
Advances in Immunotherapy and the TGF-β Resistance Pathway in Metastatic Bladder Cancer


Abstract
:Simple Summary
Abstract
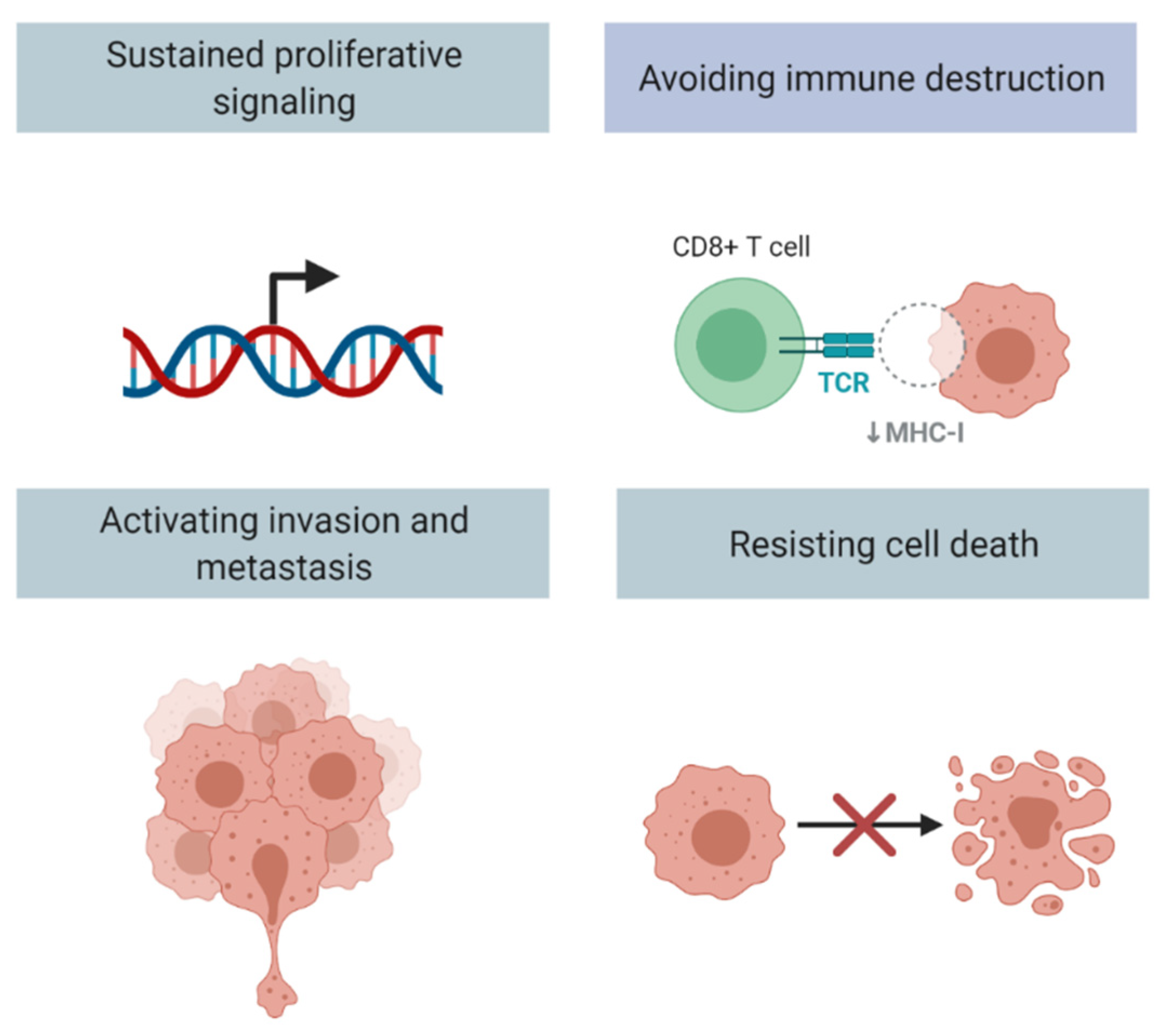
1. Introduction
2. TGF-B Signaling and Cancer Progression
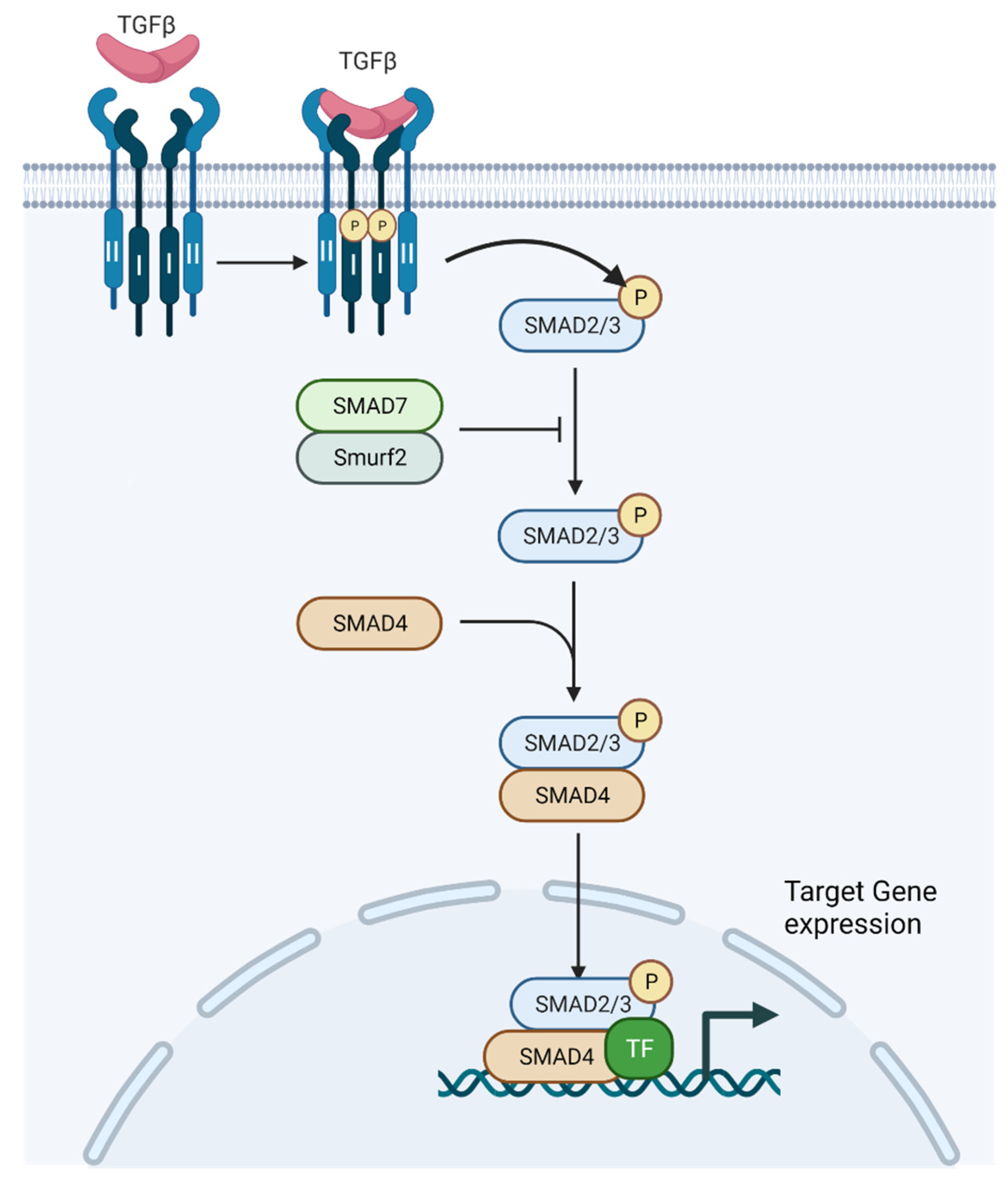
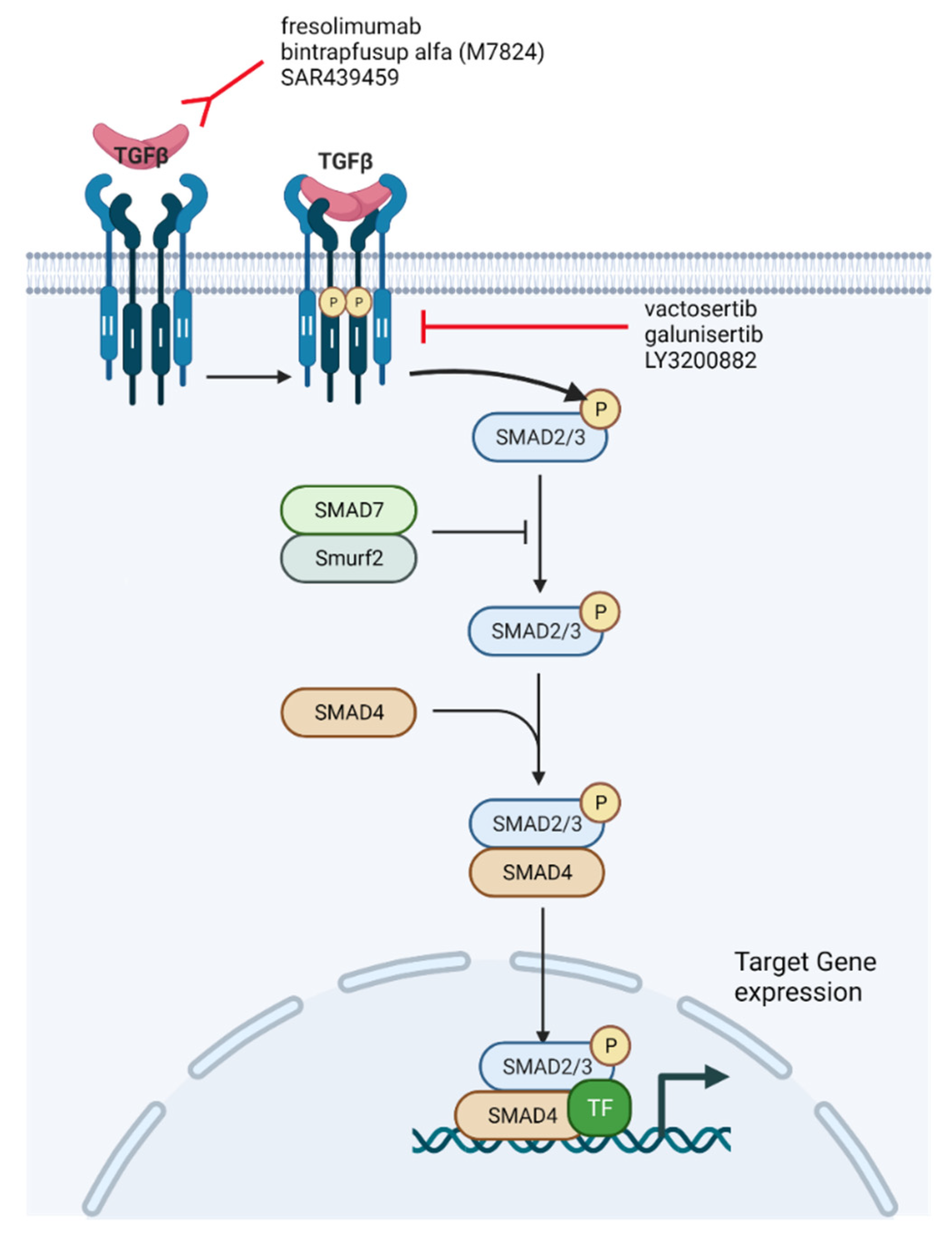
2.1. TGF-β Signaling
2.2. TGF-β Pathway and Fibroblasts
2.3. TGF-β Pathway Can Drive Cancer Metastases and Drug Resistance via Epithelial Mesenchymal Transformation (EMT)
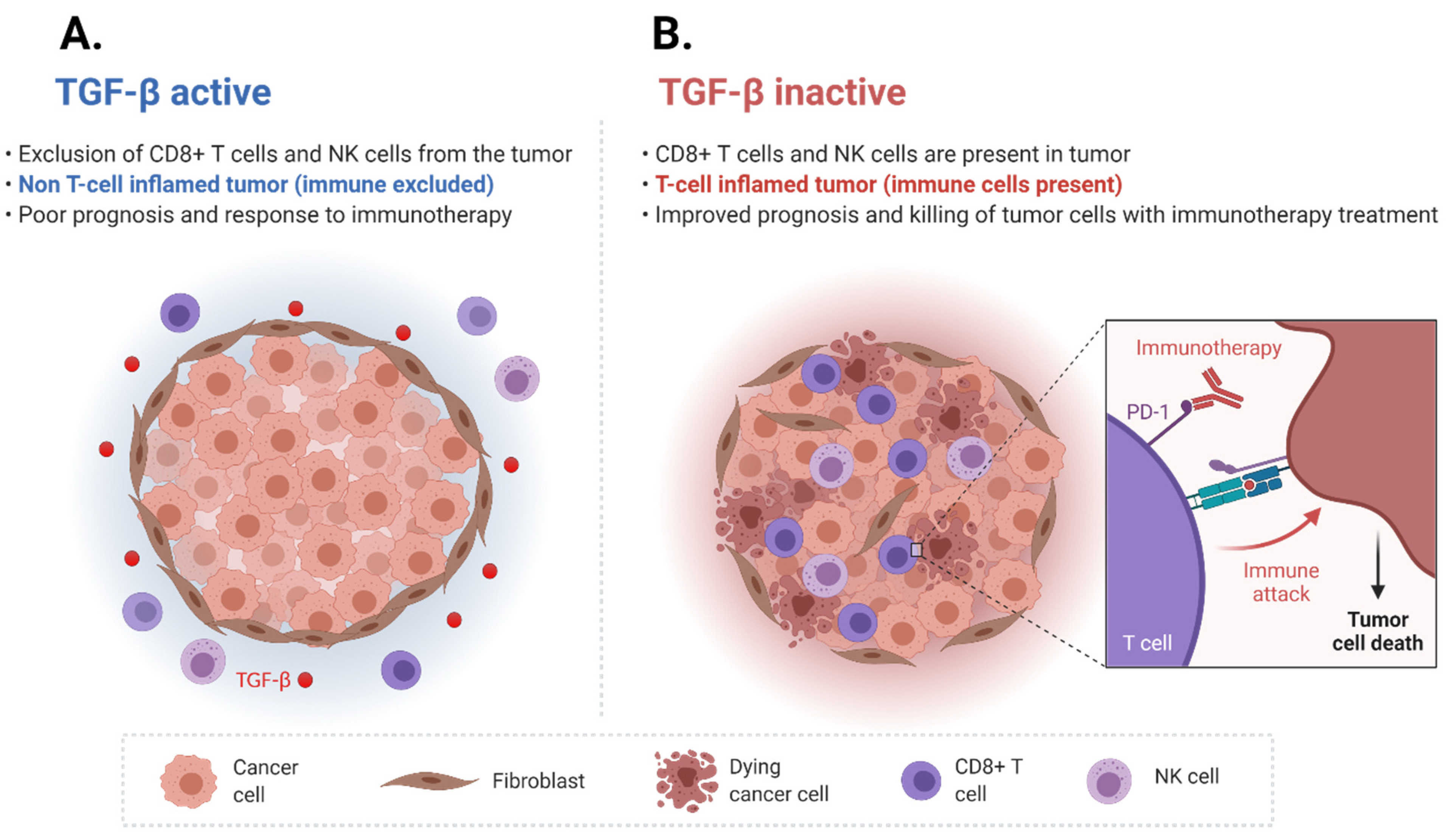
3. TGF-β Signaling Affects the Tumor Immune Microenvironment
4. TGF-β Pathway Induced Immune Cell Exclusion and Immune Checkpoint Inhibitor Resistance
5. Overcoming ICI Resistance through the Use of TGF-β Inhibition

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Setting | Anti-TGF-β Pathway Agent | Mechanism of Action of Anti-TGF-β Agent | ICI | Results | Citation |
|---|---|---|---|---|---|
| Phase I in patients with metastatic melanoma and metastatic renal cell carcinoma | Fresolimumab | Monoclonal antibody | N/A | 1 partial response (1/29 = 3.4%), 6 stable disease (6/29 = 20.7%). Median PFS of 24 weeks | Morris JC, et al. PloS One. 2014;9(3):e90353. [91] |
| Phase I in patients with glioma | Galunisertib | Small-molecule inhibitor | N/A | Clinical response (complete response + partial response + stable disease) in 12/56 (21.4%) patients | Rodon J, et al. Clin Cancer Res Off J Am Assoc Cancer Res. 2015 Feb 1;21(3):553–60. [98] |
| Phase Ib study in patients with unresectable HCC | Galunisertib | Small-molecule inhibitor | N/A | 1 partial response (1/14 = 7.1%), 11 stable disease (11/14 = 78.6%) | Ikeda M, et al. Invest New Drugs. 2019 Feb;37(1):118–26. [99] |
| Phase I in patients with several malignancies, including platinum-refractory NSCLC | Bintrafusp alfa | Bifunctional fusion protein | N/A | Overall response rate was 21.3% (17 out of 80 patients). | Paz-Ares L, et al. J Thorac Oncol Off Publ Int Assoc Study Lung Cancer. 2020 Jul;15(7):1210–22. [101] |
| Setting | Anti-TGF-β Pathway Agent | Mechanism of Action of Anti-TGF-β Agent | ICI | ClinicalTrials.gov Identifier | Status |
|---|---|---|---|---|---|
| Phase I study in patients with advanced solid tumors | SAR439459 | Pan-neutralizing anti-TGF-β antibody | With or without cemiplimab | NCT03192345 | Recruiting |
| Phase Ib study in advanced/unresectable solid tumors (TACTIC Trial) | SAR439459 | Pan-neutralizing anti-TGF-β antibody | Cemiplimab | NCT04729725 | Recruiting |
| Phase I study in advanced malignancies | NIS793 | Monoclonal antibody | PDR001 | NCT02947165 | Active, not recruiting |
| Phase II, open-label study in mUC failing ICI | Vactosertib | Oral inhibitor | Durvalumab | NCT04064190 | Not yet recruiting |
| Phase II study in ICI naïve and refractory mUC | Bintrafusp alfa (M7824) | Bifunctional fusion protein | N/A | NCT04501094 | Recruiting |
| Phase I study in adult patients with locally advanced or metastatic tumors | SRK-181 | Monoclonal antibody | Approved anti-PD-L1 therapy for each tumor type | NCT04291079 | Recruiting |
| Patients with advanced cancer | LY3200882 | Small-molecule inhibitor | Pembrolizumab | NCT04158700 | Withdrawn |
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Saginala, K.; Barsouk, A.; Aluru, J.S.; Rawla, P.; Padala, S.A.; Barsouk, A. Epidemiology of Bladder Cancer. Med. Sci. 2020, 8, 15. [Google Scholar] [CrossRef] [Green Version]
