
Temozolomide-Acquired Resistance Is Associated with Modulation of the Integrin Repertoire in Glioblastoma, Impact of α5β1 Integrin

 , , , , , , and
, , , , , , and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Drugs
2.2. Cell Culture
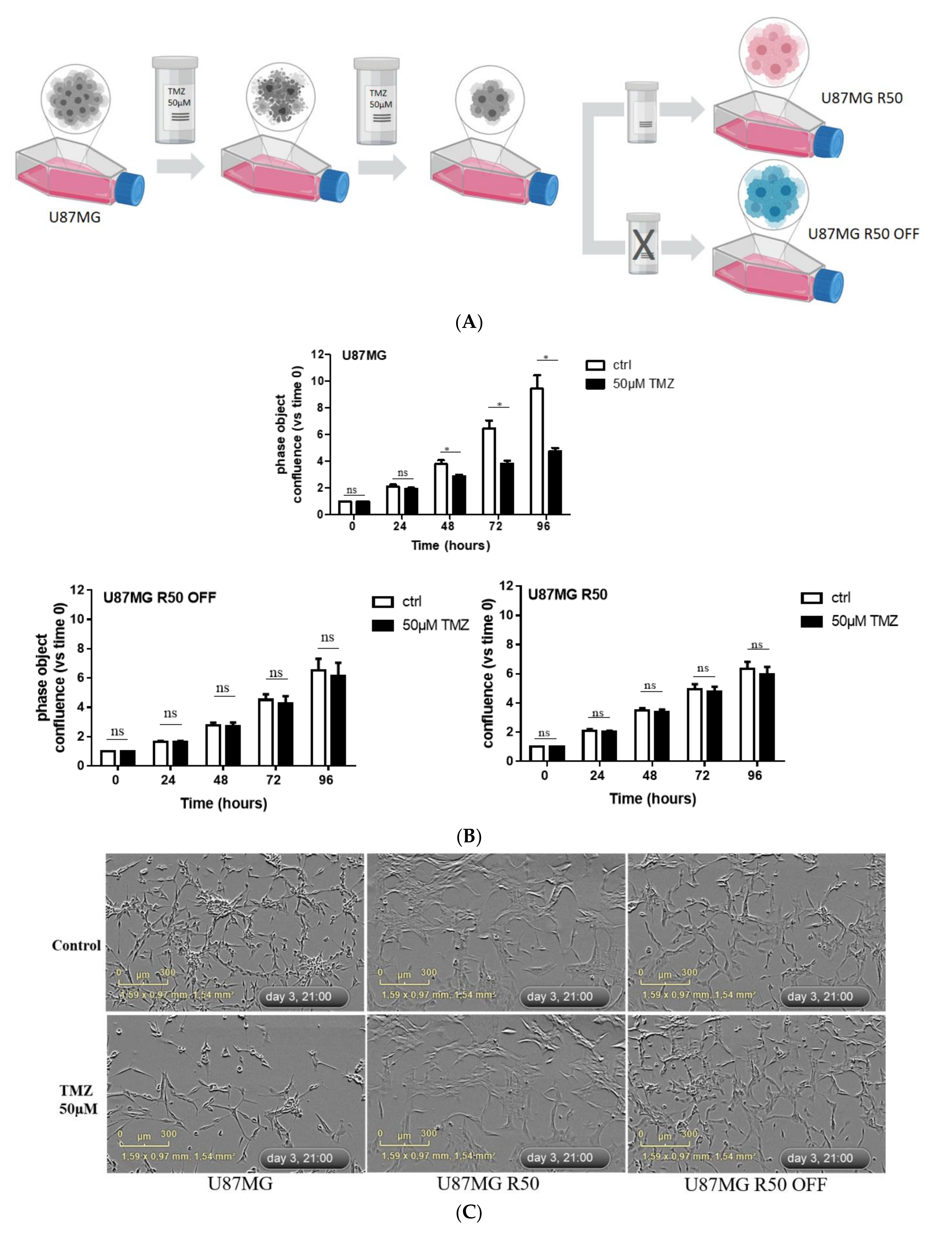
