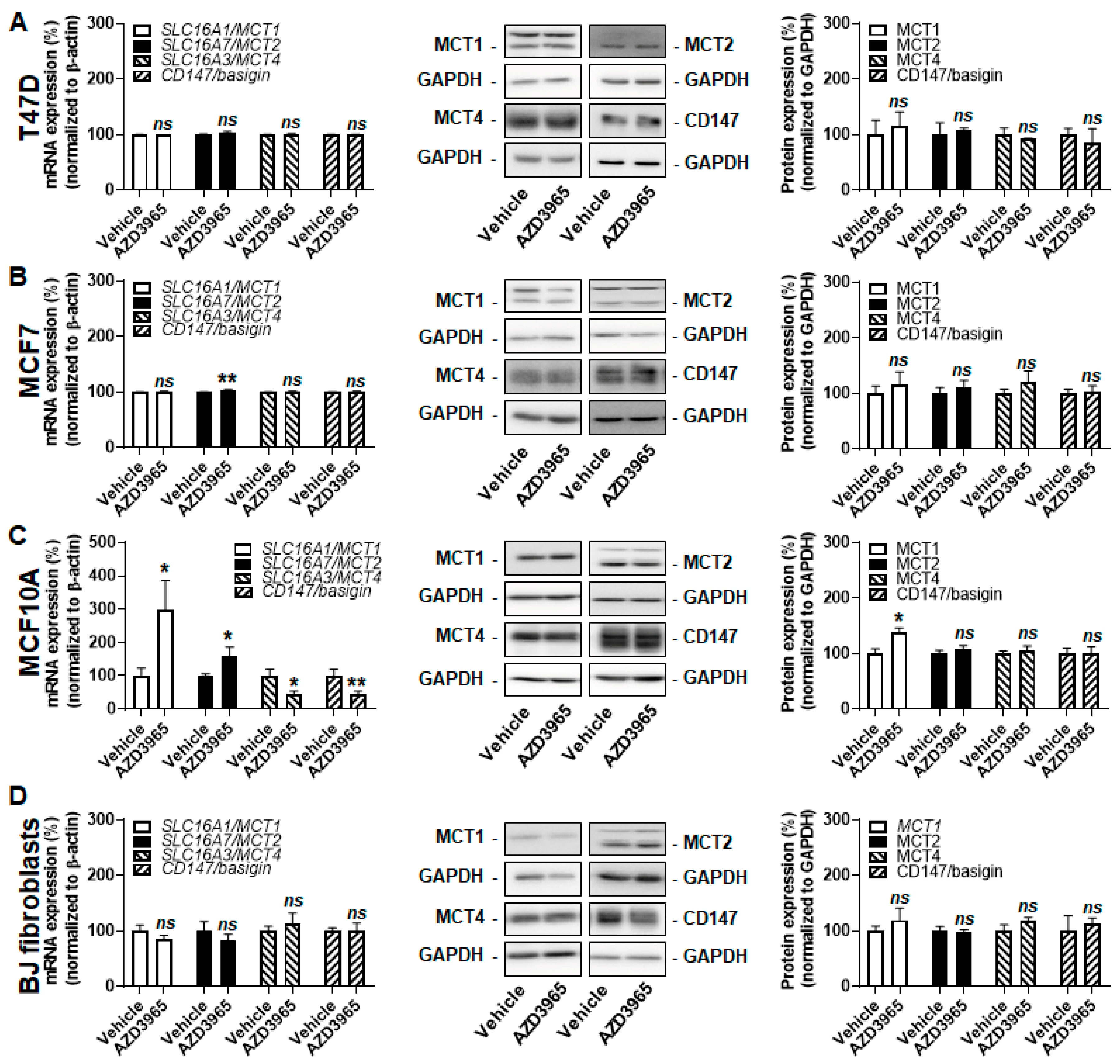
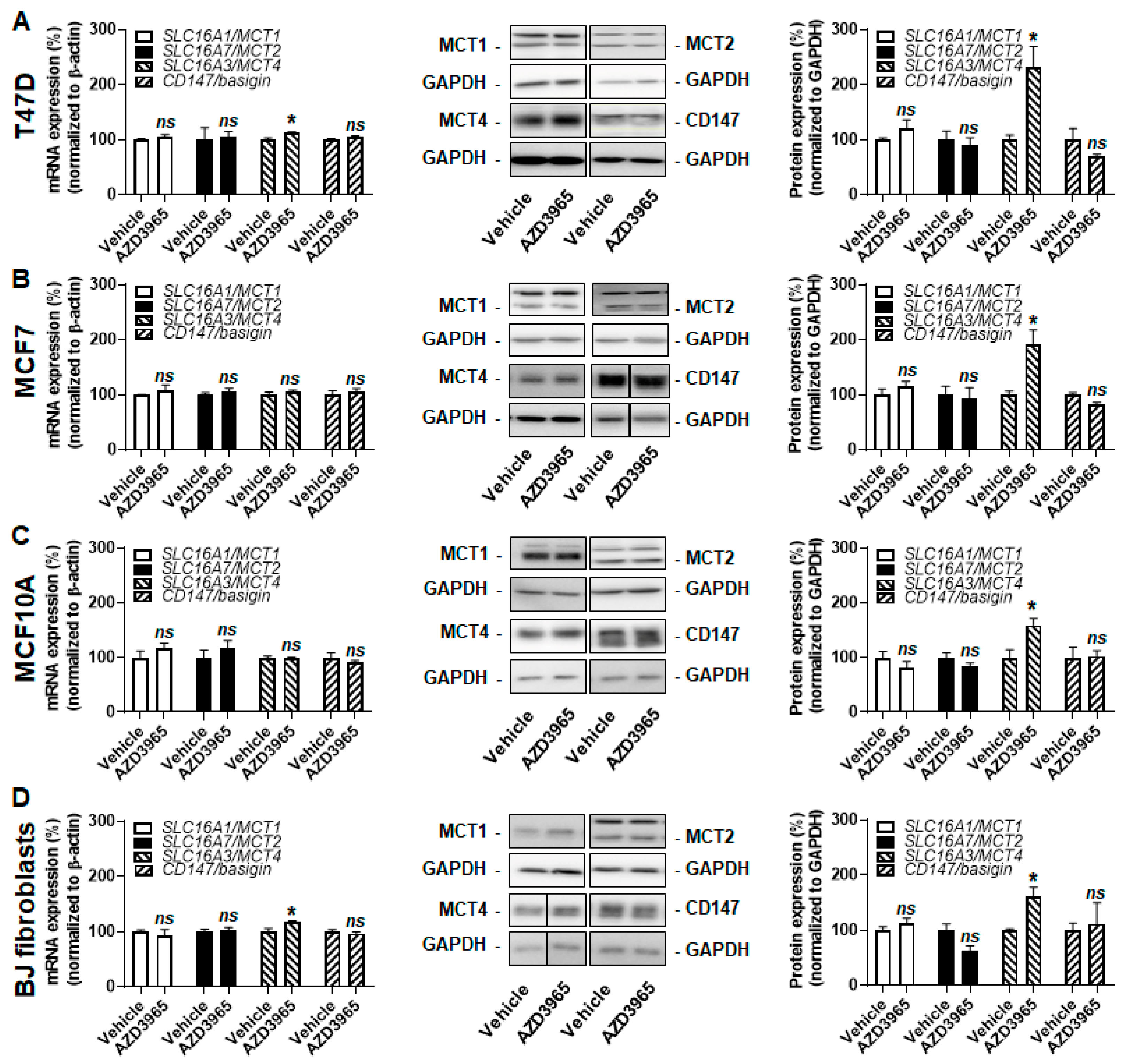
2.1. Model Selection Based on the Characterization of MCT1 and CD147 Expression
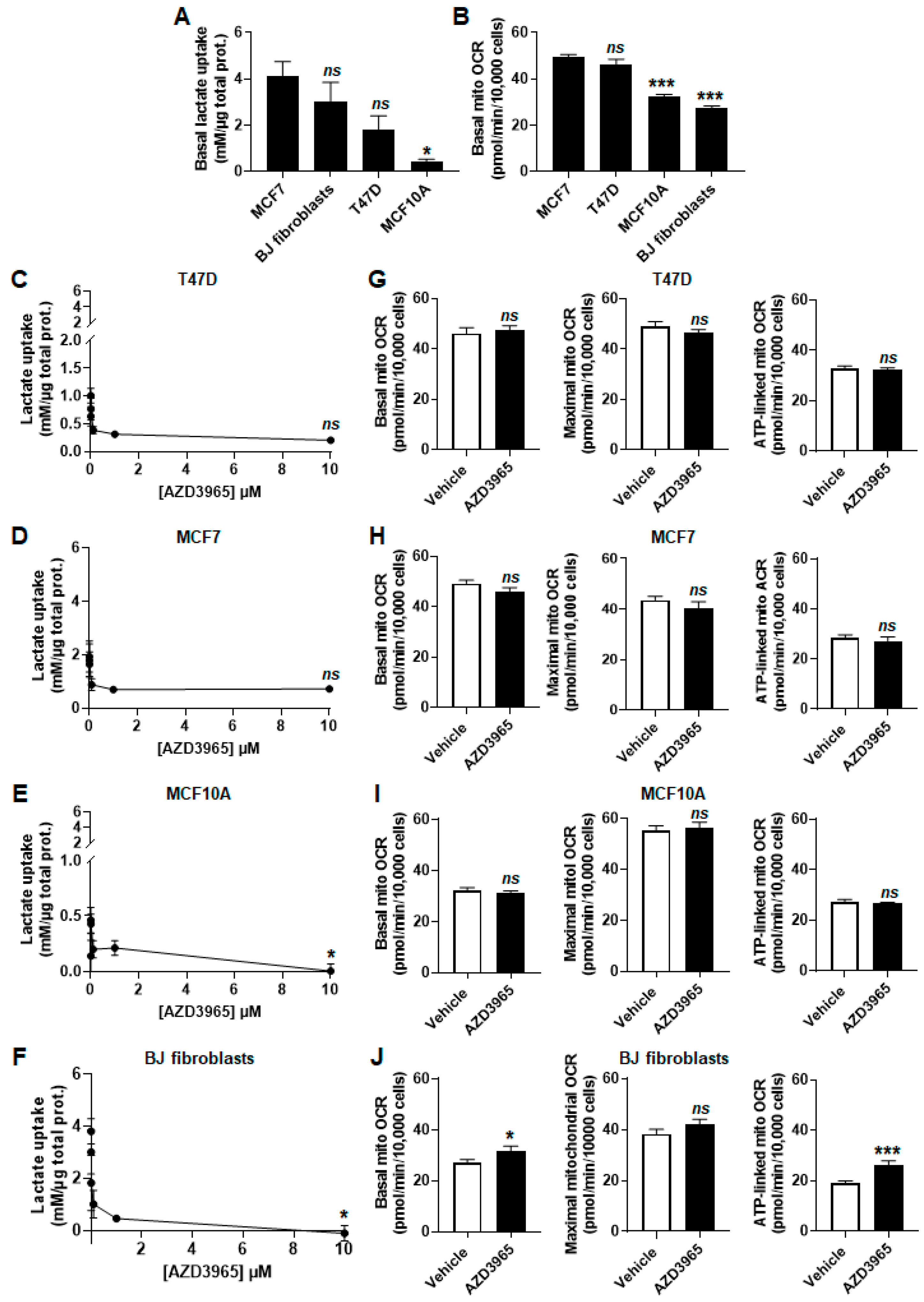
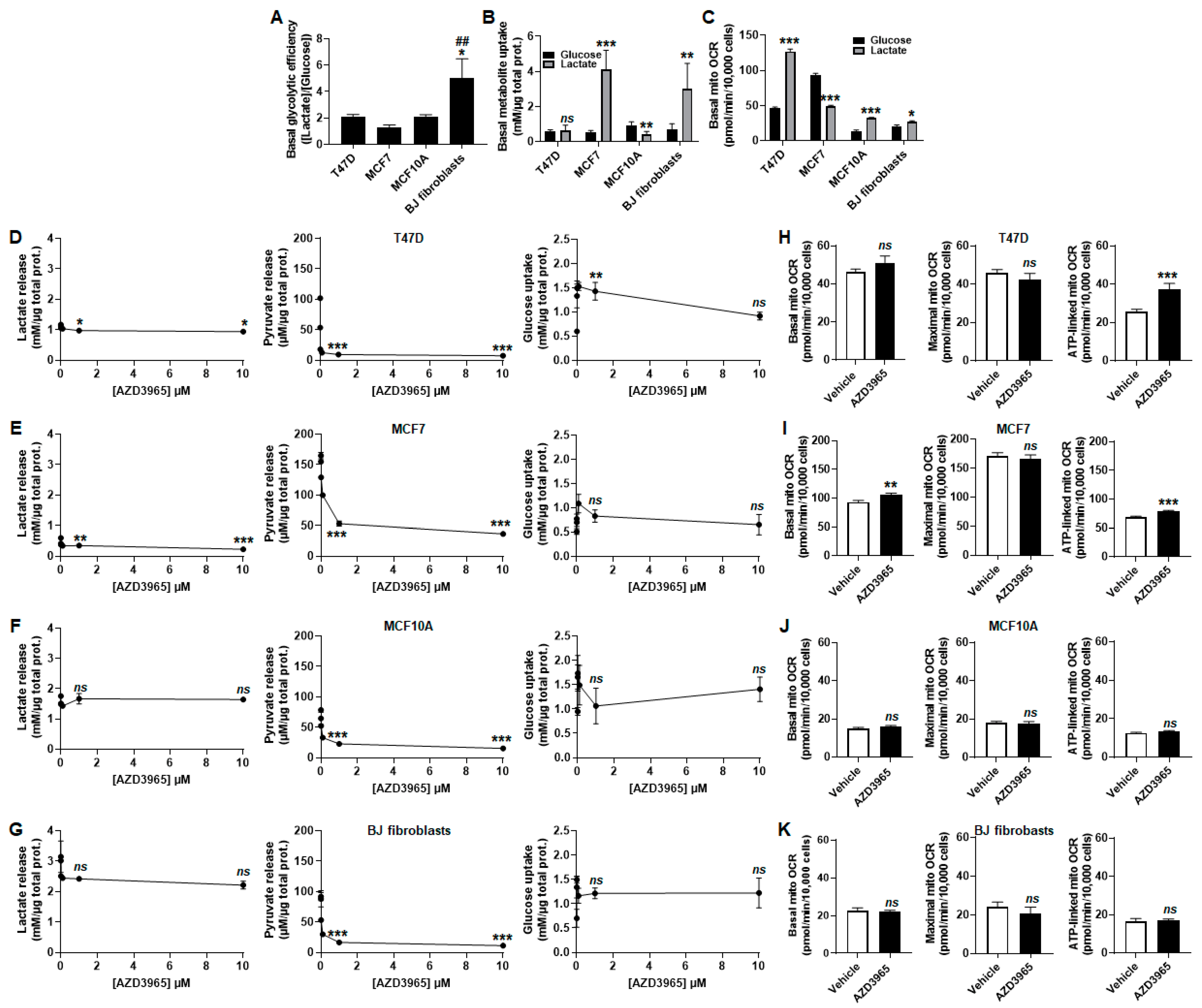
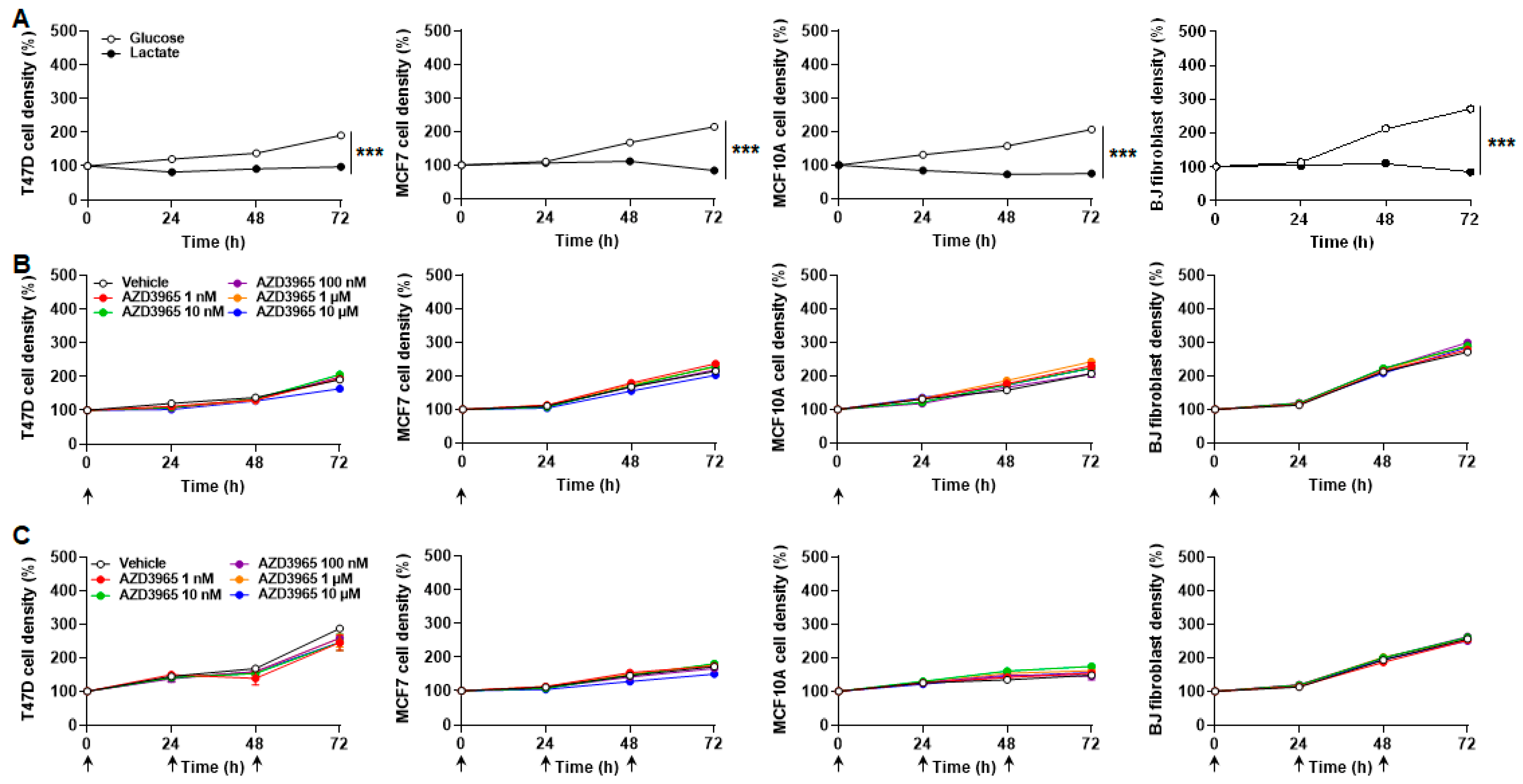
2.2. MCT1 Is the Main Facilitator of Lactate Uptake by Breast Cancer and Breast-Associated Nonmalignant Cells
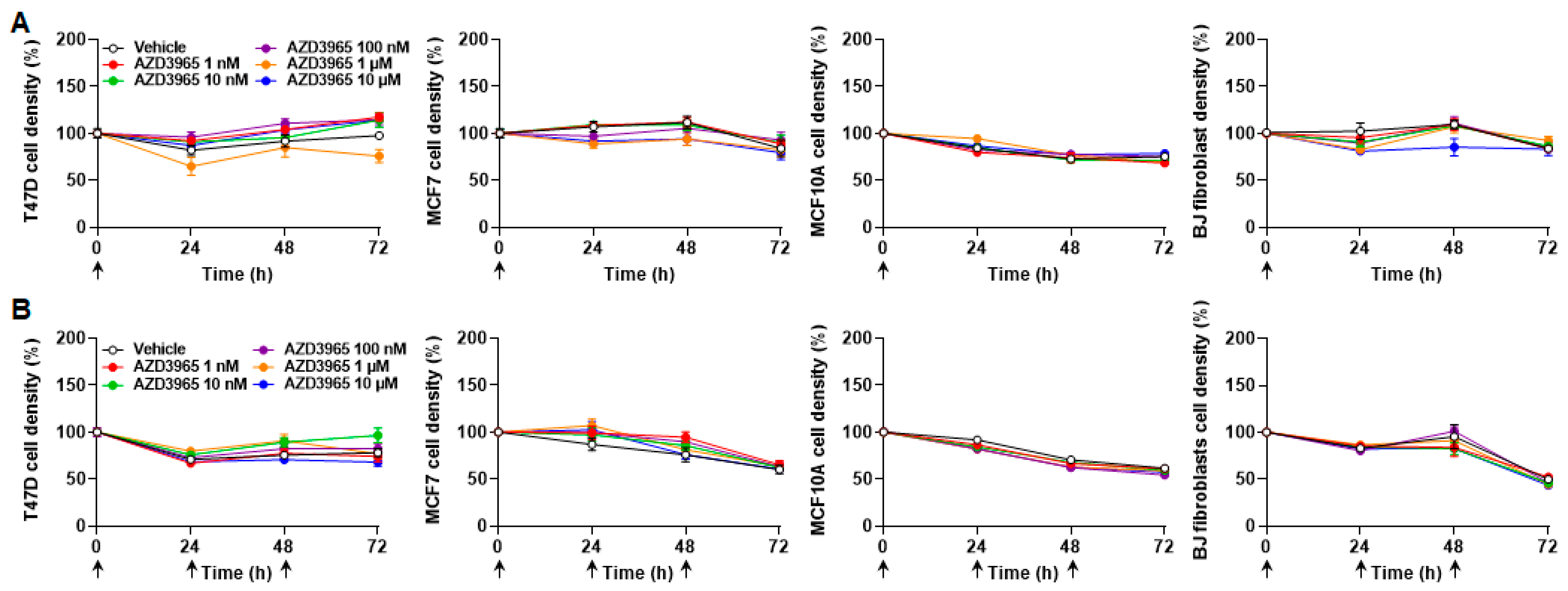
2.3. Inhibition of MCT1-Dependent Lactate Uptake by AZD3965 Does Not Kill Malignant and Nonmalignant Breast-Associated Cells
2.4. Inhibition of Lactate and Pyruvate Export by AZD3965 Induces an Oxidative Switch in Human Breast Cancer Cells, But Not in Human Breast-Associated Nonmalignant Cells
2.5. Inhibition of MCT1-Dependent Lactate Export by AZD3965 Does Not Kill Malignant and Nonmalignant Breast-Associated Cells When Glucose and Glutamine Are Available
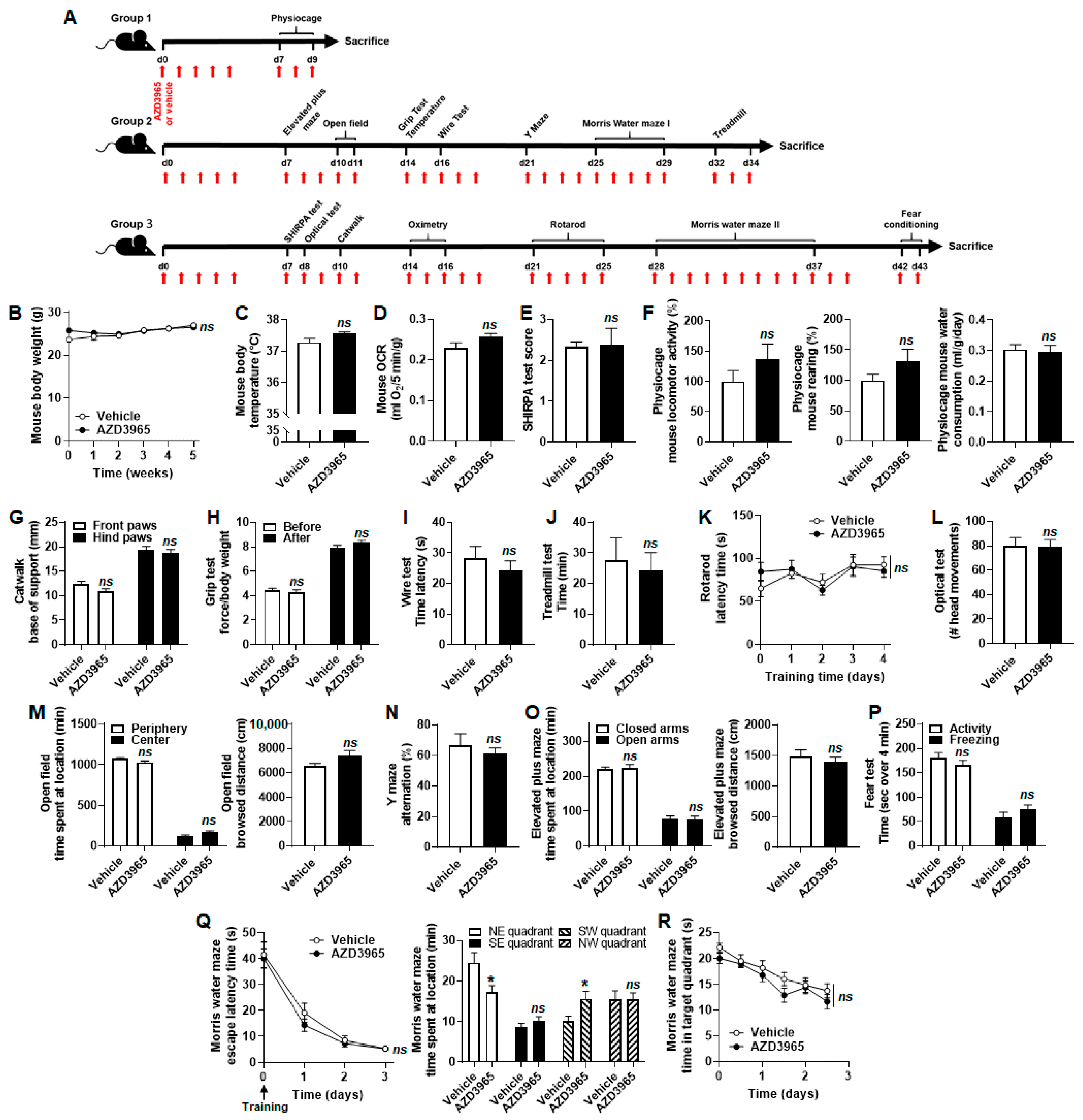
2.6. AZD3965 Does Not Alter Muscle and Brain Physiology in Mice, to the Exception of Short-Term Memory Retention
3. Discussion
4. Materials and Methods
4.1. Cells, Cell Culture and Treatments
4.2. Lactate, Pyruvate and Glucose Measurements
4.3. Cell Oximetry
4.4. Reverse Transcription and Quantitative Polymerase Chain Reaction
4.5. Western Blotting and Immunocytochemistry
4.6. Cell Density
4.7. Flow Cytometry
4.8. Mouse Experiments
4.9. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Appendix A. Detailed Materials and Methods of Mouse Experiments
Appendix A.1. Mouse Oximetry
Appendix A.2. Modified SHIRPA Test
Appendix A.3. Physiocage Assay
Appendix A.4. Catwalk
Appendix A.5. Grip Test
Appendix A.6. Wire Test
Appendix A.7. Treadmill Test
Appendix A.8. Rotarod Test
Appendix A.9. Optical Test
Appendix A.10. Open Field
Appendix A.11. Y Maze
Appendix A.12 Elevated Plus Maze
Appendix A.13 Contextual Fear-Conditioning Test
Appendix A.14. Morris Water Maze
References
- Giatromanolaki, A.; Harris, A.L. Tumour hypoxia, hypoxia signaling pathways and hypoxia inducible factor expression in human cancer. Anticancer Res. 2001, 21, 4317–4324. [Google Scholar] [PubMed]
- Thomlinson, R.H.; Gray, L.H. The histological structure of some human lung cancers and the possible implications for radiotherapy. Br. J. Cancer 1955, 9, 539–549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Groebe, K.; Vaupel, P. Evaluation of oxygen diffusion distances in human breast cancer xenografts using tumor-specific in vivo data: Role of various mechanisms in the development of tumor hypoxia. Int. J. Radiat. Oncol. Biol. Phys. 1988, 15, 691–697. [Google Scholar] [CrossRef]