- Ferlay, J.; Soerjomataram, I.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.M.; Forman, D.; Bray, F. Cancer incidence and mortality worldwide: Sources, methods and major patterns in GLOBOCAN 2012. Int. J. Cancer 2015, 136, E359–E386. [Google Scholar] [CrossRef] [PubMed]
- Burger, M.; Catto, J.; Dalbagni, G.; Grossman, H.B.; Herr, H.; Karakiewicz, P.; Kassouf, W.; Kiemeney, L.; La Vecchia, C.; Shariat, S.; et al. Epidemiology and Risk Factors of Urothelial Bladder Cancer. Eur. Urol. 2012, 63, 234–241. [Google Scholar] [CrossRef] [PubMed]
- Von Der Maase, H.; Hansen, S.W.; Roberts, J.T.; Dogliotti, L.; Oliver, T.; Moore, M.J.; Bodrogi, I.; Albers, P.; Knuth, A.; Lippert, C.M.; et al. Gemcitabine and Cisplatin Versus Methotrexate, Vinblastine, Doxorubicin, and Cisplatin in Advanced or Metastatic Bladder Cancer: Results of a Large, Randomized, Multinational, Multicenter, Phase III Study. J. Clin. Oncol. 2000, 18, 3068–3077. [Google Scholar] [CrossRef]
- Powles, T.; Park, S.H.; Voog, E.; Caserta, C.; Valderrama, B.P.; Gurney, H.; Kalofonos, H.; Radulović, S.; Demey, W.; Ullén, A.; et al. Avelumab Maintenance Therapy for Advanced or Metastatic Urothelial Carcinoma. N. Engl. J. Med. 2020, 383, 1218–1230. [Google Scholar] [CrossRef]
- Flaig, T.W.; Spiess, P.E.; Agarwal, N.; Bangs, R.; Boorjian, S.A.; Buyyounouski, M.K.; Chang, S.; Downs, T.M.; Efstathiou, J.A.; Friedlander, T.; et al. Bladder Cancer, Version 3.2020, NCCN Clinical Practice Guidelines in Oncology. J. Natl. Compr. Cancer Netw. 2020, 18, 329–354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheetham, P.J.; Petrylak, D.P. New Agents for the Treatment of Advanced Bladder Cancer. Oncology 2016, 30, 571. [Google Scholar]
- Bellmunt, J.; De Wit, R.; Vaughn, D.J.; Fradet, Y.; Lee, J.-L.; Fong, L.; Vogelzang, N.J.; Climent, M.A.; Petrylak, D.P.; Choueiri, T.K.; et al. Pembrolizumab as Second-Line Therapy for Advanced Urothelial Carcinoma. N. Engl. J. Med. 2017, 376, 1015–1026. [Google Scholar] [CrossRef] [Green Version]
- Rosenberg, J.E.; Hoffman-Censits, J.; Powles, T.; van der Heijden, M.S.; Balar, A.V.; Necchi, A.; Dawson, N.; O’Donnell, P.H.; Balmanoukian, A.; Loriot, Y.; et al. Atezolizumab in patients with locally advanced and metastatic urothelial carcinoma who have progressed following treatment with platinum-based chemotherapy: A single-arm, multicentre, phase 2 trial. Lancet 2016, 387, 1909–1920. [Google Scholar] [CrossRef] [Green Version]
- Voluntary Withdrawal Imfinzi US Bladder Indication—AstraZeneca. Available online: https://www.astrazeneca.com/media-centre/press-releases/2021/voluntary-withdrawal-imfinzi-us-bladder-indication.html (accessed on 7 May 2021).
- Roche Provides Update on Tecentriq US Indication in Prior-Platinum Treated Metastatic Bladder Cancer. Available online: https://www.roche.com/media/releases/med-cor-2021-03-08.htm (accessed on 7 May 2021).
- Lopez-Beltran, A.; Cimadamore, A.; Blanca, A.; Massari, F.; Vau, N.; Scarpelli, M.; Cheng, L.; Montironi, R. Immune Checkpoint Inhibitors for the Treatment of Bladder Cancer. Cancers 2021, 13, 131. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Hu-Lieskovan, S.; Wargo, J.A.; Ribas, A. Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell 2017, 168, 707–723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spranger, S.; Gajewski, T.F. Impact of oncogenic pathways on evasion of antitumour immune responses. Nat. Rev. Cancer 2018, 18, 139–147. [Google Scholar] [CrossRef] [PubMed]
