
Remodeling the ECM: Implications for Metastasis and Tumor Dormancy


{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. ECM and Cancer Progression
3. ECM and Stem Cells Quiescence
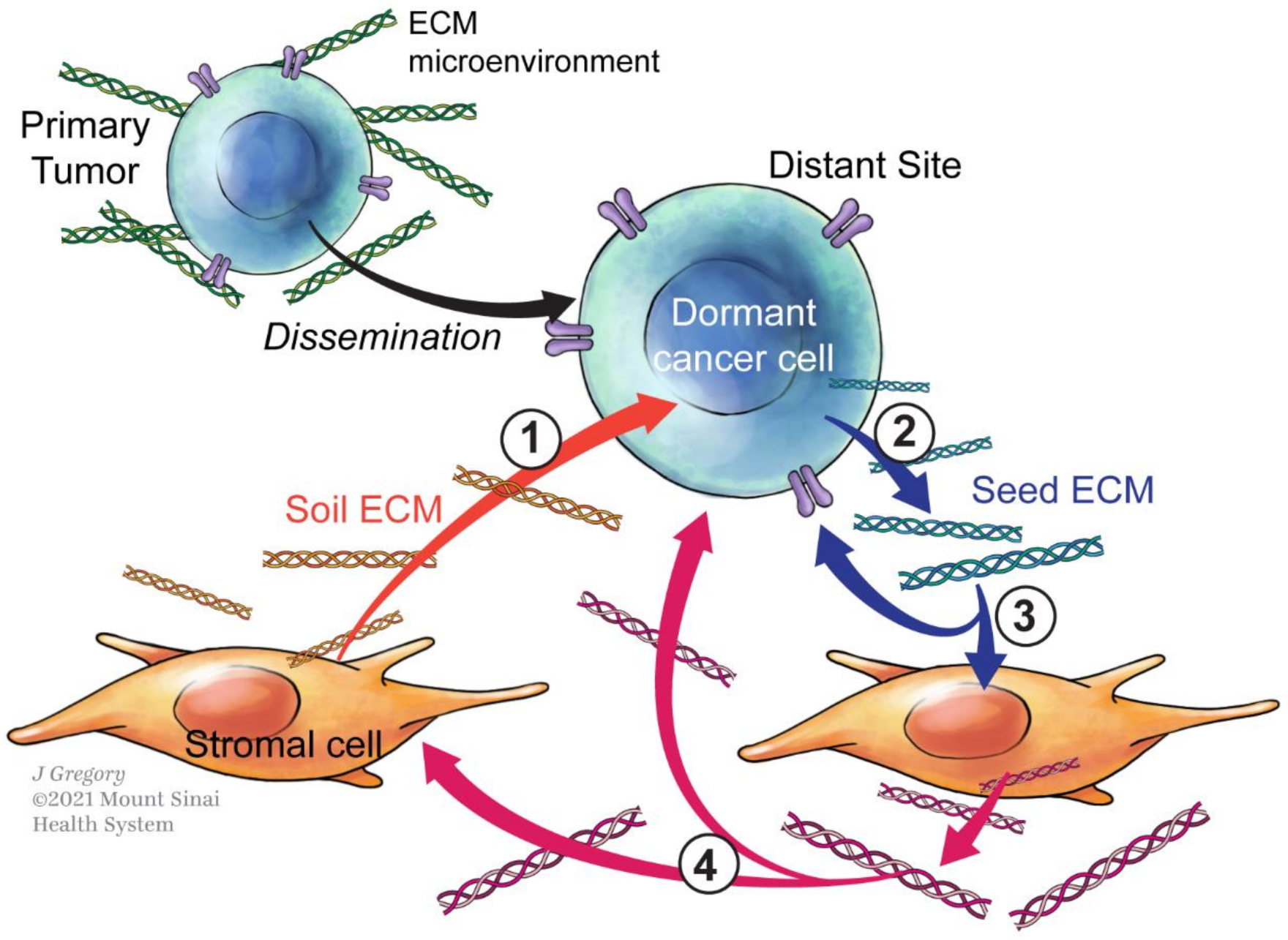
4. Role of ECM on Tumor Dormancy: What Do We Know?
5. Outstanding Questions and Discussion
5.1. What Receptors Are Critical to Sustain Dormancy?
5.2. What Is the Composition of a Dormancy ECM Niche?
5.3. How Does the Matrisome Change with Cancer Treatments?
5.4. How Does Aging Affect the Matrisome and the Scape from Dormancy?
5.5. Will Remodeling the ECM Microenvironment Prevent Metastasis?
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Phan, T.G.; Croucher, P.I. The dormant cancer cell life cycle. Nat. Rev. Cancer 2020, 20, 398–411. [Google Scholar] [CrossRef] [PubMed]
- Sosa, M.S.; Bragado, P.; Aguirre-Ghiso, J.A. Mechanisms of disseminated cancer cell dormancy: An awakening field. Nat. Rev. Cancer 2014, 14, 611–622. [Google Scholar] [CrossRef] [PubMed]
- Sosa, M.S.; Parikh, F.; Maia, A.G.; Estrada, Y.; Bosch, A.; Bragado, P.; Ekpin, E.; George, A.; Zheng, Y.; Lam, H.-M.; et al. NR2F1 controls tumour cell dormancy via SOX9- and RARβ-driven quiescence programmes. Nat. Commun. 2015, 6, 6170. [Google Scholar] [CrossRef] [Green Version]
- Linde, N.; Fluegen, G.; Aguirre-Ghiso, J.A. The Relationship Between Dormant Cancer Cells and Their Microenvironment. Adv. Cancer Res. 2016, 132, 45–71. [Google Scholar] [CrossRef] [Green Version]
- Risson, E.; Nobre, A.R.; Maguer-Satta, V.; Aguirre-Ghiso, J.A. The current paradigm and challenges ahead for the dormancy of disseminated tumor cells. Nat. Cancer 2020, 1, 672–680. [Google Scholar] [CrossRef] [PubMed]
- Aguirre-Ghiso, J.A. Models, mechanisms and clinical evidence for cancer dormancy. Nat. Rev. Cancer 2007, 7, 834–846. [Google Scholar] [CrossRef] [Green Version]
- Wilson, A.; Laurenti, E.; Trumpp, A. Balancing dormant and self-renewing hematopoietic stem cells. Curr. Opin. Genet. Dev. 2009, 19, 461–468. [Google Scholar] [CrossRef]
- Baccelli, I.; Trumpp, A. The evolving concept of cancer and metastasis stem cells. J. Cell Biol. 2012, 198, 281–293. [Google Scholar] [CrossRef]
- Ranganathan, A.C.; Zhang, L.; Adam, A.P.; Aguirre-Ghiso, J.A. Functional coupling of p38-induced up-regulation of BiP and activation of RNA-dependent protein kinase-like endoplasmic reticulum kinase to drug resistance of dormant carcinoma cells. Cancer Res. 2006, 66, 1702–1711. [Google Scholar] [CrossRef] [Green Version]
- Price, T.T.; Burness, M.L.; Sivan, A.; Warner, M.J.; Cheng, R.; Lee, C.H.; Olivere, L.; Comatas, K.; Magnani, J.; Lyerly, H.K.; et al. Dormant breast cancer micrometastases reside in specific bone marrow niches that regulate their transit to and from bone. Sci. Transl. Med. 2016, 8, 340ra73. [Google Scholar] [CrossRef]