- Dewhirst, M.W. Intermittent hypoxia furthers the rationale for hypoxia-inducible factor-1 targeting. Cancer Res. 2007, 67, 854–855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Semenza, G.L. HIF-1: Upstream and downstream of cancer metabolism. Curr. Opin. Genet. Dev. 2010, 20, 51–56. [Google Scholar] [CrossRef] [Green Version]
- Parks, S.K.; Pouyssegur, J. Targeting pH regulating proteins for cancer therapy-progress and limitations. Semin. Cancer Biol. 2017, 43, 66–73. [Google Scholar] [CrossRef]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [Green Version]
- Mullen, A.R.; Wheaton, W.W.; Jin, E.S.; Chen, P.H.; Sullivan, L.B.; Cheng, T.; Yang, Y.; Linehan, W.M.; Chandel, N.S.; DeBerardinis, R.J. Reductive carboxylation supports growth in tumour cells with defective mitochondria. Nature 2011, 481, 385–388. [Google Scholar] [CrossRef] [Green Version]
- Prakasam, G.; Iqbal, M.A.; Bamezai, R.N.K.; Mazurek, S. Posttranslational modifications of pyruvate kinase M2: Tweaks that benefit cancer. Front. Oncol. 2018, 8, 22. [Google Scholar] [CrossRef] [Green Version]
- Romero, Q.; Bendahl, P.O.; Klintman, M.; Loman, N.; Ingvar, C.; Ryden, L.; Rose, C.; Grabau, D.; Borgquist, S. Ki67 proliferation in core biopsies versus surgical samples—A model for neo-adjuvant breast cancer studies. BMC Cancer 2011, 11, 341. [Google Scholar] [CrossRef] [Green Version]
- Melling, N.; Kowitz, C.M.; Simon, R.; Bokemeyer, C.; Terracciano, L.; Sauter, G.; Izbicki, J.R.; Marx, A.H. High Ki67 expression is an independent good prognostic marker in colorectal cancer. J. Clin. Pathol. 2016, 69, 209–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mucci, N.R.; Rubin, M.A.; Strawderman, M.S.; Montie, J.E.; Smith, D.C.; Pienta, K.J. Expression of nuclear antigen Ki-67 in prostate cancer needle biopsy and radical prostatectomy specimens. J. Natl. Cancer Inst. 2000, 92, 1941–1942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sonveaux, P.; Vegran, F.; Schroeder, T.; Wergin, M.C.; Verrax, J.; Rabbani, Z.N.; De Saedeleer, C.J.; Kennedy, K.M.; Diepart, C.; Jordan, B.F.; et al. Targeting lactate-fueled respiration selectively kills hypoxic tumor cells in mice. J. Clin. Investig. 2008, 118, 3930–3942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brisson, L.; Banski, P.; Sboarina, M.; Dethier, C.; Danhier, P.; Fontenille, M.J.; Van Hee, V.F.; Vazeille, T.; Tardy, M.; Falces, J.; et al. Lactate dehydrogenase B controls lysosome activity and autophagy in cancer. Cancer Cell 2016, 30, 418–431. [Google Scholar] [CrossRef] [Green Version]
- Halestrap, A.P. The SLC16 gene family—Structure, role and regulation in health and disease. Mol. Asp. Med. 2013, 34, 337–349. [Google Scholar] [CrossRef]
- Pereira-Nunes, A.; Afonso, J.; Granja, S.; Baltazar, F. Lactate and Lactate Transporters as Key Players in the Maintenance of the Warburg Effect. Adv. Exp. Med. Biol. 2020, 1219, 51–74. [Google Scholar]