2.3. Generation of Temozolomide-Resistant Glioblastoma Cells
2.4. IncuCyte Cell Confluence Assay as an Index of Proliferation
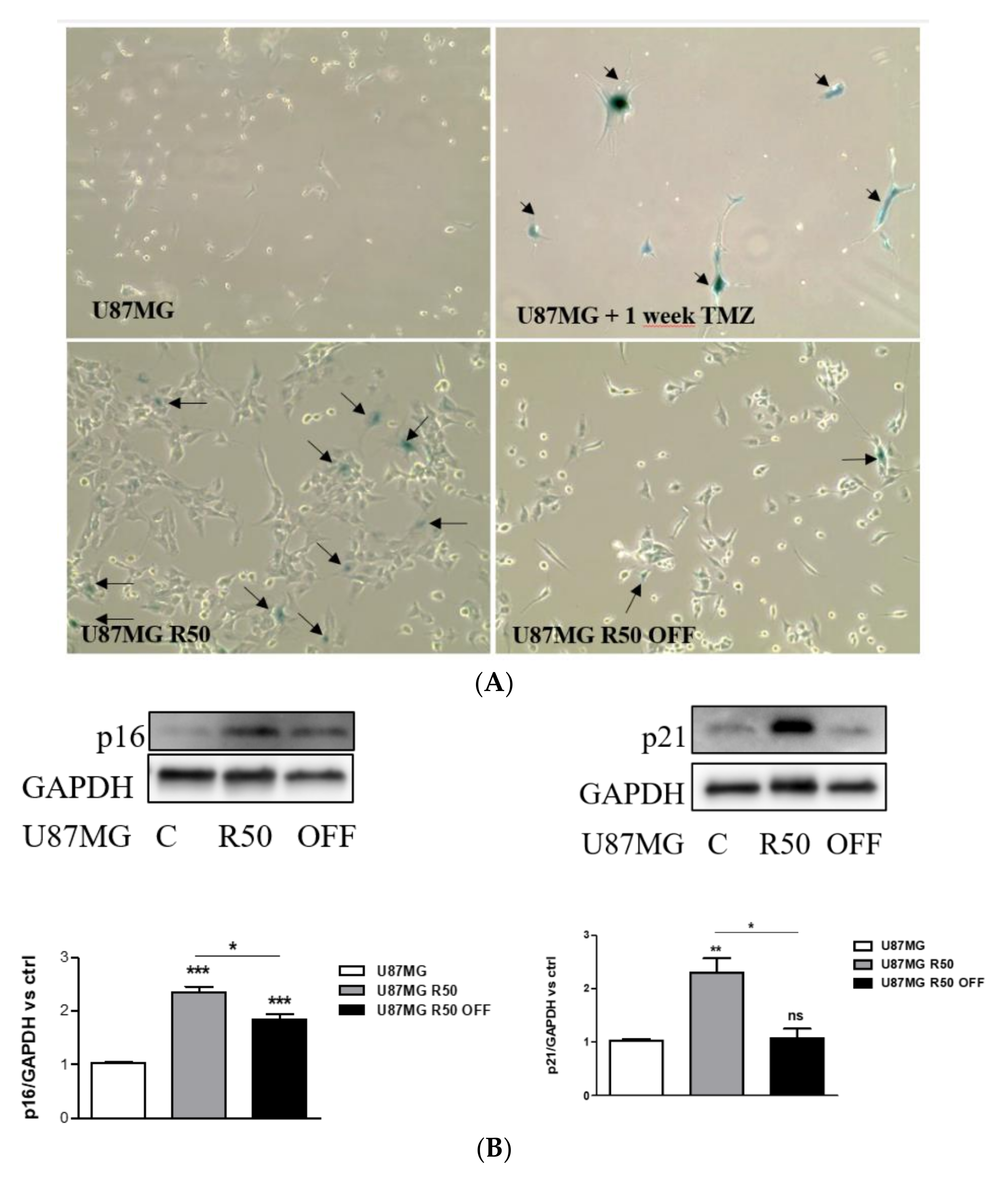
2.5. Senescence Assay
2.6. Western Blotting
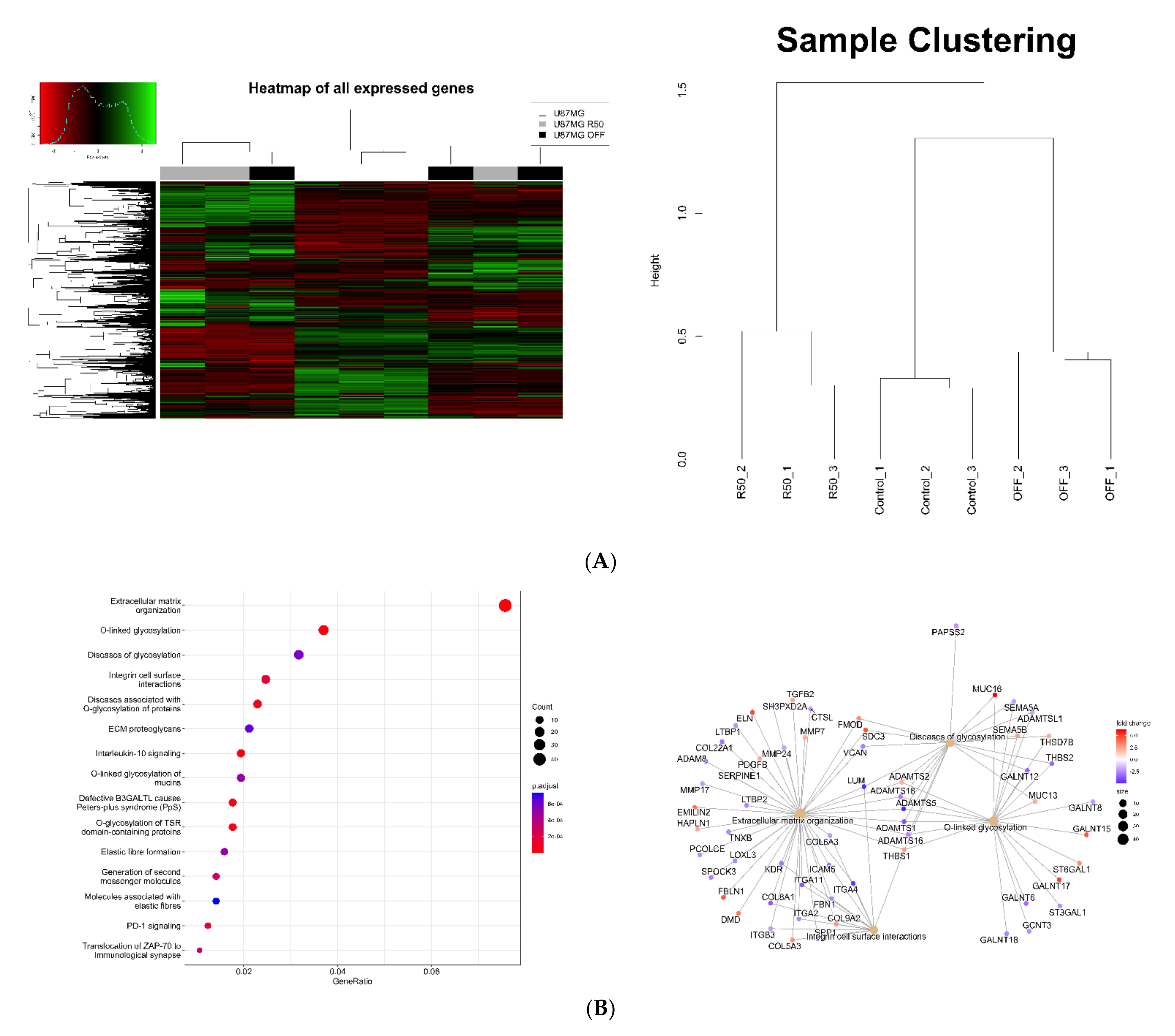
