
p38 MAPK Inhibition Mitigates Hypoxia-Induced AR Signaling in Castration-Resistant Prostate Cancer


Abstract
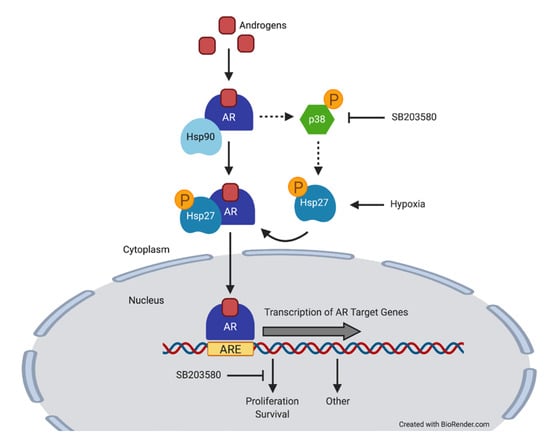
:Simple Summary
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. p38 MAPK Inhibition Decreases Cell Proliferation in Prostate Cancer Cells Expressing AR
2.2. p38 MAPK Inhibition Decreases Hsp27 Phosphorylation, AR Activity and Expression of AR Target Genes under Normoxia and Hypoxia
2.3. Hypoxia and AR Signaling Activate Hsp27
2.4. p38 MAPK Inhibition Prolongs Survival in Mice Bearing CRPC Xenografts
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Chemicals
4.3. Cell proliferation Assay
4.4. Western Blotting
4.5. AR Activity Luciferase Assay
4.6. RNA Extraction and Quantitative PCR
4.7. Clonogenic Assay
4.8. siRNA Transfections
4.9. Xenograft Experiments
4.10. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [Green Version]
- Kirby, M.; Hirst, C.; Crawford, E.D. Characterising the castration-resistant prostate cancer population: A systematic review. Int. J. Clin. Pract. 2011, 65, 1180–1192. [Google Scholar] [CrossRef]
- Lin, T.T.; Chen, Y.H.; Wu, Y.P.; Chen, S.Z.; Li, X.D.; Lin, Y.Z.; Chen, S.H.; Zheng, Q.S.; Wei, Y.; Xu, N.; et al. Risk factors for progression to castration-resistant prostate cancer in metastatic prostate cancer patients. J. Cancer 2019, 10, 5608–5613. [Google Scholar] [CrossRef]
- Lalonde, E.; Ishkanian, A.S.; Sykes, J.; Fraser, M.; Ross-Adams, H.; Erho, N.; Dunning, M.J.; Halim, S.; Lamb, A.D.; Moon, N.C.; et al. Tumour genomic and microenvironmental heterogeneity for integrated prediction of 5-year biochemical recurrence of prostate cancer: A retrospective cohort study. Lancet Oncol. 2014, 15, 1521–1532. [Google Scholar] [CrossRef]
- Milosevic, M.; Warde, P.; Menard, C.; Chung, P.; Toi, A.; Ishkanian, A.; McLean, M.; Pintilie, M.; Sykes, J.; Gospodarowicz, M.; et al. Tumor hypoxia predicts biochemical failure following radiotherapy for clinically localized prostate cancer. Clin. Cancer Res. 2012, 18, 2108–2114. [Google Scholar] [CrossRef] [Green Version]
- Wilson, W.R.; Hay, M.P. Targeting hypoxia in cancer therapy. Nat. Rev. Cancer 2011, 11, 393–410. [Google Scholar] [CrossRef]
- Brown, J.M.; Wilson, W.R. Exploiting tumour hypoxia in cancer treatment. Nat. Rev. Cancer 2004, 4, 437–447. [Google Scholar] [CrossRef]
- Mitani, T.; Yamaji, R.; Higashimura, Y.; Harada, N.; Nakano, Y.; Inui, H. Hypoxia enhances transcriptional activity of androgen receptor through hypoxia-inducible factor-1alpha in a low androgen environment. J. Steroid. Biochem. Mol. Biol. 2011, 123, 58–64. [Google Scholar] [CrossRef]
- Park, C.; Kim, Y.; Shim, M.; Lee, Y. Hypoxia enhances ligand-occupied androgen receptor activity. Biochem. Biophys. Res. Commun. 2012, 418, 319–323. [Google Scholar] [CrossRef]
- Park, S.Y.; Kim, Y.J.; Gao, A.C.; Mohler, J.L.; Onate, S.A.; Hidalgo, A.A.; Ip, C.; Park, E.M.; Yoon, S.Y.; Park, Y.M. Hypoxia increases androgen receptor activity in prostate cancer cells. Cancer Res. 2006, 66, 5121–5129. [Google Scholar] [CrossRef] [Green Version]