- Payen, V.L.; Mina, E.; Van Hee, V.F.; Porporato, P.E.; Sonveaux, P. Monocarboxylate transporters in cancer. Mol. Metab. 2020, 33, 48–66. [Google Scholar] [CrossRef]
- Ullah, M.S.; Davies, A.J.; Halestrap, A.P. The plasma membrane lactate transporter MCT4, but not MCT1, is up-regulated by hypoxia through a HIF-1alpha-dependent mechanism. J. Biol. Chem. 2006, 281, 9030–9037. [Google Scholar] [CrossRef] [Green Version]
- Dimmer, K.S.; Friedrich, B.; Lang, F.; Deitmer, J.W.; Broer, S. The low-affinity monocarboxylate transporter MCT4 is adapted to the export of lactate in highly glycolytic cells. Biochem. J. 2000, 350 Pt 1, 219–227. [Google Scholar] [CrossRef]
- Pertega-Gomes, N.; Vizcaino, J.R.; Gouveia, C.; Jeronimo, C.; Henrique, R.M.; Lopes, C.; Baltazar, F. Monocarboxylate transporter 2 (MCT2) as putative biomarker in prostate cancer. Prostate 2013, 73, 763–769. [Google Scholar] [CrossRef] [Green Version]
- Philp, N.J.; Yoon, H.; Lombardi, L. Mouse MCT3 gene is expressed preferentially in retinal pigment and choroid plexus epithelia. Am. J. Physiol. Cell Physiol. 2001, 280, C1319–C1326. [Google Scholar] [CrossRef] [PubMed]
- Polanski, R.; Hodgkinson, C.L.; Fusi, A.; Nonaka, D.; Priest, L.; Kelly, P.; Trapani, F.; Bishop, P.W.; White, A.; Critchlow, S.E.; et al. Activity of the monocarboxylate transporter 1 inhibitor AZD3965 in small cell lung cancer. Clin. Cancer Res. 2014, 20, 926–937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le Floch, R.; Chiche, J.; Marchiq, I.; Naiken, T.; Ilc, K.; Murray, C.M.; Critchlow, S.E.; Roux, D.; Simon, M.P.; Pouyssegur, J. CD147 subunit of lactate/H+ symporters MCT1 and hypoxia-inducible MCT4 is critical for energetics and growth of glycolytic tumors. Proc. Natl. Acad. Sci. USA 2011, 108, 16663–16668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Curtis, N.J.; Mooney, L.; Hopcroft, L.; Michopoulos, F.; Whalley, N.; Zhong, H.; Murray, C.; Logie, A.; Revill, M.; Byth, K.F.; et al. Pre-clinical pharmacology of AZD3965, a selective inhibitor of MCT1: DLBCL, NHL and Burkitt’s lymphoma anti-tumor activity. Oncotarget 2017, 8, 69219–69236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nancolas, B.; Sessions, R.B.; Halestrap, A.P. Identification of key binding site residues of MCT1 for AR-C155858 reveals the molecular basis of its isoform selectivity. Biochem. J. 2015, 466, 177–188. [Google Scholar] [CrossRef] [Green Version]
- Ovens, M.J.; Manoharan, C.; Wilson, M.C.; Murray, C.M.; Halestrap, A.P. The inhibition of monocarboxylate transporter 2 (MCT2) by AR-C155858 is modulated by the associated ancillary protein. Biochem. J. 2010, 431, 217–225. [Google Scholar] [CrossRef] [Green Version]