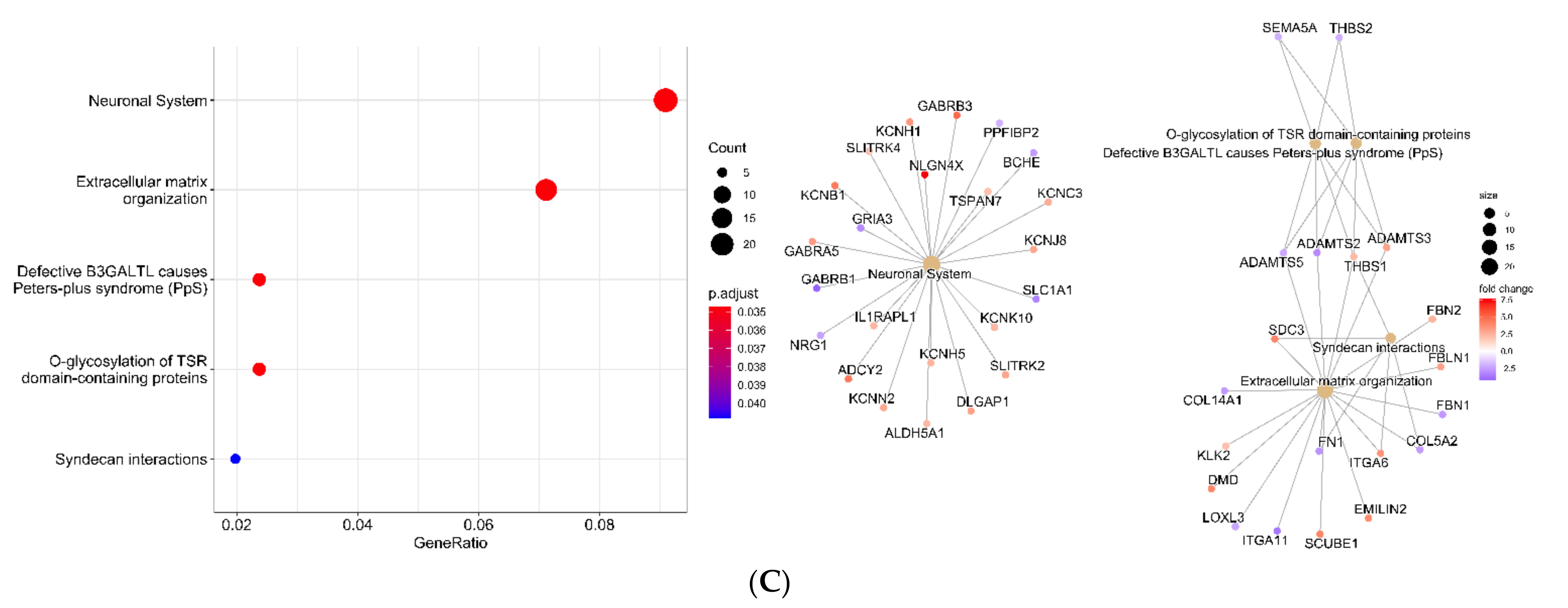
2.7. RNAseq Data
2.8. Confocal Microscopy and Image Analysis
2.9. Spheroid Migration Assays
2.10. Statistical Analysis