- Chehrazi-Raffle, A.; Dorff, T.; Pal, S.; Lyou, Y. Wnt/β-Catenin Signaling and Immunotherapy Resistance: Lessons for the Treatment of Urothelial Carcinoma. Cancers 2021, 13, 889. [Google Scholar] [CrossRef] [PubMed]
- Mariathasan, S.; Turley, S.J.; Nickles, D.; Castiglioni, A.; Yuen, K.; Wang, Y.; Kadel, E.E., III; Koeppen, H.; Astarita, J.L.; Cubas, R.; et al. TGFβ attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature 2018, 554, 544–548. [Google Scholar] [CrossRef] [PubMed]
- Hahn, S.A.; Schutte, M.; Hoque, A.T.M.S.; Moskaluk, C.A.; da Costa, L.T.; Rozenblum, E.; Weinstein, C.L.; Fischer, A.; Yeo, C.J.; Hruban, R.H.; et al. DPC4, A Candidate Tumor Suppressor Gene at Human Chromosome 18q21.1. Science 1996, 271, 350–353. [Google Scholar] [CrossRef]
- Yan, Z.; Deng, X.; Friedman, E. Oncogenic Ki-ras Confers a More Aggressive Colon Cancer Phenotype through Modification of Transforming Growth Factor-β Receptor III. J. Biol. Chem. 2001, 276, 1555–1563. [Google Scholar] [CrossRef] [Green Version]
- Desruisseau, S.; Ghazarossian-Ragni, E.; Chinot, O.; Martin, P.M. Divergent effect of TGFbeta1 on growth and proteolytic modulation of human prostatic-cancer cell lines. Int. J. Cancer 1996, 66, 796–801. [Google Scholar] [CrossRef]
- Katakura, Y.; Nakata, E.; Miura, T.; Shirahata, S. Transforming Growth Factor β Triggers Two Independent-Senescence Programs in Cancer Cells. Biochem. Biophys. Res. Commun. 1999, 255, 110–115. [Google Scholar] [CrossRef]
- Perry, R.; Kang, Y.; Greaves, B. Relationship between tamoxifen-induced transforming growth factor beta 1 expression, cytostasis and apoptosis in human breast cancer cells. Br. J. Cancer 1995, 72, 1441–1446. [Google Scholar] [CrossRef] [Green Version]
- Ananth, S.; Knebelmann, B.; Grüning, W.; Dhanabal, M.; Walz, G.; E Stillman, I.; Sukhatme, V.P. Transforming growth factor beta1 is a target for the von Hippel-Lindau tumor suppressor and a critical growth factor for clear cell renal carcinoma. Cancer Res. 1999, 59, 226–231. [Google Scholar]
- Torre-Amione, G.; Beauchamp, R.D.; Koeppen, H.; Park, B.H.; Schreiber, H.; Moses, H.L.; Rowley, D.A. A highly immunogenic tumor transfected with a murine transforming growth factor type beta 1 cDNA escapes immune surveillance. Proc. Natl. Acad. Sci. USA 1990, 87, 1486–1490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- David, C.J.; Massagué, J. Contextual determinants of TGFβ action in development, immunity and cancer. Nat. Rev. Mol. Cell Biol. 2018, 19, 419–435. [Google Scholar] [CrossRef] [PubMed]
- Mullen, A.C.; Wrana, J.L. TGF-β Family Signaling in Embryonic and Somatic Stem-Cell Renewal and Differentiation. Cold Spring Harb. Perspect. Biol. 2017, 9, a022186. [Google Scholar] [CrossRef]
- Derynck, R.; Turley, S.J.; Akhurst, R.J. TGFβ biology in cancer progression and immunotherapy. Nat. Rev. Clin. Oncol. 2020, 18, 9–34. [Google Scholar] [CrossRef]
- Robertson, I.B.; Rifkin, D.B. Regulation of the Bioavailability of TGF-β and TGF-β-Related Proteins. Cold Spring Harb. Perspect. Biol. 2016, 8, a021907. [Google Scholar] [CrossRef]
- Henderson, N.C.; Sheppard, D. Integrin-mediated regulation of TGFβ in fibrosis. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2012, 1832, 891–896. [Google Scholar] [CrossRef] [Green Version]
- Brown, N.F.; Marshall, J.F. Integrin-Mediated TGFβ Activation Modulates the Tumour Microenvironment. Cancers 2019, 11, 1221. [Google Scholar] [CrossRef] [Green Version]