- Wilson, M.C.; Meredith, D.; Fox, J.E.; Manoharan, C.; Davies, A.J.; Halestrap, A.P. Basigin (CD147) is the target for organomercurial inhibition of monocarboxylate transporter isoforms 1 and 4: The ancillary protein for the insensitive MCT2 is EMBIGIN (gp70). J. Biol. Chem. 2005, 280, 27213–27221. [Google Scholar] [CrossRef] [Green Version]
- Juel, C. Current aspects of lactate exchange: Lactate/H+ transport in human skeletal muscle. Eur. J. Appl. Physiol. 2001, 86, 12–16. [Google Scholar]
- Debernardi, R.; Pierre, K.; Lengacher, S.; Magistretti, P.J.; Pellerin, L. Cell-specific expression pattern of monocarboxylate transporters in astrocytes and neurons observed in different mouse brain cortical cell cultures. J. Neurosci. Res. 2003, 73, 141–155. [Google Scholar] [CrossRef]
- Hui, S.; Ghergurovich, J.M.; Morscher, R.J.; Jang, C.; Teng, X.; Lu, W.; Esparza, L.A.; Reya, T.; Le, Z.; Yanxiang Guo, J.; et al. Glucose feeds the TCA cycle via circulating lactate. Nature 2017, 551, 115–118. [Google Scholar] [CrossRef] [Green Version]
- Hong, C.S.; Graham, N.A.; Gu, W.; Espindola Camacho, C.; Mah, V.; Maresh, E.L.; Alavi, M.; Bagryanova, L.; Krotee, P.A.L.; Gardner, B.K.; et al. MCT1 modulates cancer cell pyruvate export and growth of tumors that co-express MCT1 and MCT4. Cell Rep. 2016, 14, 1590–1601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaupel, P. Is there a critical tissue oxygen tension for bioenergetic status and cellular pH regulation in solid tumors? Experientia 1996, 52, 464–468. [Google Scholar] [CrossRef] [PubMed]
- Guan, X.; Ruszaj, D.; Morris, M.E. Development and validation of a liquid chromatography tandem mass spectrometry assay for AZD3965 in mouse plasma and tumor tissue: Application to pharmacokinetic and breast tumor xenograft studies. J. Pharm. Biomed. Anal. 2018, 155, 270–275. [Google Scholar] [CrossRef] [PubMed]
- Guan, X.; Morris, M.E. Pharmacokinetics of the monocarboxylate transporter 1 inhibitor AZD3965 in mice: Potential enterohepatic circulation and target-mediated disposition. Pharm. Res. 2019, 37, 5. [Google Scholar] [CrossRef]
- Masuya, H.; Inoue, M.; Wada, Y.; Shimizu, A.; Nagano, J.; Kawai, A.; Inoue, A.; Kagami, T.; Hirayama, T.; Yamaga, A.; et al. Implementation of the modified-SHIRPA protocol for screening of dominant phenotypes in a large-scale ENU mutagenesis program. Mamm. Genome 2005, 16, 829–837. [Google Scholar] [CrossRef]
- Polet, F.; Feron, O. Endothelial cell metabolism and tumour angiogenesis: Glucose and glutamine as essential fuels and lactate as the driving force. J. Intern. Med. 2013, 273, 156–165. [Google Scholar] [CrossRef]