- Cuenda, A.; Rousseau, S. p38 MAP-kinases pathway regulation, function and role in human diseases. Biochim. Biophys. Acta 2007, 1773, 1358–1375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zarubin, T.; Han, J. Activation and signaling of the p38 MAP kinase pathway. Cell Res. 2005, 15, 11–18. [Google Scholar] [CrossRef] [Green Version]
- Zoubeidi, A.; Zardan, A.; Beraldi, E.; Fazli, L.; Sowery, R.; Rennie, P.; Nelson, C.; Gleave, M. Cooperative interactions between androgen receptor (AR) and heat-shock protein 27 facilitate AR transcriptional activity. Cancer Res. 2007, 67, 10455–10465. [Google Scholar] [CrossRef] [Green Version]
- Lin, D.L.; Whitney, M.C.; Yao, Z.; Keller, E.T. Interleukin-6 induces androgen responsiveness in prostate cancer cells through up-regulation of androgen receptor expression. Clin. Cancer Res. 2001, 7, 1773–1781. [Google Scholar]
- Khandrika, L.; Lieberman, R.; Koul, S.; Kumar, B.; Maroni, P.; Chandhoke, R.; Meacham, R.B.; Koul, H.K. Hypoxia-associated p38 mitogen-activated protein kinase-mediated androgen receptor activation and increased HIF-1alpha levels contribute to emergence of an aggressive phenotype in prostate cancer. Oncogene 2009, 28, 1248–1260. [Google Scholar] [CrossRef] [Green Version]
- Ricote, M.; Garcia-Tunon, I.; Fraile, B.; Fernandez, C.; Aller, P.; Paniagua, R.; Royuela, M. P38 MAPK protects against TNF-alpha-provoked apoptosis in LNCaP prostatic cancer cells. Apoptosis 2006, 11, 1969–1975. [Google Scholar] [CrossRef]
- Zhang, Y.; Guo, Z.; Du, T.; Chen, J.; Wang, W.; Xu, K.; Lin, T.; Huang, H. Prostate specific membrane antigen (PSMA): A novel modulator of p38 for proliferation, migration, and survival in prostate cancer cells. Prostate 2013, 73, 835–841. [Google Scholar] [CrossRef]
- Cui, Y.; Sun, Y.; Hu, S.; Luo, J.; Li, L.; Li, X.; Yeh, S.; Jin, J.; Chang, C. Neuroendocrine prostate cancer (NEPCa) increased the neighboring PCa chemoresistance via altering the PTHrP/p38/Hsp27/androgen receptor (AR)/p21 signals. Oncogene 2016, 35, 6065–6076. [Google Scholar] [CrossRef] [Green Version]
- Royuela, M.; Arenas, M.I.; Bethencourt, F.R.; Sanchez-Chapado, M.; Fraile, B.; Paniagua, R. Regulation of proliferation/apoptosis equilibrium by mitogen-activated protein kinases in normal, hyperplastic, and carcinomatous human prostate. Hum. Pathol. 2002, 33, 299–306. [Google Scholar] [CrossRef]
- Emerling, B.M.; Platanias, L.C.; Black, E.; Nebreda, A.R.; Davis, R.J.; Chandel, N.S. Mitochondrial reactive oxygen species activation of p38 mitogen-activated protein kinase is required for hypoxia signaling. Mol. Cell Biol. 2005, 25, 4853–4862. [Google Scholar] [CrossRef] [Green Version]
- Shi, Q.; Shi, J.; Luo, F.; Song, G.; He, X.; Xia, Y. Major Differences in Hypoxia Tolerance and P38 Regulation Among Different Renal Cells. Cell Physiol. Biochem. 2018, 46, 1483–1492. [Google Scholar] [CrossRef]