- Takasaka, N.; Seed, R.I.; Cormier, A.; Bondesson, A.J.; Lou, J.; Elattma, A.; Ito, S.; Yanagisawa, H.; Hashimoto, M.; Ma, R.; et al. Integrin αvβ8–expressing tumor cells evade host immunity by regulating TGF-β activation in immune cells. JCI Insight 2018, 3, e122591. [Google Scholar] [CrossRef] [Green Version]
- Derynck, R.; Budi, E.H. Specificity, versatility, and control of TGF-β family signaling. Sci. Signal. 2019, 12, eaav5183. [Google Scholar] [CrossRef] [Green Version]
- Hata, A.; Chen, Y.-G. TGF-β Signaling from Receptors to Smads. Cold Spring Harb. Perspect. Biol. 2016, 8, a022061. [Google Scholar] [CrossRef]
- Hill, C.S. Transcriptional Control by the SMADs. Cold Spring Harb. Perspect. Biol. 2016, 8, a022079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, L.; Zhang, Y.; Zhang, L.; Huang, F.; Li, J.; Wang, S. MicroRNAs, TGF-β signaling, and the inflammatory microenvironment in cancer. Tumor Biol. 2015, 37, 115–125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blahna, M.T.; Hata, A. Regulation of miRNA biogenesis as an integrated component of growth factor signaling. Curr. Opin. Cell Biol. 2013, 25, 233–240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanchez-Vega, F.; Mina, M.; Armenia, J.; Chatila, W.K.; Luna, A.; La, K.C.; Dimitriadoy, S.; Liu, D.L.; Kantheti, H.S.; Saghafinia, S.; et al. Oncogenic Signaling Pathways in The Cancer Genome Atlas. Cell 2018, 173, 321–337. [Google Scholar] [CrossRef] [Green Version]
- Gao, R.; Zhang, N.; Yang, J.; Zhu, Y.; Zhang, Z.; Wang, J.; Xu, X.; Li, Z.; Liu, X.; Li, Z.; et al. Long non-coding RNA ZEB1-AS1 regulates miR-200b/FSCN1 signaling and enhances migration and invasion induced by TGF-β1 in bladder cancer cells. J. Exp. Clin. Cancer Res. 2019, 38, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Cao, J.; Zhao, X. miR-221 facilitates the TGFbeta1-induced epithelial-mesenchymal transition in human bladder cancer cells by targeting STMN1. BMC Urol. 2015, 15, 36. [Google Scholar] [CrossRef] [Green Version]
- Shaker, O.; Hammam, O.; Wishahi, M.; Roshdi, M. TGF-B1 pathway as biological marker of bladder carcinoma schistosomal and non-schistosomal. Urol. Oncol. Semin. Orig. Investig. 2013, 31, 372–378. [Google Scholar] [CrossRef]
- Monteran, L.; Erez, N. The Dark Side of Fibroblasts: Cancer-Associated Fibroblasts as Mediators of Immunosuppression in the Tumor Microenvironment. Front. Immunol. 2019, 10, 1835. [Google Scholar] [CrossRef] [Green Version]
- Sahai, E.; Astsaturov, I.; Cukierman, E.; DeNardo, D.G.; Egeblad, M.; Evans, R.M.; Fearon, D.; Greten, F.R.; Hingorani, S.R.; Hunter, T.; et al. A framework for advancing our understanding of cancer-associated fibroblasts. Nat. Rev. Cancer 2020, 20, 174–186. [Google Scholar] [CrossRef] [Green Version]
- Galbo, P.M.; Zang, X.; Zheng, D. Molecular Features of Cancer-associated Fibroblast Subtypes and their Implication on Cancer Pathogenesis, Prognosis, and Immunotherapy Resistance. Clin. Cancer Res. 2021, 27, 2636–2647. [Google Scholar] [CrossRef] [PubMed]
- LeBleu, V.S.; Kalluri, R. A peek into cancer-associated fibroblasts: Origins, functions and translational impact. Dis. Model. Mech. 2018, 11, dmm029447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calon, A.; Tauriello, D.; Batlle, E. TGF-beta in CAF-mediated tumor growth and metastasis. Semin. Cancer Biol. 2014, 25, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Yin, M.; Soikkeli, J.; Jahkola, T.; Virolainen, S.; Saksela, O.; Hölttä, E. TGF-β Signaling, Activated Stromal Fibroblasts, and Cysteine Cathepsins B and L Drive the Invasive Growth of Human Melanoma Cells. Am. J. Pathol. 2012, 181, 2202–2216. [Google Scholar] [CrossRef]