- Allen, E.; Mieville, P.; Warren, C.M.; Saghafinia, S.; Li, L.; Peng, M.W.; Hanahan, D. Metabolic symbiosis enables adaptive resistance to anti-angiogenic therapy that is dependent on mTOR signaling. Cell Rep. 2016, 15, 1144–1160. [Google Scholar] [CrossRef] [Green Version]
- Pisarsky, L.; Bill, R.; Fagiani, E.; Dimeloe, S.; Goosen, R.W.; Hagmann, J.; Hess, C.; Christofori, G. Targeting metabolic symbiosis to overcome resistance to anti-angiogenic therapy. Cell Rep. 2016, 15, 1161–1174. [Google Scholar] [CrossRef] [Green Version]
- Jimenez-Valerio, G.; Martinez-Lozano, M.; Bassani, N.; Vidal, A.; Ochoa-de-Olza, M.; Suarez, C.; Garcia-Del-Muro, X.; Carles, J.; Vinals, F.; Graupera, M.; et al. Resistance to antiangiogenic therapies by metabolic symbiosis in renal cell carcinoma PDX models and patients. Cell Rep. 2016, 15, 1134–1143. [Google Scholar] [CrossRef] [Green Version]
- Martinez-Outschoorn, U.E.; Lin, Z.; Whitaker-Menezes, D.; Howell, A.; Lisanti, M.P.; Sotgia, F. Ketone bodies and two-compartment tumor metabolism: Stromal ketone production fuels mitochondrial biogenesis in epithelial cancer cells. Cell Cycle 2012, 11, 3956–3963. [Google Scholar] [CrossRef]
- Puri, S.; Juvale, K. Monocarboxylate transporter 1 and 4 inhibitors as potential therapeutics for treating solid tumours: A review with structure-activity relationship insights. Eur. J. Med. Chem. 2020, 199, 112393. [Google Scholar] [CrossRef] [PubMed]
- Pinheiro, C.; Albergaria, A.; Paredes, J.; Sousa, B.; Dufloth, R.; Vieira, D.; Schmitt, F.; Baltazar, F. Monocarboxylate transporter 1 is up-regulated in basal-like breast carcinoma. Histopathology 2010, 56, 860–867. [Google Scholar] [CrossRef] [PubMed]
- Soule, H.D.; Maloney, T.M.; Wolman, S.R.; Peterson, W.D., Jr.; Brenz, R.; McGrath, C.M.; Russo, J.; Pauley, R.J.; Jones, R.F.; Brooks, S.C. Isolation and characterization of a spontaneously immortalized human breast epithelial cell line, MCF-10. Cancer Res. 1990, 50, 6075–6086. [Google Scholar] [PubMed]
- Bodnar, A.G.; Ouellette, M.; Frolkis, M.; Holt, S.E.; Chiu, C.P.; Morin, G.B.; Harley, C.B.; Shay, J.W.; Lichtsteiner, S.; Wright, W.E. Extension of life-span by introduction of telomerase into normal human cells. Science 1998, 279, 349–352. [Google Scholar] [CrossRef] [Green Version]
- Walenta, S.; Snyder, S.; Haroon, Z.A.; Braun, R.D.; Amin, K.; Brizel, D.; Mueller-Klieser, W.; Chance, B.; Dewhirst, M.W. Tissue gradients of energy metabolites mirror oxygen tension gradients in a rat mammary carcinoma model. Int. J. Radiat. Oncol. Biol. Phys. 2001, 51, 840–848. [Google Scholar] [CrossRef]