- Goddard, E.T.; Bozic, I.; Riddell, S.R.; Ghajar, C.M. Dormant tumour cells, their niches and the influence of immunity. Nat. Cell Biol. 2018, 20, 1240–1249. [Google Scholar] [CrossRef]
- Byrne, N.M.; Summers, M.A.; McDonald, M.M. Tumor Cell Dormancy and Reactivation in Bone: Skeletal Biology and Therapeutic Opportunities. JBMR Plus 2019, 3, e10125. [Google Scholar] [CrossRef]
- Ghajar, C.M. Metastasis prevention by targeting the dormant niche. Nat. Rev. Cancer 2015, 15, 238–247. [Google Scholar] [CrossRef]
- Barkan, D.; El Touny, L.H.; Michalowski, A.M.; Smith, J.A.; Chu, I.; Davis, A.S.; Webster, J.D.; Hoover, S.; Simpson, R.M.; Gauldie, J.; et al. Metastatic growth from dormant cells induced by a Col-I-enriched fibrotic environment. Cancer Res. 2010, 70, 5706–5716. [Google Scholar] [CrossRef] [Green Version]
- MacKie, R.M.; Reid, R.; Junor, B. Fatal melanoma transferred in a donated kidney 16 years after melanoma surgery. N. Engl. J. Med. 2003, 348, 567–568. [Google Scholar] [CrossRef]
- Ison, M.G.; Nalesnik, M.A. An update on donor-derived disease transmission in organ transplantation. Am. J. Transplant. 2011, 11, 1123–1130. [Google Scholar] [CrossRef]
- Matser, Y.A.H.; Terpstra, M.L.; Nadalin, S.; Nossent, G.D.; de Boer, J.; van Bemmel, B.C.; van Eeden, S.; Budde, K.; Brakemeier, S.; Bemelman, F.J. Transmission of breast cancer by a single multiorgan donor to 4 transplant recipients. Am. J. Transplant. 2018, 18, 1810–1814. [Google Scholar] [CrossRef] [Green Version]
- Morris, B.A.; Burkel, B.; Ponik, S.M.; Fan, J.; Condeelis, J.S.; Aguirre-Ghiso, J.A.; Castracane, J.; Denu, J.M.; Keely, P.J. Collagen Matrix Density Drives the Metabolic Shift in Breast Cancer Cells. EBioMedicine 2016, 13, 146–156. [Google Scholar] [CrossRef] [Green Version]
- Nagao, T.; Inoue, S.; Goto, S.; Mizuta, T.; Omori, Y.; Kawano, N.; Morioka, Y. Hepatic resection for hepatocellular carcinoma: Clinical features and long-term prognosis. Ann. Surg. 1987, 205, 33–40. [Google Scholar] [CrossRef]
- Huang, J.; Li, B.-K.; Chen, G.-H.; Li, J.-Q.; Zhang, Y.-Q.; Li, G.-H.; Yuan, Y.-F. Long-term outcomes and prognostic factors of elderly patients with hepatocellular carcinoma undergoing hepatectomy. J. Gastrointest. Surg. Off. J. Soc. Surg. Aliment. Tract 2009, 13, 1627–1635. [Google Scholar] [CrossRef]
- Lamandé, S.R.; Bateman, J.F. Genetic Disorders of the Extracellular Matrix. Anat. Rec. 2020, 303, 1527–1542. [Google Scholar] [CrossRef]
- Naba, A.; Clauser, K.R.; Hoersch, S.; Liu, H.; Carr, S.A.; Hynes, R.O. The Matrisome: In Silico Definition and In Vivo Characterization by Proteomics of Normal and Tumor Extracellular Matrices. Mol. Cell. Proteom. 2012, 11, M111.014647. [Google Scholar] [CrossRef] [Green Version]