- Acharya, P.S.; Majumdar, S.; Jacob, M.; Hayden, J.; Mrass, P.; Weninger, W.; Assoian, R.K.; Puré, E. Fibroblast migration is mediated by CD44-dependent TGFβ activation. J. Cell Sci. 2008, 121, 1393–1402. [Google Scholar] [CrossRef] [Green Version]
- E Postlethwaite, A.; Keski-Oja, J.; Moses, H.L.; Kang, A.H. Stimulation of the chemotactic migration of human fibroblasts by transforming growth factor beta. J. Exp. Med. 1987, 165, 251–256. [Google Scholar] [CrossRef] [Green Version]
- Huang, H.; Zhang, Y.; Gallegos, V.; Sorrelle, N.; Zaid, M.M.; Toombs, J.; Du, W.; Wright, S.; Hagopian, M.; Wang, Z.; et al. Targeting TGF βR2-mutant tumors exposes vulnerabilities to stromal TGF β blockade in pancreatic cancer. EMBO Mol. Med. 2019, 11, e10515. [Google Scholar] [CrossRef]
- Giussani, M.; Triulzi, T.; Sozzi, G.; Tagliabue, E. Tumor Extracellular Matrix Remodeling: New Perspectives as a Circulating Tool in the Diagnosis and Prognosis of Solid Tumors. Cells 2019, 8, 81. [Google Scholar] [CrossRef] [Green Version]
- Ribatti, D.; Tamma, R.; Annese, T. Epithelial-Mesenchymal Transition in Cancer: A Historical Overview. Transl. Oncol. 2020, 13, 100773. [Google Scholar] [CrossRef]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial–mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef] [Green Version]
- Derynck, R.; Muthusamy, B.P.; Saeteurn, K.Y. Signaling pathway cooperation in TGF-β-induced epithelial–mesenchymal transition. Curr. Opin. Cell Biol. 2014, 31, 56–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zou, J.; Huang, R.; Li, H.; Wang, B.; Chen, Y.; Chen, S.; Ou, K.; Wang, X. Secreted TGF-beta-induced protein promotes aggressive progression in bladder cancer cells. Cancer Manag. Res. 2019, 11, 6995–7006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Z.; He, S.; Zhan, Y.; He, A.; Fang, D.; Gong, Y.; Li, X.; Zhou, L. TGF-β-induced transgelin promotes bladder cancer metastasis by regulating epithelial-mesenchymal transition and invadopodia formation. EBioMedicine 2019, 47, 208–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, Y.-L.; Wang, S.-S.; Jiang, J.; Liang, X.-H. Links between cancer stem cells and epithelial– mesenchymal transition. OncoTargets Ther. 2015, 8, 2973–2980. [Google Scholar] [CrossRef] [Green Version]
- Clarke, M.F. Clinical and Therapeutic Implications of Cancer Stem Cells. N. Engl. J. Med. 2019, 380, 2237–2245. [Google Scholar] [CrossRef]
- Radvanyi, L. Immunotherapy Exposes Cancer Stem Cell Resistance and a New Synthetic Lethality. Mol. Ther. 2013, 21, 1472–1474. [Google Scholar] [CrossRef] [Green Version]
- Soundararajan, R.; Fradette, J.; Konen, J.M.; Moulder, S.; Zhang, X.; Gibbons, D.L.; Varadarajan, N.; Wistuba, I.I.; Tripathy, D.; Bernatchez, C.; et al. Targeting the Interplay between Epithelial-to-Mesenchymal-Transition and the Immune System for Effective Immunotherapy. Cancers 2019, 11, 714. [Google Scholar] [CrossRef] [Green Version]
- Terry, S.; Savagner, P.; Ortiz-Cuaran, S.; Mahjoubi, L.; Saintigny, P.; Thiery, J.-P.; Chouaib, S. New insights into the role of EMT in tumor immune escape. Mol. Oncol. 2017, 11, 824–846. [Google Scholar] [CrossRef] [Green Version]
- Liang, Y.; Zhu, F.; Zhang, H.; Chen, D.; Zhang, X.; Gao, Q.; Li, Y. Conditional ablation of TGF-β signaling inhibits tumor progression and invasion in an induced mouse bladder cancer model. Sci. Rep. 2016, 6, 29479. [Google Scholar] [CrossRef]