- Thangaraju, M.; Karunakaran, S.K.; Itagaki, S.; Gopal, E.; Elangovan, S.; Prasad, P.D.; Ganapathy, V. Transport by SLC5A8 with subsequent inhibition of histone deacetylase 1 (HDAC1) and HDAC3 underlies the antitumor activity of 3-bromopyruvate. Cancer 2009, 115, 4655–4666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sonveaux, P.; Copetti, T.; De Saedeleer, C.J.; Vegran, F.; Verrax, J.; Kennedy, K.M.; Moon, E.J.; Dhup, S.; Danhier, P.; Frerart, F.; et al. Targeting the lactate transporter MCT1 in endothelial cells inhibits lactate-induced HIF-1 activation and tumor angiogenesis. PLoS ONE 2012, 7, e33418. [Google Scholar] [CrossRef]
- Lizardo, D.Y.; Lin, Y.L.; Gokcumen, O.; Atilla-Gokcumen, G.E. Regulation of lipids is central to replicative senescence. Mol. Biosyst. 2017, 13, 498–509. [Google Scholar] [CrossRef] [PubMed]
- DeBerardinis, R.J.; Mancuso, A.; Daikhin, E.; Nissim, I.; Yudkoff, M.; Wehrli, S.; Thompson, C.B. Beyond aerobic glycolysis: Transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc. Natl. Acad. Sci. USA 2007, 104, 19345–19350. [Google Scholar] [CrossRef] [Green Version]
- Landon, J.; Fawcett, J.K.; Wynn, V. Blood pyruvate concentration measured by a specific method in control subjects. J. Clin. Pathol. 1962, 15, 579–584. [Google Scholar] [CrossRef]
- Doherty, J.R.; Yang, C.; Scott, K.E.; Cameron, M.D.; Fallahi, M.; Li, W.; Hall, M.A.; Amelio, A.L.; Mishra, J.K.; Li, F.; et al. Blocking lactate export by inhibiting the Myc target MCT1 disables glycolysis and glutathione synthesis. Cancer Res. 2014, 74, 908–920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manning Fox, J.E.; Meredith, D.; Halestrap, A.P. Characterisation of human monocarboxylate transporter 4 substantiates its role in lactic acid efflux from skeletal muscle. J. Physiol. 2000, 529 Pt 2, 285–293. [Google Scholar]
- Singleton, D.C.; Dechaume, A.L.; Murray, P.M.; Katt, W.P.; Baguley, B.C.; Leung, E.Y. Pyruvate anaplerosis is a mechanism of resistance to pharmacological glutaminase inhibition in triple-receptor negative breast cancer. BMC Cancer 2020, 20, 470. [Google Scholar] [CrossRef] [PubMed]
- Murray, C.M.; Hutchinson, R.; Bantick, J.R.; Belfield, G.P.; Benjamin, A.D.; Brazma, D.; Bundick, R.V.; Cook, I.D.; Craggs, R.I.; Edwards, S.; et al. Monocarboxylate transporter MCT1 is a target for immunosuppression. Nat. Chem. Biol. 2005, 1, 371–376. [Google Scholar] [CrossRef] [PubMed]
- Huang, T.; Feng, Q.; Wang, Z.; Li, W.; Sun, Z.; Wilhelm, J.; Huang, G.; Vo, T.; Sumer, B.D.; Gao, J. Tumor-targeted inhibition of monocarboxylate transporter 1 improves T-cell immunotherapy of solid tumors. Adv. Healthc. Mater. 2020. [Google Scholar] [CrossRef]
- Allen, A.E.; Martin, E.A.; Greenwood, K.; Grant, C.; Vince, P.; Lucas, R.J.; Redfern, W.S. Effects of a monocarboxylate transport 1 inhibitor, AZD3965, on retinal and visual function in the rat. Br. J. Pharmacol. 2020, 177, 4734–4749. [Google Scholar] [CrossRef]