- Shao, X.; Taha, I.N.; Clauser, K.R.; Gao, Y.T.; Naba, A. MatrisomeDB: The ECM-protein knowledge database. Nucleic Acids Res. 2020, 48, D1136–D1144. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Weaver, V.M.; Petersen, O.W.; Wang, F.; Larabell, C.A.; Briand, P.; Damsky, C.; Bissell, M.J. Reversion of the malignant phenotype of human breast cells in three-dimensional culture and in vivo by integrin blocking antibodies. J. Cell Biol. 1997, 137, 231–245. [Google Scholar] [CrossRef] [Green Version]
- Conklin, M.W.; Eickhoff, J.C.; Riching, K.M.; Pehlke, C.A.; Eliceiri, K.W.; Provenzano, P.P.; Friedl, A.; Keely, P.J. Aligned collagen is a prognostic signature for survival in human breast carcinoma. Am. J. Pathol. 2011, 178, 1221–1232. [Google Scholar] [CrossRef]
- Alkmin, S.; Brodziski, R.; Simon, H.; Hinton, D.; Goldsmith, R.H.; Patankar, M.; Campagnola, P.J. Role of Collagen Fiber Morphology on Ovarian Cancer Cell Migration Using Image-Based Models of the Extracellular Matrix. Cancers 2020, 12, 1390. [Google Scholar] [CrossRef]
- Han, W.; Chen, S.; Yuan, W.; Fan, Q.; Tian, J.; Wang, X.; Chen, L.; Zhang, X.; Wei, W.; Liu, R.; et al. Oriented collagen fibers direct tumor cell intravasation. Proc. Natl. Acad. Sci. USA 2016, 113, 11208–11213. [Google Scholar] [CrossRef] [Green Version]
- Levental, K.R.; Yu, H.; Kass, L.; Lakins, J.N.; Egeblad, M.; Erler, J.T.; Fong, S.F.T.; Csiszar, K.; Giaccia, A.; Weninger, W.; et al. “Matrix Crosslinking Forces Tumor Progression by Enhancing Integrin Signaling. Cell 2009, 139, 891–906. [Google Scholar] [CrossRef] [Green Version]
- O’Rourke, J.M.; Sagar, V.M.; Shah, T.; Shetty, S. Carcinogenesis on the background of liver fibrosis: Implications for the management of hepatocellular cancer. World J. Gastroenterol. 2018, 24, 4436–4447. [Google Scholar] [CrossRef]
- Juin, A.; Billottet, C.; Moreau, V.; Destaing, O.; Albiges-Rizo, C.; Rosenbaum, J.; Genot, E.; Saltel, F. Physiological type I collagen organization induces the formation of a novel class of linear invadosomes. Mol. Biol. Cell 2012, 23, 297–309. [Google Scholar] [CrossRef]
- Zhang, G.; Miyake, M.; Lawton, A.; Goodison, S.; Rosser, C.J. Matrix metalloproteinase-10 promotes tumor progression through regulation of angiogenic and apoptotic pathways in cervical tumors. BMC Cancer 2014, 14, 310. [Google Scholar] [CrossRef] [Green Version]
- Mehner, C.; Hockla, A.; Miller, E.; Ran, S.; Radisky, D.C.; Radisky, E.S. Tumor cell-produced matrix metalloproteinase 9 (MMP-9) drives malignant progression and metastasis of basal-like triple negative breast cancer. Oncotarget 2014, 5, 2736–2749. [Google Scholar] [CrossRef] [Green Version]
- Owyong, M.; Chou, J.; van den Bijgaart, R.J.; Kong, N.; Efe, G.; Maynard, C.; Talmi-Frank, D.; Solomonov, I.; Koopman, C.; Hadler-Olsen, E.; et al. MMP9 modulates the metastatic cascade and immune landscape for breast cancer anti-metastatic therapy. Life Sci. Alliance 2019, 2, e201800226. [Google Scholar] [CrossRef] [Green Version]