- Singh, A.; Settleman, J. EMT, cancer stem cells and drug resistance: An emerging axis of evil in the war on cancer. Oncogene 2010, 29, 4741–4751. [Google Scholar] [CrossRef] [Green Version]
- Van Staalduinen, J.; Baker, D.; Dijke, P.T.; van Dam, H. Epithelial–mesenchymal-transition-inducing transcription factors: New targets for tackling chemoresistance in cancer? Oncogene 2018, 37, 6195–6211. [Google Scholar] [CrossRef] [PubMed]
- Fischer, K.R.; Durrans, A.; Lee, S.; Sheng, J.; Li, F.; Wong, S.T.C.; Choi, H.; El Rayes, T.; Ryu, S.; Troeger, J.; et al. Epithelial-to-mesenchymal transition is not required for lung metastasis but contributes to chemoresistance. Nature 2015, 527, 472–476. [Google Scholar] [CrossRef] [PubMed]
- Dongre, A.; Weinberg, R.A. New insights into the mechanisms of epithelial–mesenchymal transition and implications for cancer. Nat. Rev. Mol. Cell Biol. 2018, 20, 69–84. [Google Scholar] [CrossRef]
- Jiang, Y.; Zhan, H. Communication between EMT and PD-L1 signaling: New insights into tumor immune evasion. Cancer Lett. 2019, 468, 72–81. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Saci, A.; Szabo, P.M.; Chasalow, S.D.; Castillo-Martin, M.; Domingo-Domenech, J.; Siefker-Radtke, A.; Sharma, P.; Sfakianos, J.P.; Gong, Y.; et al. EMT- and stroma-related gene expression and resistance to PD-1 blockade in urothelial cancer. Nat. Commun. 2018, 9, 3503. [Google Scholar] [CrossRef] [PubMed]
- de Olza, M.O.; Rodrigo, B.N.; Zimmermann, S.; Coukos, G. Turning up the heat on non-immunoreactive tumours: Opportunities for clinical development. Lancet Oncol. 2020, 21, e419–e430. [Google Scholar] [CrossRef]
- Le Trinh, T.; Kandell, W.M.; Donatelli, S.S.; Tu, N.; Tejera, M.M.; Gilvary, D.L.; Eksioglu, E.A.; Burnette, A.; Adams, W.A.; Liu, J.; et al. Immune evasion by TGFβ-induced miR-183 repression of MICA/B expression in human lung tumor cells. OncoImmunology 2019, 8, e1557372. [Google Scholar] [CrossRef] [Green Version]
- Nam, J.-S.; Terabe, M.; Mamura, M.; Kang, M.-J.; Chae, H.; Stuelten, C.; A Kohn, E.; Tang, B.; Sabzevari, H.; Anver, M.R.; et al. An Anti–Transforming Growth Factor β Antibody Suppresses Metastasis via Cooperative Effects on Multiple Cell Compartments. Cancer Res. 2008, 68, 3835–3843. [Google Scholar] [CrossRef] [Green Version]
- Sadallah, S.; Schmied, L.; Eken, C.; Charoudeh, H.N.; Amicarella, F.; Schifferli, J.A. Platelet-Derived Ectosomes Reduce NK Cell Function. J. Immunol. 2016, 197, 1663–1671. [Google Scholar] [CrossRef] [Green Version]
- Lazarova, M.; Steinle, A. Impairment of NKG2D-Mediated Tumor Immunity by TGF-β. Front. Immunol. 2019, 10, 2689. [Google Scholar] [CrossRef] [Green Version]
- Laouar, Y.; Sutterwala, F.S.; Gorelik, L.; A Flavell, R. Transforming growth factor-β controls T helper type 1 cell development through regulation of natural killer cell interferon-γ. Nat. Immunol. 2005, 6, 600–607. [Google Scholar] [CrossRef] [PubMed]
- Naganuma, H.; Sasaki, A.; Satoh, E.; Nagasaka, M.; Nakano, S.; Isoe, S.; Tasaka, K.; Nukui, H. Transforming Growth Factor-β Inhibits Interferon-γ Secretion by Lymph okine-activated Killer Cells Stimulated with Tumor Cells. Neurol. Medico-Chirurgica 1996, 36, 789–795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takeuchi, M.; Kosiewicz, M.M.; Alard, P.; Streilein, J.W. On the mechanisms by which transforming growth factor-β2 alters antigen-presenting abilities of macrophages on T cell activation. Eur. J. Immunol. 1997, 27, 1648–1656. [Google Scholar] [CrossRef] [PubMed]