- McNeillis, R.; Greystoke, A.; Walton, J.; Bacon, C.; Keun, H.; Siskos, A.; Petrides, G.; Leech, N.; Jenkinson, F.; Bowron, A.; et al. Correction: A case of malignant hyperlactaemic acidosis appearing upon treatment with the mono-carboxylase transporter 1 inhibitor AZD3965. Br. J. Cancer 2020, 122, 1272. [Google Scholar] [CrossRef] [Green Version]
- Lengacher, S.; Nehiri-Sitayeb, T.; Steiner, N.; Carneiro, L.; Favrod, C.; Preitner, F.; Thorens, B.; Stehle, J.C.; Dix, L.; Pralong, F.; et al. Resistance to diet-induced obesity and associated metabolic perturbations in haploinsufficient monocarboxylate transporter 1 mice. PLoS ONE 2013, 8, e82505. [Google Scholar] [CrossRef] [Green Version]
- Guan, X.; Rodriguez-Cruz, V.; Morris, M.E. Cellular Uptake of MCT1 inhibitors AR-C155858 and AZD3965 and their effects on MCT-mediated transport of L-lactate in murine 4T1 breast tumor cancer cells. AAPS J. 2019, 21, 13. [Google Scholar] [CrossRef]
- Suzuki, A.; Stern, S.A.; Bozdagi, O.; Huntley, G.W.; Walker, R.H.; Magistretti, P.J.; Alberini, C.M. Astrocyte-neuron lactate transport is required for long-term memory formation. Cell 2011, 144, 810–823. [Google Scholar] [CrossRef] [Green Version]
- Mota, A.L.; Evangelista, A.F.; Macedo, T.; Oliveira, R.; Scapulatempo-Neto, C.; Vieira, R.A.; Marques, M.M.C. Molecular characterization of breast cancer cell lines by clinical immunohistochemical markers. Oncol. Lett. 2017, 13, 4708–4712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Saedeleer, C.J.; Porporato, P.E.; Copetti, T.; Perez-Escuredo, J.; Payen, V.L.; Brisson, L.; Feron, O.; Sonveaux, P. Glucose deprivation increases monocarboxylate transporter 1 (MCT1) expression and MCT1-dependent tumor cell migration. Oncogene 2014, 33, 4060–4068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corbet, C.; Draoui, N.; Polet, F.; Pinto, A.; Drozak, X.; Riant, O.; Feron, O. The SIRT1/HIF2alpha axis drives reductive glutamine metabolism under chronic acidosis and alters tumor response to therapy. Cancer Res. 2014, 74, 5507–5519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Hee, V.F.; Perez-Escuredo, J.; Cacace, A.; Copetti, T.; Sonveaux, P. Lactate does not activate NF-kappaB in oxidative tumor cells. Front. Pharmacol. 2015, 6, 228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zampieri, L.X.; Grasso, D.; Bouzin, C.; Brusa, D.; Rossignol, R.; Sonveaux, P. Mitochondria participate in chemoresistance to cisplatin in human ovarian cancer cells. Mol. Cancer Res. 2020, 18, 1379–1391. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| [AZD3965], 1 dose, 72 h | |||||||
| Vehicle | 1 nmol/L | 10 nmol/L | 100 nmol/L | 1 µmol/L | 10 µmol/L | n | |