- Naba, A.; Clauser, K.R.; Lamar, J.M.; Carr, S.A.; Hynes, R.O. Extracellular matrix signatures of human mammary carcinoma identify novel metastasis promoters. eLife 2014, 2014, 1–23. [Google Scholar] [CrossRef] [Green Version]
- Le Borgne-Rochet, M.; Angevin, L.; Bazellières, E.; Ordas, L.; Comunale, F.; Denisov, E.V.; Tashireva, L.A.; Perelmuter, V.M.; Bièche, I.; Vacher, S.; et al. P-cadherin-induced decorin secretion is required for collagen fiber alignment and directional collective cell migration. J. Cell Sci. 2019, 132, jcs233189. [Google Scholar] [CrossRef]
- Kimata, K.; Honma, Y.; Okayama, M.; Oguri, K.; Hozumi, M.; Suzuki, S. Increased synthesis of hyaluronic acid by mouse mammary carcinoma cell variants with high metastatic potential. Cancer Res. 1983, 43, 1347–1354. [Google Scholar]
- Zhang, L.; Underhill, C.B.; Chen, L. Hyaluronan on the surface of tumor cells is correlated with metastatic behavior. Cancer Res. 1995, 55, 428–433. [Google Scholar]
- Patarroyo, M.; Tryggvason, K.; Virtanen, I. Laminin isoforms in tumor invasion, angiogenesis and metastasis. Semin. Cancer Biol. 2002, 12, 197–207. [Google Scholar] [CrossRef]
- Pyke, C.; Rømer, J.; Kallunki, P.; Lund, L.R.; Ralfkiaer, E.; Danø, K.; Tryggvason, K. The gamma 2 chain of kalinin/laminin 5 is preferentially expressed in invading malignant cells in human cancers. Am. J. Pathol. 1994, 145, 782–791. [Google Scholar]
- Ferraro, F.; Celso, C.L.; Scadden, D. Adult stem cels and their niches. Adv. Exp. Med. Biol. 2010, 695, 155–168. [Google Scholar] [CrossRef] [Green Version]
- Gattazzo, F.; Urciuolo, A.; Bonaldo, P. Extracellular matrix: A dynamic microenvironment for stem cell niche. Biochim. Biophys. Acta—Gen. Subj. 2014, 1840, 2506–2519. [Google Scholar] [CrossRef]
- Shiozawa, Y.; Pedersen, E.A.; Havens, A.M.; Jung, Y.; Mishra, A.; Joseph, J.; Kim, J.K.; Patel, L.R.; Ying, C.; Ziegler, A.M.; et al. Human prostate cancer metastases target the hematopoietic stem cell niche to establish footholds in mouse bone marrow. J. Clin. Investig. 2011, 121, 1298–1312. [Google Scholar] [CrossRef] [Green Version]
- Carlson, P.; Dasgupta, A.; Grzelak, C.A.; Kim, J.; Barrett, A.; Coleman, I.M.; Shor, R.E.; Goddard, E.T.; Dai, J.; Schweitzer, E.M.; et al. Targeting the perivascular niche sensitizes disseminated tumour cells to chemotherapy. Nat. Cell Biol. 2019, 21, 238–250. [Google Scholar] [CrossRef]
- Urciuolo, A.; Quarta, M.; Morbidoni, V.; Gattazzo, F.; Molon, S.; Grumati, P.; Montemurro, F.; Tedesco, F.S.; Blaauw, B.; Cossu, G.; et al. Collagen VI regulates satellite cell self-renewal and muscle regeneration. Nat. Commun. 2013, 4, 1964. [Google Scholar] [CrossRef] [Green Version]
- Ghajar, C.M.; Peinado, H.; Mori, H.; Matei, I.R.; Evason, K.J.; Brazier, H.; Almeida, D.; Koller, A.; Hajjar, K.A.; Stainier, D.Y.R.; et al. The perivascular niche regulates breast tumour dormancy. Nat. Cell Biol. 2013, 15, 807–817. [Google Scholar] [CrossRef]