- Demidem, A.; Taylor, J.R.; Grammer, S.F.; Streilein, J.W. Comparison of Effects of Transforming Growth Factor-Beta and Cyclosporin A on Antigen-Presenting Cells of Blood and Epidermis. J. Investig. Dermatol. 1991, 96, 401–407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seeger, P.; Musso, T.; Sozzani, S. The TGF-β superfamily in dendritic cell biology. Cytokine Growth Factor Rev. 2015, 26, 647–657. [Google Scholar] [CrossRef]
- Lee, Y.S.; Park, J.S.; Kim, J.H.; Jung, S.M.; Lee, J.Y.; Kim, S.-J.; Park, S.H. Smad6-specific recruitment of Smurf E3 ligases mediates TGF-β1-induced degradation of MyD88 in TLR4 signalling. Nat. Commun. 2011, 2, 460. [Google Scholar] [CrossRef] [Green Version]
- Batlle, E.; Massagué, J. Transforming Growth Factor-β Signaling in Immunity and Cancer. Immunity 2019, 50, 924–940. [Google Scholar] [CrossRef]
- Siegert, A.; Denkert, C.; LeClere, A.; Hauptmann, S. Suppression of the reactive oxygen intermediates production of human macrophages by colorectal adenocarcinoma cell lines. Immunology 1999, 98, 551–556. [Google Scholar] [CrossRef]
- Tan, H.-Y.; Wang, N.; Li, S.; Hong, M.; Wang, X.; Feng, Y. The Reactive Oxygen Species in Macrophage Polarization: Reflecting Its Dual Role in Progression and Treatment of Human Diseases. Oxidative Med. Cell. Longev. 2016, 2016, 2795090. [Google Scholar] [CrossRef] [Green Version]
- Kobie, J.J.; Wu, R.S.; A Kurt, R.; Lou, S.; Adelman, M.K.; Whitesell, L.J.; Ramanathapuram, L.V.; Arteaga, C.L.; Akporiaye, E.T. Transforming growth factor beta inhibits the antigen-presenting functions and antitumor activity of dendritic cell vaccines. Cancer Res. 2003, 63, 1860–1864. [Google Scholar]
- Novitskiy, S.V.; Pickup, M.W.; Chytil, A.; Polosukhina, D.; Owens, P.; Moses, H.L. Deletion of TGF- signaling in myeloid cells enhances their anti-tumorigenic properties. J. Leukoc. Biol. 2012, 92, 641–651. [Google Scholar] [CrossRef] [Green Version]
- Das, L.; Levine, A.D. TGF-β Inhibits IL-2 Production and Promotes Cell Cycle Arrest in TCR-Activated Effector/Memory T Cells in the Presence of Sustained TCR Signal Transduction. J. Immunol. 2008, 180, 1490–1498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, D.A.; Massagué, J. TGF-β directly targets cytotoxic T cell functions during tumor evasion of immune surveillance. Cancer Cell 2005, 8, 369–380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mangan, P.R.; Harrington, L.E.; O’Quinn, D.B.; Helms, W.S.; Bullard, D.C.; Elson, C.O.; Hatton, R.D.; Wahl, S.M.; Schoeb, T.R.; Weaver, C.T. Transforming growth factor-β induces development of the TH17 lineage. Nature 2006, 441, 231–234. [Google Scholar] [CrossRef] [PubMed]
- Tay, R.E.; Richardson, E.K.; Toh, H.C. Revisiting the role of CD4+ T cells in cancer immunotherapy—new insights into old paradigms. Cancer Gene Ther. 2020, 28, 5–17. [Google Scholar] [CrossRef] [PubMed]
- Bai, X.; Yi, M.; Jiao, Y.; Chu, Q.; Wu, K. Blocking TGF-β Signaling To Enhance The Efficacy Of Immune Checkpoint Inhibitor. OncoTargets Ther. 2019, 12, 9527–9538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Atezolizumab as First-Line Treatment in Cisplatin-Ineligible Patients with Locally Advanced and Metastatic Urothelial Car-cinoma: A Single-arm, Multicentre, Phase 2 Trial—The Lancet. Available online: https://www.thelancet.com/journals/lancet/article/PIIS0140-6736(16)32455-2/fulltext (accessed on 7 May 2021).