| T47D | No cell death 1 | No cell death | No cell death | No cell death | No cell death | No cell death | 15–16 |
| MCF7 | No cell death | No cell death | No cell death | No cell death | No cell death | No cell death | 15–16 |
| MCF10A | 194 ± 20 | 152 ± 15 | 171 ± 19 | 241 ± 45 | 165 ± 21 | 222 ± 26 | 15–16 |
| BJ fibro | 460 ± 96 | 299 ± 34 | 396 ± 130 | 283 ± 25 | No cell death | 141 ±36 | 15–16 |
| [AZD3965], 3 daily doses, 72 h | |||||||
| Vehicle | 1 nmol/L | 10 nmol/L | 100 nmol/L | 1 µmol/L | 10 µmol/L | n | |
| T47D | 181 ± 31 | No cell death | No cell death | No cell death | 187 ± 42 | 138 ±51 | 15–16 |
| MCF7 | 103 ± 15 | No cell death | 138 ± 25 | 153 ± 30 | 68 ± 35 | 106 ± 8 | 15–16 |
| MCF10A | 119 ± 13 | 120 ± 14 | 109 ± 11 | 88 ± 7 | 104 ± 10 | 95 ± 7 | 16 |
| BJ fibro | 73 ± 3 | 82 ± 7 | 66 ± 4 | 68 ± 6 | 63 ± 2 | 59 ± 1 | 15–16 |
| [AZD3965], 1 dose, 72 h | |||||||
| Vehicle | 1 nmol/L | 10 nmol/L | 100 nmol/L | 1 µmol/L | 10 µmol/L | n | |
| T47D | 78 ± 3 | 77 ± 3 | 71 ± 4 | 75 ± 5 | 74 ± 4 | 103 ± 5 *** | 8 |
| MCF7 | 62 ± 3 | 58 ± 2 | 57 ± 1 | 64 ± 1 | 62 ± 2 | 72 ± 3 * | 7–8 |
| MCF10A | 70 ± 3 | 60 ± 1 | 63 ± 2 | 72 ± 6 | 57 ± 2 * | 63 ± 3 | 8 |
| BJ fibroblasts | 50 ± 1 | 49 ± 1 | 47 ± 1 | 46 ± 1 * | 47 ± 1 | 48 ± 1 | 8 |
| [AZD3965], 3 daily doses, 72 h | |||||||
| Vehicle | 1 nmol/L | 10 nmol/L | 100 nmol/L | 1 µmol/L | 10 µmol/L | n | |
| T47D | 49 ± 5 | 52 ± 6 | 52 ± 2 | 53 ± 3 | 51 ± 4 | 59 ± 7 | 7–8 |
| MCF7 | 87 ± 6 | 98 ± 11 | 86 ± 4 | 100 ± 4 | 88 ± 4 | 129 ± 4 ** | 7–8 |
| MCF10A | 132 ± 15 | 127 ± 16 | 91 ± 3 | 164 ± 24 | 108 ± 8 | 123 ± 13 | 7–8 |
| BJ fibroblasts | 53 ± 1 | 54 ± 1 | 51 ± 0.5 | 52 ± 0.5 | 53 ± 0.5 | 55 ± 1 | 7–8 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Benyahia, Z.; Blackman, M.C.N.M.; Hamelin, L.; Zampieri, L.X.; Capeloa, T.; Bedin, M.L.; Vazeille, T.; Schakman, O.; Sonveaux, P. In Vitro and In Vivo Characterization of MCT1 Inhibitor AZD3965 Confirms Preclinical Safety Compatible with Breast Cancer Treatment. Cancers 2021, 13, 569. https://doi.org/10.3390/cancers13030569
Benyahia Z, Blackman MCNM, Hamelin L, Zampieri LX, Capeloa T, Bedin ML, Vazeille T, Schakman O, Sonveaux P. In Vitro and In Vivo Characterization of MCT1 Inhibitor AZD3965 Confirms Preclinical Safety Compatible with Breast Cancer Treatment. Cancers. 2021; 13(3):569. https://doi.org/10.3390/cancers13030569
Chicago/Turabian StyleBenyahia, Zohra, Marine C. N. M. Blackman, Loïc Hamelin, Luca X. Zampieri, Tania Capeloa, Marie L. Bedin, Thibaut Vazeille, Olivier Schakman, and Pierre Sonveaux. 2021. "In Vitro and In Vivo Characterization of MCT1 Inhibitor AZD3965 Confirms Preclinical Safety Compatible with Breast Cancer Treatment" Cancers 13, no. 3: 569. https://doi.org/10.3390/cancers13030569