- Baghdadi, M.B.; Castel, D.; Machado, L.; Fukada, S.-I.; Birk, D.E.; Relaix, F.; Tajbakhsh, S.; Mourikis, P. Reciprocal signalling by Notch-Collagen V-CALCR retains muscle stem cells in their niche. Nature 2018, 557, 714–718. [Google Scholar] [CrossRef]
- Khurana, S.; Schouteden, S.; Manesia, J.K.; Santamaria-Martínez, A.; Huelsken, J.; Lacy-Hulbert, A.; Verfaillie, C.M. Outside-in integrin signalling regulates haematopoietic stem cell function via Periostin-Itgav axis. Nat. Commun. 2016, 7, 13500. [Google Scholar] [CrossRef] [Green Version]
- Morgner, J.; Ghatak, S.; Jakobi, T.; Dieterich, C.; Aumailley, M.; Wickström, S.A. Integrin-linked kinase regulates the niche of quiescent epidermal stem cells. Nat. Commun. 2015, 6, 8198. [Google Scholar] [CrossRef] [Green Version]
- Méndez-Ferrer, S.; Michurina, T.V.; Ferraro, F.; Mazloom, A.R.; Macarthur, B.D.; Lira, S.A.; Scadden, D.T.; Ma’ayan, A.; Enikolopov, G.N.; Frenette, P.S. Mesenchymal and haematopoietic stem cells form a unique bone marrow niche. Nature 2010, 466, 829–834. [Google Scholar] [CrossRef]
- Bragado, P.; Estrada, Y.; Parikh, F.; Krause, S.; Capobianco, C.; Farina, H.G.; Schewe, D.M.; Aguirre-Ghiso, J.A. TGF-β2 dictates disseminated tumour cell fate in target organs through TGF-β-RIII and p38α/β signalling. Nat. Cell Biol. 2013, 15, 1351–1361. [Google Scholar] [CrossRef] [Green Version]
- Malladi, S.; Macalinao, D.G.; Jin, X.; He, L.; Basnet, H.; Zou, Y.; de Stanchina, E.; Massagué, J. Metastatic Latency and Immune Evasion through Autocrine Inhibition of WNT. Cell 2016, 165, 45–60. [Google Scholar] [CrossRef] [Green Version]
- Harper, K.L.; Sosa, M.S.; Entenberg, D.; Hosseini, H.; Cheung, J.F.; Nobre, R.; Avivar-Valderas, A.; Nagi, C.; Girnius, N.; Davis, R.J.; et al. Mechanism of early dissemination and metastasis in Her2(+) mammary cancer. Nature 2016, 540, 588–592. [Google Scholar] [CrossRef] [PubMed]
- Nobre, A.R.; Risson, E.; Singh, D.K.; di Martino, J.; Cheung, J.F.; Wang, J.; Johnson, J.; Russnes, H.G.; Bravo-Cordero, J.J.; Birbrair, A.; et al. Bone MarrowNG2+/Nestin+ mesenchymal stem cells drive DTC dormancy via TGFβ2. Nature Cancer 2021, 2, 327–339. [Google Scholar] [CrossRef]
- Oskarsson, T.; Acharyya, S.; Zhang, X.H.F.; Vanharanta, S.; Tavazoie, S.F.; Morris, P.G.; Downey, R.J.; Manova-Todorova, K.; Brogi, E.; Massagué, J. Breast cancer cells produce tenascin C as a metastatic niche component to colonize the lungs. Nat. Med. 2011, 17, 867–874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, J.; Garva, R.; Pickard, A.; Yeung, C.-Y.C.; Mallikarjun, V.; Swift, J.; Holmes, D.F.; Calverley, B.; Lu, Y.; Adamson, A.; et al. Circadian control of the secretory pathway maintains collagen homeostasis. Nat. Cell Biol. 2020, 22, 74–86. [Google Scholar] [CrossRef]