- Gorczynski, R.M.; Chen, Z.; Erin, N.; Khatri, I.; Podnos, A. Comparison of Immunity in Mice Cured of Primary/Metastatic Growth of EMT6 or 4THM Breast Cancer by Chemotherapy or Immunotherapy. PLOS ONE 2014, 9, e113597. [Google Scholar] [CrossRef] [Green Version]
- Morris, J.C.; Tan, A.R.; Olencki, T.E.; Shapiro, G.I.; Dezube, B.J.; Reiss, M.; Hsu, F.J.; Berzofsky, J.A.; Lawrence, D.P. Phase I Study of GC1008 (Fresolimumab): A Human Anti-Transforming Growth Factor-Beta (TGFβ) Monoclonal Antibody in Patients with Advanced Malignant Melanoma or Renal Cell Carcinoma. PLOS ONE 2014, 9, e90353. [Google Scholar] [CrossRef]
- Greco, R.; Qu, H.; Qu, H.; Theilhaber, J.; Shapiro, G.; Gregory, R.; Winter, C.; Malkova, N.; Sun, F.; Jaworski, J.; et al. Pan-TGFβ inhibition by SAR439459 relieves immunosuppression and improves antitumor efficacy of PD-1 blockade. OncoImmunology 2020, 9, 1811605. [Google Scholar] [CrossRef]
- Laverty, H.; Wakefield, L.; Occleston, N.; O’Kane, S.; Ferguson, M. TGF-β3 and cancer: A review. Cytokine Growth Factor Rev. 2009, 20, 305–317. [Google Scholar] [CrossRef]
- Yingling, J.M.; McMillen, W.T.; Yan, L.; Huang, H.; Sawyer, J.S.; Graff, J.; Clawson, D.K.; Britt, K.S.; Anderson, B.D.; Beight, D.W.; et al. Preclinical assessment of galunisertib (LY2157299 monohydrate), a first-in-class transforming growth factor-β receptor type I inhibitor. Oncotarget 2017, 9, 6659–6677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, S.Y.; Hwang, S.; Clarke, J.M.; Bauer, T.M.; Keedy, V.L.; Lee, H.; Park, N.; Kim, S.-J.; Lee, J.I. Pharmacokinetic characteristics of vactosertib, a new activin receptor-like kinase 5 inhibitor, in patients with advanced solid tumors in a first-in-human phase 1 study. Investig. New Drugs 2019, 38, 812–820. [Google Scholar] [CrossRef] [PubMed]
- Brandes, A.A.; Carpentier, A.F.; Kesari, S.; Sepulveda-Sanchez, J.M.; Wheeler, H.R.; Chinot, O.; Cher, L.; Steinbach, J.P.; Capper, D.; Specenier, P.; et al. A Phase II randomized study of galunisertib monotherapy or galunisertib plus lomustine compared with lomustine monotherapy in patients with recurrent glioblastoma. Neuro-Oncology 2016, 18, 1146–1156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tijeras-Raballand, A.; Hobeika, C.; Martinet, M.; Astorgues-Xerri, L.; Paven, E.; Le Bitoux, M.-A.; Maillard, A.; Eveno, C.; Pocard, M.; Bonnin, P.; et al. Abstract 2944: TGF-β inhibitor galunisertib combined with antiangio-genic therapies showed antitumor effects in vitro and in vivo in hepatocellular carcinoma (HCC). Cancer Res. 2018, 78 (Suppl. 13), 2944. [Google Scholar]
- Rodon, J.; Carducci, M.A.; Sepulveda-Sánchez, J.M.; Azaro, A.; Calvo, E.; Seoane, J.; Brana, I.; Sicart, E.; Gueorguieva, I.; Cleverly, A.L.; et al. First-in-Human Dose Study of the Novel Transforming Growth Factor-β Receptor I Kinase Inhibitor LY2157299 Monohydrate in Patients with Advanced Cancer and Glioma. Clin. Cancer Res. 2014, 21, 553–560. [Google Scholar] [CrossRef] [Green Version]
- Ikeda, M.; Morimoto, M.; Tajimi, M.; Inoue, K.; Benhadji, K.A.; Lahn, M.M.F.; Sakai, D. A phase 1b study of transforming growth factor-beta receptor I inhibitor galunisertib in combination with sorafenib in Japanese patients with unresectable hepatocellular carcinoma. Investig. New Drugs 2018, 37, 118–126. [Google Scholar] [CrossRef] [Green Version]
- Ravi, R.; Noonan, K.A.; Pham, V.; Bedi, R.; Zhavoronkov, A.; Ozerov, I.V.; Makarev, E.; Artemov, A.V.; Wysocki, P.; Mehra, R.; et al. Bifunctional immune checkpoint-targeted antibody-ligand traps that simultaneously disable TGFβ enhance the efficacy of cancer immunotherapy. Nat. Commun. 2018, 9, 741. [Google Scholar] [CrossRef]
- Paz-Ares, L.; Kim, T.M.; Vicente, D.; Felip, E.; Lee, D.H.; Lee, K.H.; Lin, C.-C.; Flor, M.J.; Di Nicola, M.; Alvarez, R.M.; et al. Bintrafusp Alfa, a Bifunctional Fusion Protein Targeting TGF-β and PD-L1, in Second-Line Treatment of Patients With NSCLC: Results From an Expansion Cohort of a Phase 1 Trial. J. Thorac. Oncol. 2020, 15, 1210–1222. [Google Scholar] [CrossRef]
- Dias, N.; A Stein, C. Antisense oligonucleotides: Basic concepts and mechanisms. Mol. Cancer Ther. 2002, 1, 347–355. [Google Scholar]
- Jaschinski, F.; Rothhammer, T.; Jachimczak, P.; Seitz, C.; Schneider, A.; Schlingensiepen, K.-H. The antisense oligonucleotide trabedersen (AP 12009) for the targeted inhibition of TGF-β2. Curr. Pharm. Biotechnol. 2011, 12, 2203–2213. [Google Scholar] [CrossRef]
- Kim, B.-G.; Malek, E.; Choi, S.H.; Ignatz-Hoover, J.J.; Driscoll, J.J. Novel therapies emerging in oncology to target the TGF-β pathway. J. Hematol. Oncol. 2021, 14, 55. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Benjamin, D.J.; Lyou, Y. Advances in Immunotherapy and the TGF-β Resistance Pathway in Metastatic Bladder Cancer. Cancers 2021, 13, 5724. https://doi.org/10.3390/cancers13225724
Benjamin DJ, Lyou Y. Advances in Immunotherapy and the TGF-β Resistance Pathway in Metastatic Bladder Cancer. Cancers. 2021; 13(22):5724. https://doi.org/10.3390/cancers13225724
Chicago/Turabian StyleBenjamin, David J., and Yung Lyou. 2021. "Advances in Immunotherapy and the TGF-β Resistance Pathway in Metastatic Bladder Cancer" Cancers 13, no. 22: 5724. https://doi.org/10.3390/cancers13225724