- Xu, L.; Pathak, P.S.; Fukumura, D. Hypoxia-induced activation of p38 mitogen-activated protein kinase and phosphatidylinositol 3’-kinase signaling pathways contributes to expression of interleukin 8 in human ovarian carcinoma cells. Clin. Cancer Res. 2004, 10, 701–707. [Google Scholar] [CrossRef] [Green Version]
- Graham, L.; Schweizer, M.T. Targeting persistent androgen receptor signaling in castration-resistant prostate cancer. Med. Oncol. 2016, 33, 44. [Google Scholar] [CrossRef]
- Ranasinghe, W.K.; Xiao, L.; Kovac, S.; Chang, M.; Michiels, C.; Bolton, D.; Shulkes, A.; Baldwin, G.S.; Patel, O. The role of hypoxia-inducible factor 1alpha in determining the properties of castrate-resistant prostate cancers. PLoS ONE 2013, 8, e54251. [Google Scholar] [CrossRef]
- Gioeli, D.; Black, B.E.; Gordon, V.; Spencer, A.; Kesler, C.T.; Eblen, S.T.; Paschal, B.M.; Weber, M.J. Stress kinase signaling regulates androgen receptor phosphorylation, transcription, and localization. Mol. Endocrinol. 2006, 20, 503–515. [Google Scholar] [CrossRef] [Green Version]
- Ni, H.; Wang, X.S.; Diener, K.; Yao, Z. MAPKAPK5, a novel mitogen-activated protein kinase (MAPK)-activated protein kinase, is a substrate of the extracellular-regulated kinase (ERK) and p38 kinase. Biochem. Biophys. Res. Commun. 1998, 243, 492–496. [Google Scholar] [CrossRef]
- New, L.; Jiang, Y.; Zhao, M.; Liu, K.; Zhu, W.; Flood, L.J.; Kato, Y.; Parry, G.C.; Han, J. PRAK, a novel protein kinase regulated by the p38 MAP kinase. EMBO J. 1998, 17, 3372–3384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, P.; Yoshizuka, N.; New, L.; Moser, B.A.; Li, Y.; Liao, R.; Xie, C.; Chen, J.; Deng, Q.; Yamout, M.; et al. PRAK is essential for ras-induced senescence and tumor suppression. Cell 2007, 128, 295–308. [Google Scholar] [CrossRef]
- Tran, M.G.B.; Bibby, B.A.S.; Yang, L.; Lo, F.; Warren, A.Y.; Shukla, D.; Osborne, M.; Hadfield, J.; Carroll, T.; Stark, R.; et al. Independence of HIF1a and androgen signaling pathways in prostate cancer. BMC Cancer 2020, 20, 469. [Google Scholar] [CrossRef]
- Zhai, W.; Sun, Y.; Jiang, M.; Wang, M.; Gasiewicz, T.A.; Zheng, J.; Chang, C. Differential regulation of LncRNA-SARCC suppresses VHL-mutant RCC cell proliferation yet promotes VHL-normal RCC cell proliferation via modulating androgen receptor/HIF-2alpha/C-MYC axis under hypoxia. Oncogene 2016, 35, 4866–4880. [Google Scholar] [CrossRef]
- Fernandez, E.V.; Reece, K.M.; Ley, A.M.; Troutman, S.M.; Sissung, T.M.; Price, D.K.; Chau, C.H.; Figg, W.D. Dual targeting of the androgen receptor and hypoxia-inducible factor 1alpha pathways synergistically inhibits castration-resistant prostate cancer cells. Mol. Pharmacol. 2015, 87, 1006–1012. [Google Scholar] [CrossRef] [Green Version]