- Aguirre-Ghiso, J.A.; Estrada, Y.; Liu, D.; Ossowski, L. ERK(MAPK) activity as a determinant of tumor growth and dormancy; regulation by p38(SAPK). Cancer Res. 2003, 63, 1684–1695. [Google Scholar] [CrossRef]
- Barney, L.E.; Hall, C.L.; Schwartz, A.D.; Parks, A.N.; Sparages, C.; Galarza, S.; Platt, M.O.; Mercurio, A.M.; Peyton, S.R. Tumor cell-organized fibronectin maintenance of a dormant breast cancer population. Sci. Adv. 2020, 6, eaaz4157. [Google Scholar] [CrossRef] [Green Version]
- Korah, R.; Boots, M.; Wieder, R. Integrin alpha5beta1 promotes survival of growth-arrested breast cancer cells: An in vitro paradigm for breast cancer dormancy in bone marrow. Cancer Res. 2004, 64, 4514–4522. [Google Scholar] [CrossRef] [Green Version]
- Barrios, J.; Wieder, R. Dual FGF-2 and intergrin alpha5beta1 signaling mediate GRAF-induced RhoA inactivation in a model of breast cancer dormancy. Cancer Microenviron. 2009, 2, 33–47. [Google Scholar] [CrossRef] [Green Version]
- Soikkeli, J.; Podlasz, P.; Yin, M.; Nummela, P.; Jahkola, T.; Virolainen, S.; Krogerus, L.; Heikkilä, P.; von Smitten, K.; Saksela, O.; et al. Metastatic outgrowth encompasses COL-I, FN1, and POSTN up-regulation and assembly to fibrillar networks regulating cell adhesion, migration, and growth. Am. J. Pathol. 2010, 177, 387–403. [Google Scholar] [CrossRef]
- Wang, Z.; Ouyang, G. Periostin: A bridge between cancer stem cells and their metastatic niche. Cell Stem Cell 2012, 10, 111–112. [Google Scholar] [CrossRef] [Green Version]
- Deng, X.; Ao, S.; Hou, J.; Li, Z.; Lei, Y.; Lyu, G. Prognostic significance of periostin in colorectal cancer. Chin. J. Cancer Res. 2019, 31, 547–556. [Google Scholar] [CrossRef] [PubMed]
- Lowy, C.M.; Oskarsson, T. Tenascin C in metastasis: A view from the invasive front. Cell Adhes. Migr. 2015, 9, 112–124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Z.; Schwenzer, A.; Rupp, T.; Murdamoothoo, D.; Vegliante, R.; Lefebvre, O.; Klein, A.; Hussenet, T.; Orend, G. Tenascin-C Promotes Tumor Cell Migration and Metastasis through Integrin α9β1-Mediated YAP Inhibition. Cancer Res. 2018, 78, 950–961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Z.; Velázquez-Quesada, I.; Murdamoothoo, D.; Ahowesso, C.; Yilmaz, A.; Spenlé, C.; Averous, G.; Erne, W.; Oberndorfer, F.; Oszwald, A.; et al. Tenascin-C increases lung metastasis by impacting blood vessel invasions. Matrix Biol. 2019, 83, 26–47. [Google Scholar] [CrossRef] [PubMed]
- Boyerinas, B.; Zafrir, M.; Yesilkanal, A.E.; Price, T.T.; Hyjek, E.M.; Sipkins, D.A. Adhesion to osteopontin in the bone marrow niche regulates lymphoblastic leukemia cell dormancy. Blood 2013, 121, 4821–4831. [Google Scholar] [CrossRef] [Green Version]
- Weidenfeld, K.; Schif-Zuck, S.; Abu-Tayeh, H.; Kang, K.; Kessler, O.; Weissmann, M.; Neufeld, G.; Barkan, D. Dormant tumor cells expressing LOXL2 acquire a stem-like phenotype mediating their transition to proliferative growth. Oncotarget 2016, 7, 71362–71377. [Google Scholar] [CrossRef] [Green Version]