3. Results
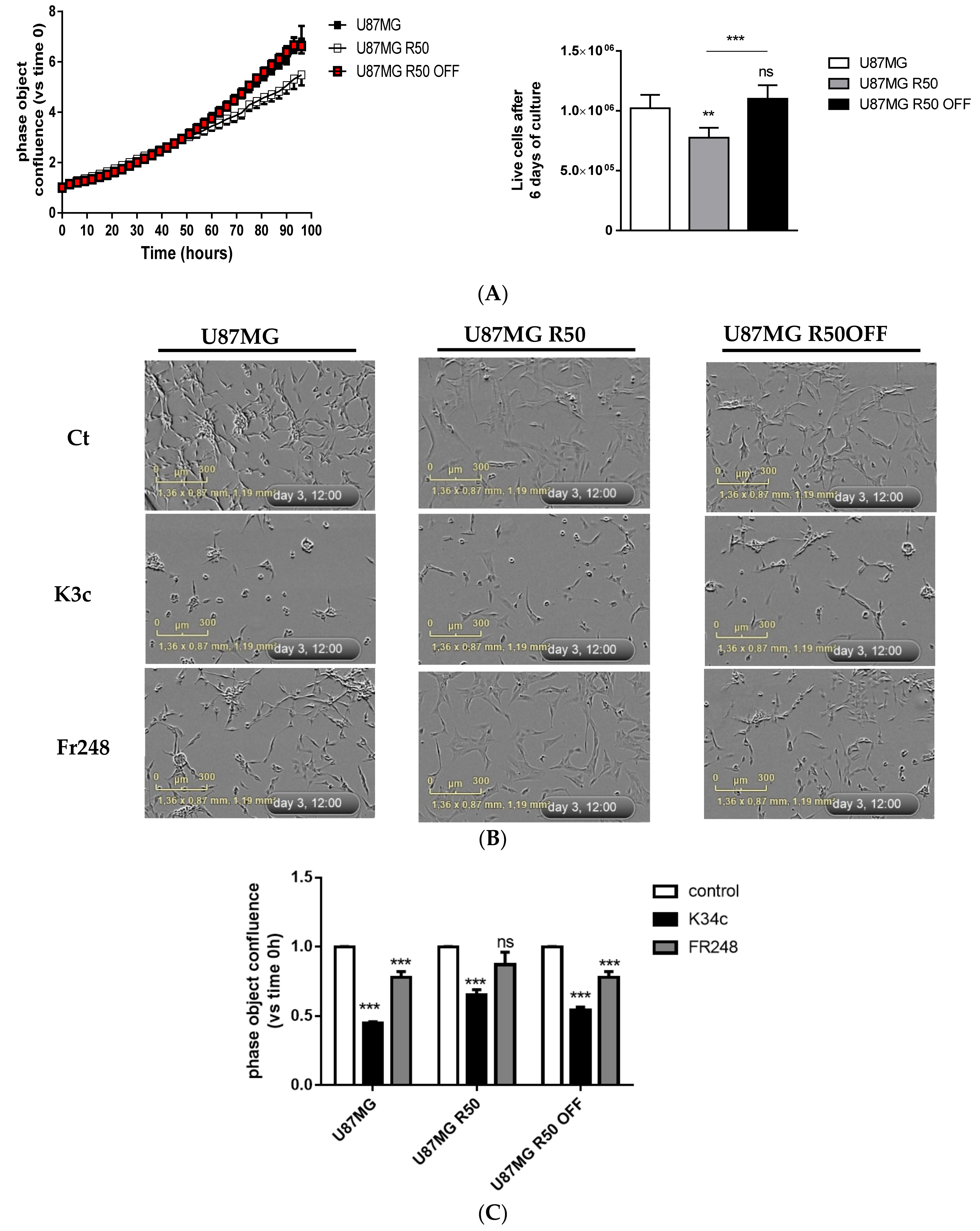
3.1. Long-Term Exposure of U87MG Cells to TMZ Generates Persistent Resistant Cells