- Vergote, I.; Heitz, F.; Buderath, P.; Powell, M.; Sehouli, J.; Lee, C.M.; Hamilton, A.; Fiorica, J.; Moore, K.N.; Teneriello, M.; et al. A randomized, double-blind, placebo-controlled phase 1b/2 study of ralimetinib, a p38 MAPK inhibitor, plus gemcitabine and carboplatin versus gemcitabine and carboplatin for women with recurrent platinum-sensitive ovarian cancer. Gynecol. Oncol. 2020, 156, 23–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biau, J.; Thivat, E.; Chautard, E.; Stefan, D.; Boone, M.; Chauffert, B.; Bourgne, C.; Richard, D.; Molna, I.; Levesque, S.; et al. Phase 1 trial of ralimetinib (LY2228820) with radiotherapy plus concomitant temozolomide in the treatment of newly diagnosed glioblastoma. Radiother. Oncol. 2020, 154, 227–234. [Google Scholar] [CrossRef] [PubMed]
- Bendell, J.C.; Bischoff, H.G.; Hwang, J.; Reinhardt, H.C.; Zander, T.; Wang, X.; Hynes, S.; Pitou, C.; Campbell, R.; Iversen, P.; et al. A phase 1 dose-escalation study of checkpoint kinase 1 (CHK1) inhibitor prexasertib in combination with p38 mitogen-activated protein kinase (p38 MAPK) inhibitor ralimetinib in patients with advanced or metastatic cancer. Investig. New Drugs 2020, 38, 1145–1155. [Google Scholar] [CrossRef]
- Song, Y.H.; Chai, Q.; Wang, N.L.; Yang, F.F.; Wang, G.H.; Hu, J.Y. X-rays induced IL-8 production in lung cancer cells via p38/MAPK and NF-kappaB pathway. Int. J. Radiat. Biol. 2020, 96, 1374–1381. [Google Scholar] [CrossRef] [PubMed]
- Yahyapour, R.; Amini, P.; Rezapoor, S.; Rezaeyan, A.; Farhood, B.; Cheki, M.; Fallah, H.; Najafi, M. Targeting of Inflammation for Radiation Protection and Mitigation. Curr. Mol. Pharmacol. 2018, 11, 203–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krishnamurthy, P.M.; Shukla, S.; Ray, P.; Mehra, R.; Nyati, M.K.; Lawrence, T.S.; Ray, D. Involvement of p38-betaTrCP-Tristetraprolin-TNFalpha axis in radiation pneumonitis. Oncotarget 2017, 8, 47767–47779. [Google Scholar] [CrossRef] [PubMed]
- Kuruma, H.; Matsumoto, H.; Shiota, M.; Bishop, J.; Lamoureux, F.; Thomas, C.; Briere, D.; Los, G.; Gleave, M.; Fanjul, A.; et al. A novel antiandrogen, Compound 30, suppresses castration-resistant and MDV3100-resistant prostate cancer growth in vitro and in vivo. Mol. Cancer Ther. 2013, 12, 567–576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neveu, B.; Jain, P.; Tetu, B.; Wu, L.; Fradet, Y.; Pouliot, F. A PCA3 gene-based transcriptional amplification system targeting primary prostate cancer. Oncotarget 2016, 7, 1300–1310. [Google Scholar] [CrossRef] [Green Version]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cheung, S.; Jain, P.; So, J.; Shahidi, S.; Chung, S.; Koritzinsky, M. p38 MAPK Inhibition Mitigates Hypoxia-Induced AR Signaling in Castration-Resistant Prostate Cancer. Cancers 2021, 13, 831. https://doi.org/10.3390/cancers13040831
Cheung S, Jain P, So J, Shahidi S, Chung S, Koritzinsky M. p38 MAPK Inhibition Mitigates Hypoxia-Induced AR Signaling in Castration-Resistant Prostate Cancer. Cancers. 2021; 13(4):831. https://doi.org/10.3390/cancers13040831
Chicago/Turabian StyleCheung, Serina, Pallavi Jain, Jonathan So, Saeid Shahidi, Stephen Chung, and Marianne Koritzinsky. 2021. "p38 MAPK Inhibition Mitigates Hypoxia-Induced AR Signaling in Castration-Resistant Prostate Cancer" Cancers 13, no. 4: 831. https://doi.org/10.3390/cancers13040831