- Peixoto, P.; Etcheverry, A.; Aubry, M.; Missey, A.; Lachat, C.; Perrard, J.; Hendrick, E.; Delage-Mourroux, R.; Mosser, J.; Borg, C.; et al. EMT is associated with an epigenetic signature of ECM remodeling genes. Cell Death Dis. 2019, 10, 205. [Google Scholar] [CrossRef] [Green Version]
- Aguirre-Ghiso, J.; Nobre, A.R.; Dalla, E.; Yang, J.; Huang, X.; Kenigsberg, E.; Wang, J. A Mesenchymal-Like Program of Dormancy Controlled by ZFP281 Serves as a Barrier to Metastatic Progression of Early Disseminated Cancer Cells; Research Square: Durham, NC, USA, 2021. [Google Scholar] [CrossRef]
- Padmanaban, V.; Krol, I.; Suhail, Y.; Szczerba, B.M.; Aceto, N.; Bader, J.S.; Ewald, A.J. E-cadherin is required for metastasis in multiple models of breast cancer. Nature 2019, 573, 439–444. [Google Scholar] [CrossRef]
- Albrengues, J.; Shields, M.A.; Ng, D.; Park, C.G.; Ambrico, A.; Poindexter, M.E.; Upadhyay, P.; Uyeminami, D.L.; Pommier, A.; Küttner, V.; et al. Neutrophil extracellular traps produced during inflammation awaken dormant cancer cells in mice. Science 2018, 361, 4227. [Google Scholar] [CrossRef] [Green Version]
- Keeratichamroen, S.; Lirdprapamongkol, K.; Svasti, J. Mechanism of ECM-induced dormancy and chemoresistance in A549 human lung carcinoma cells. Oncol. Rep. 2018, 39, 1765–1774. [Google Scholar] [CrossRef] [Green Version]
- Pogány, G.; Timár, F.; Oláh, J.; Harisi, R.; Polony, G.; Paku, S.; Bocsi, J.; Laurie, G.W. Role of the basement membrane in tumor cell dormancy and cytotoxic resistance. Oncology 2001, 60, 274–281. [Google Scholar] [CrossRef]
- Shibue, T.; Reinhardt, F.; Weinberg, R.A. Syndecan-Mediated Ligation of ECM Proteins Triggers Proliferative Arrest of Disseminated Tumor Cells. Cancer Res. 2019, 79, 5944–5957. [Google Scholar] [CrossRef] [Green Version]
- Hattar, R.; Maller, O.; McDaniel, S.; Hansen, K.C.; Hedman, K.J.; Lyons, T.R.; Lucia, S.; Wilson, R.S.J.; Schedin, P. Tamoxifen induces pleiotrophic changes in mammary stroma resulting in extracellular matrix that suppresses transformed phenotypes. Breast Cancer Res. 2009, 11, R5. [Google Scholar] [CrossRef] [Green Version]
- Lim, S.B.; Chua, M.L.K.; Yeong, J.P.S.; Tan, S.J.; Lim, W.-T.; Lim, C.T. Pan-cancer analysis connects tumor matrisome to immune response. NPJ Precis. Oncol. 2019, 3, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raavé, R.; van Kuppevelt, T.H.; Daamen, W.F. Chemotherapeutic drug delivery by tumoral extracellular matrix targeting. J. Control. Release 2018, 274, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Kaur, A.; Ecker, B.L.; Douglass, S.M.; Kugel, C.H.; Webster, M.R.; Almeida, F.V.; Somasundaram, R.; Hayden, J.; Ban, E.; Ahmadzadeh, H.; et al. Remodeling of the Collagen Matrix in Aging Skin Promotes Melanoma Metastasis and Affects Immune Cell Motility. Cancer Discov. 