3.2. TMZ Induces Senescence in U87MG Cells
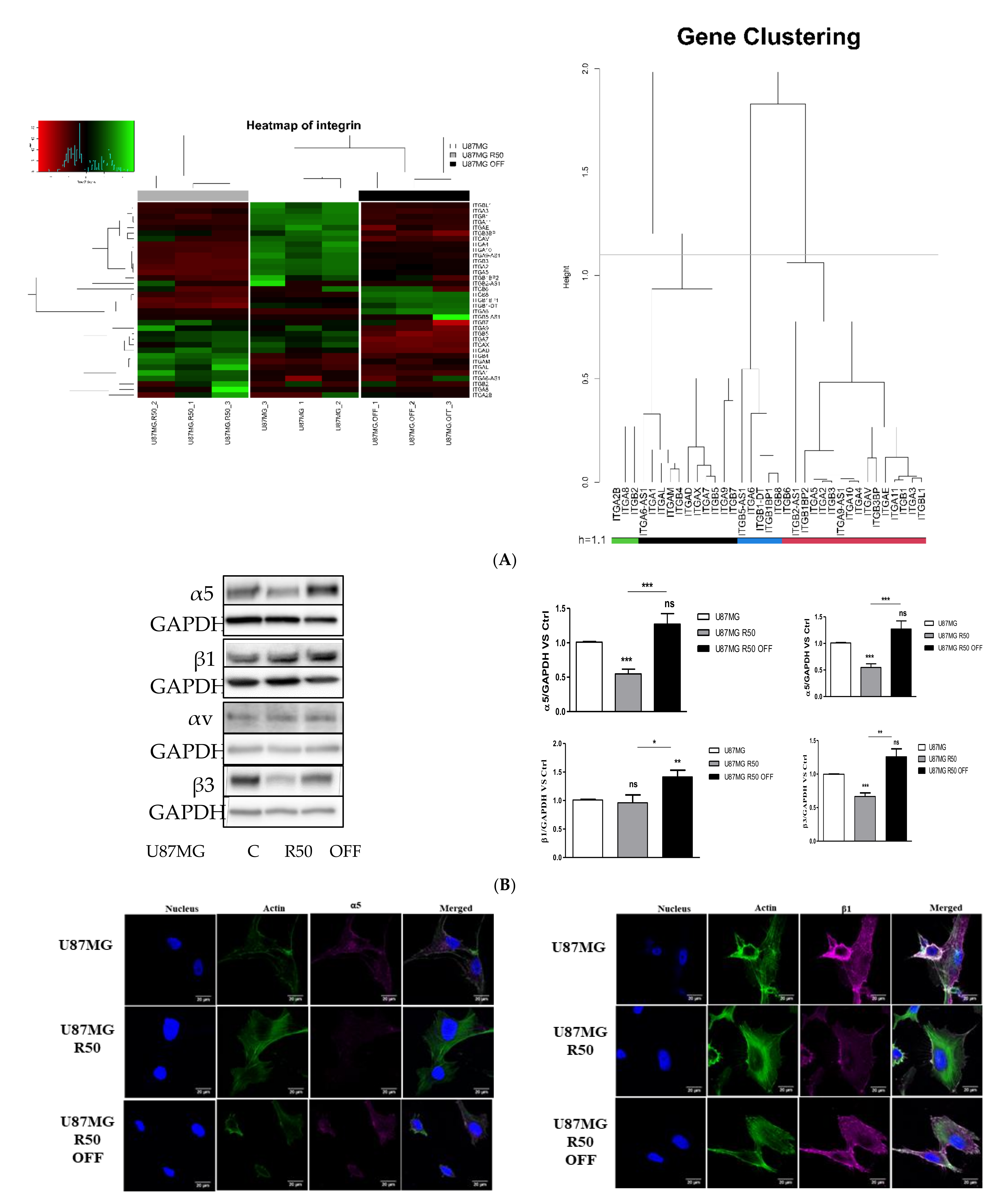
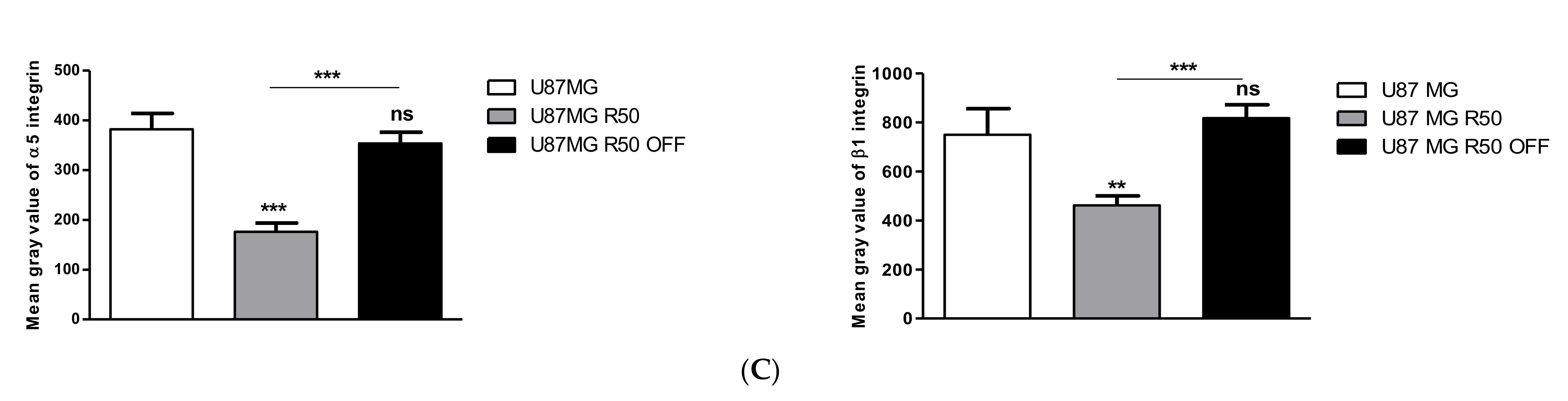