2019, 9, 64–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ecker, B.L.; Kaur, A.; Douglass, S.M.; Webster, M.R.; Almeida, F.V.; Marino, G.E.; Sinnamon, A.J.; Neuwirth, M.G.; Alicea, G.M.; Ndoye, A.; et al. Age-Related Changes in HAPLN1 Increase Lymphatic Permeability and Affect Routes of Melanoma Metastasis. Cancer Discov. 2019, 9, 82–95. [Google Scholar] [CrossRef] [Green Version]
- Langton, A.K.; Graham, H.K.; Griffiths, C.E.M.; Watson, R.E.B. Ageing significantly impacts the biomechanical function and structural composition of skin. Exp. Dermatol. 2019, 28, 981–984. [Google Scholar] [CrossRef] [Green Version]
- McCabe, M.C.; Hill, R.C.; Calderone, K.; Cui, Y.; Yan, Y.; Quan, T.; Fisher, G.J.; Hansen, K.C. Alterations in extracellular matrix composition during aging and photoaging of the skin. Matrix Biol. Plus 2020, 8, 100041. [Google Scholar] [CrossRef] [PubMed]
- Ganceviciene, R.; Liakou, A.I.; Theodoridis, A.; Makrantonaki, E.; Zouboulis, C.C. Skin anti-aging strategies. Dermatoendocrinology 2012, 4, 308–319. [Google Scholar] [CrossRef] [Green Version]
- Ramadass, S.K.; Perumal, S.; Gopinath, A.; Nisal, A.; Subramanian, S.; Madhan, B. Sol-gel assisted fabrication of collagen hydrolysate composite scaffold: A novel therapeutic alternative to the traditional collagen scaffold. ACS Appl. Mater. Interfaces 2014, 6, 15015–15025. [Google Scholar] [CrossRef] [PubMed]
- Perumal, R.K.; Gopinath, A.; Thangam, R.; Perumal, S.; Masilamani, D.; Ramadass, S.K.; Madhan, B. Collagen-silica bio-composite enriched with Cynodon dactylon extract for tissue repair and regeneration. Mater. Sci. Eng. C Mater. Biol. Appl. 2018, 92, 297–306. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, D.; Ghosh, B.; Mukhopadhyay, M. Development of nanotechnology for advancement and application in wound healing: A review. IET Nanobiotechnol. 2019, 13, 778–785. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Zhang, L.; Wan, D.; Zhou, L.; Zheng, S.; Lin, S.; Qiao, Y. Extracellular matrix and its therapeutic potential for cancer treatment. Signal Transduct. Target. Ther. 2021, 6, 153. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Di Martino, J.S.; Akhter, T.; Bravo-Cordero, J.J. Remodeling the ECM: Implications for Metastasis and Tumor Dormancy. Cancers 2021, 13, 4916. https://doi.org/10.3390/cancers13194916
Di Martino JS, Akhter T, Bravo-Cordero JJ. Remodeling the ECM: Implications for Metastasis and Tumor Dormancy. Cancers. 2021; 13(19):4916. https://doi.org/10.3390/cancers13194916
Chicago/Turabian StyleDi Martino, Julie S., Tasmiah Akhter, and Jose Javier Bravo-Cordero. 2021. "Remodeling the ECM: Implications for Metastasis and Tumor Dormancy" Cancers 13, no. 19: 4916. https://doi.org/10.3390/cancers13194916