3.3. Extracellular Matrix Organization and Integrins Are Affected by TMZ
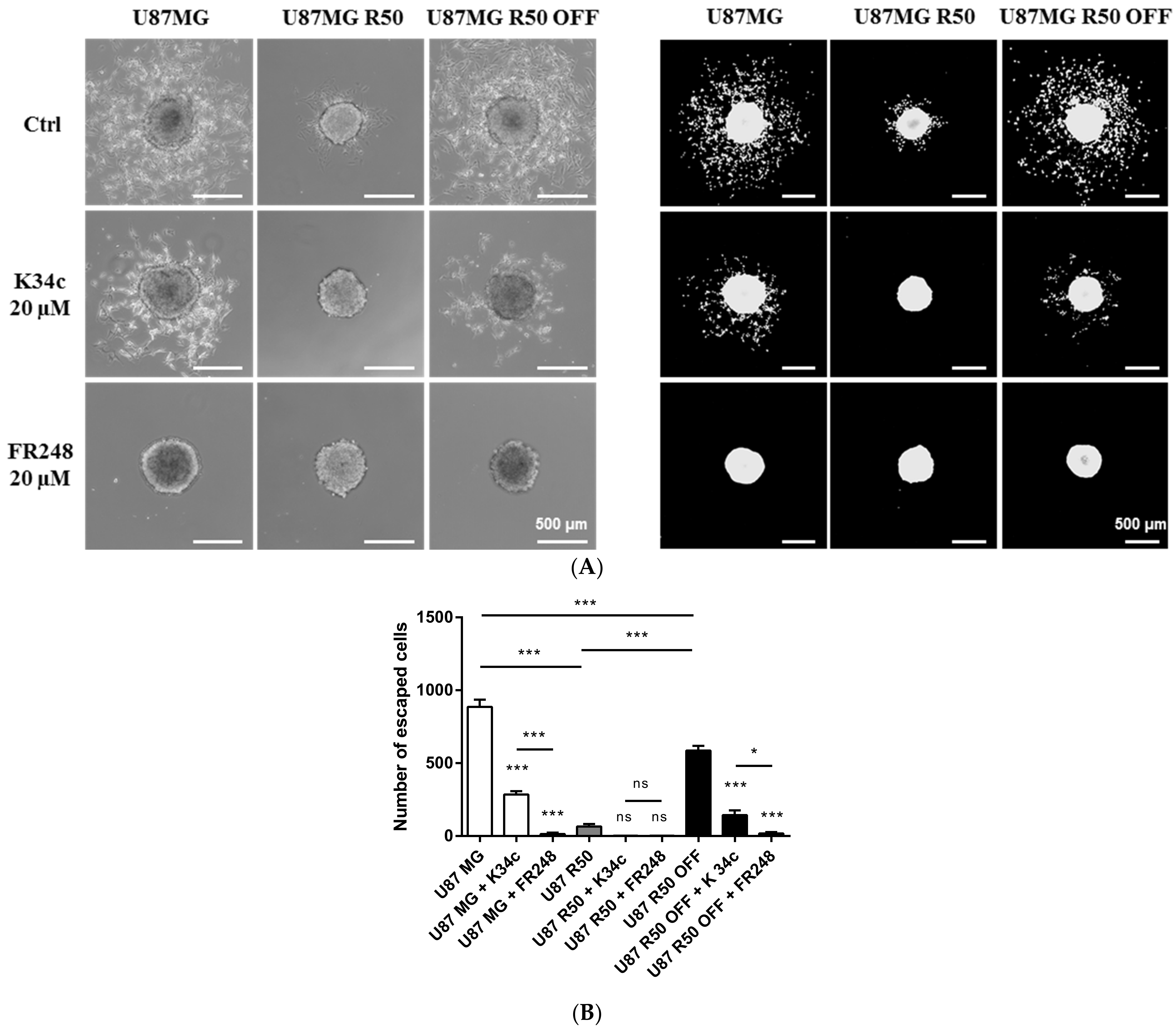
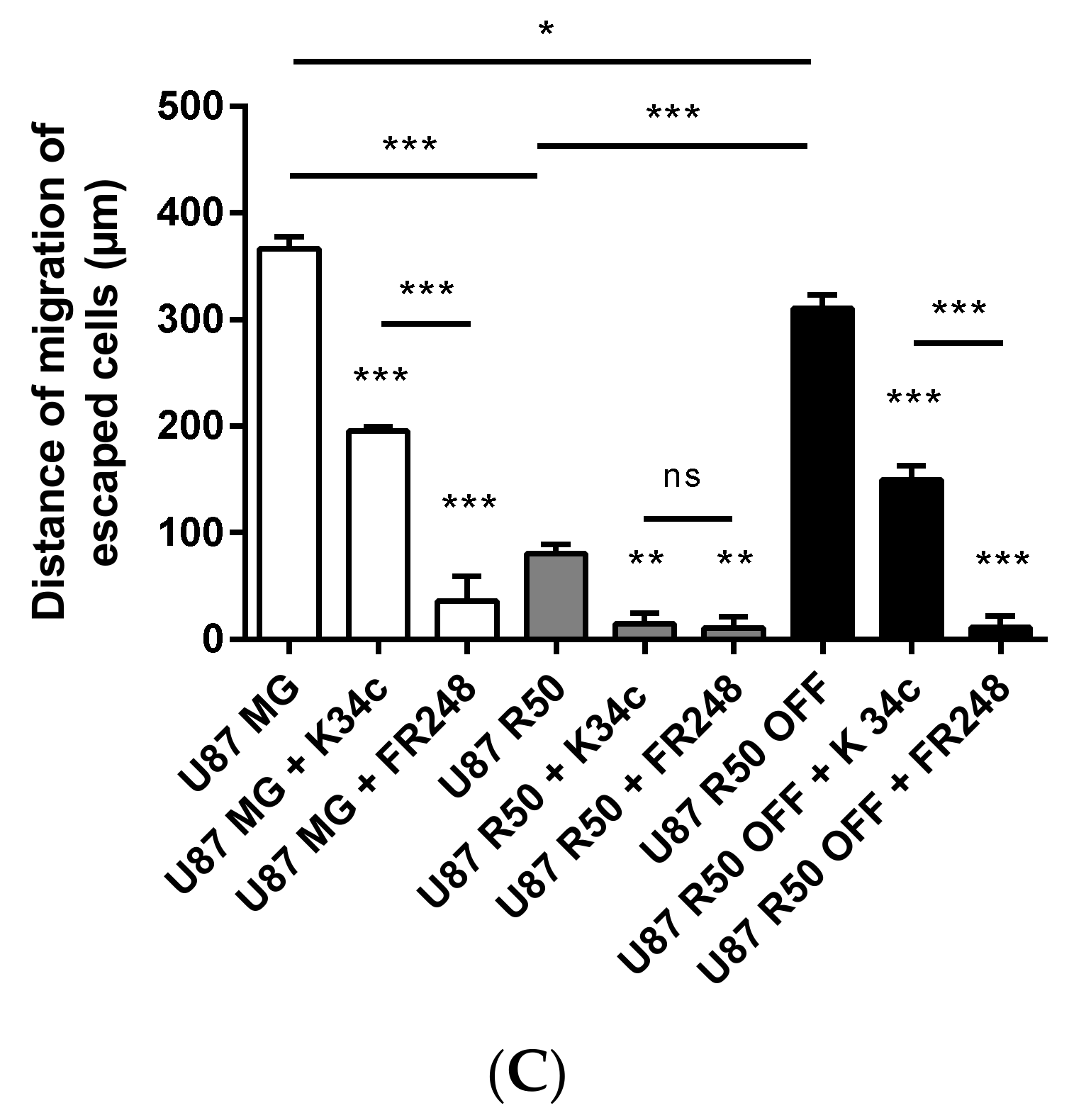
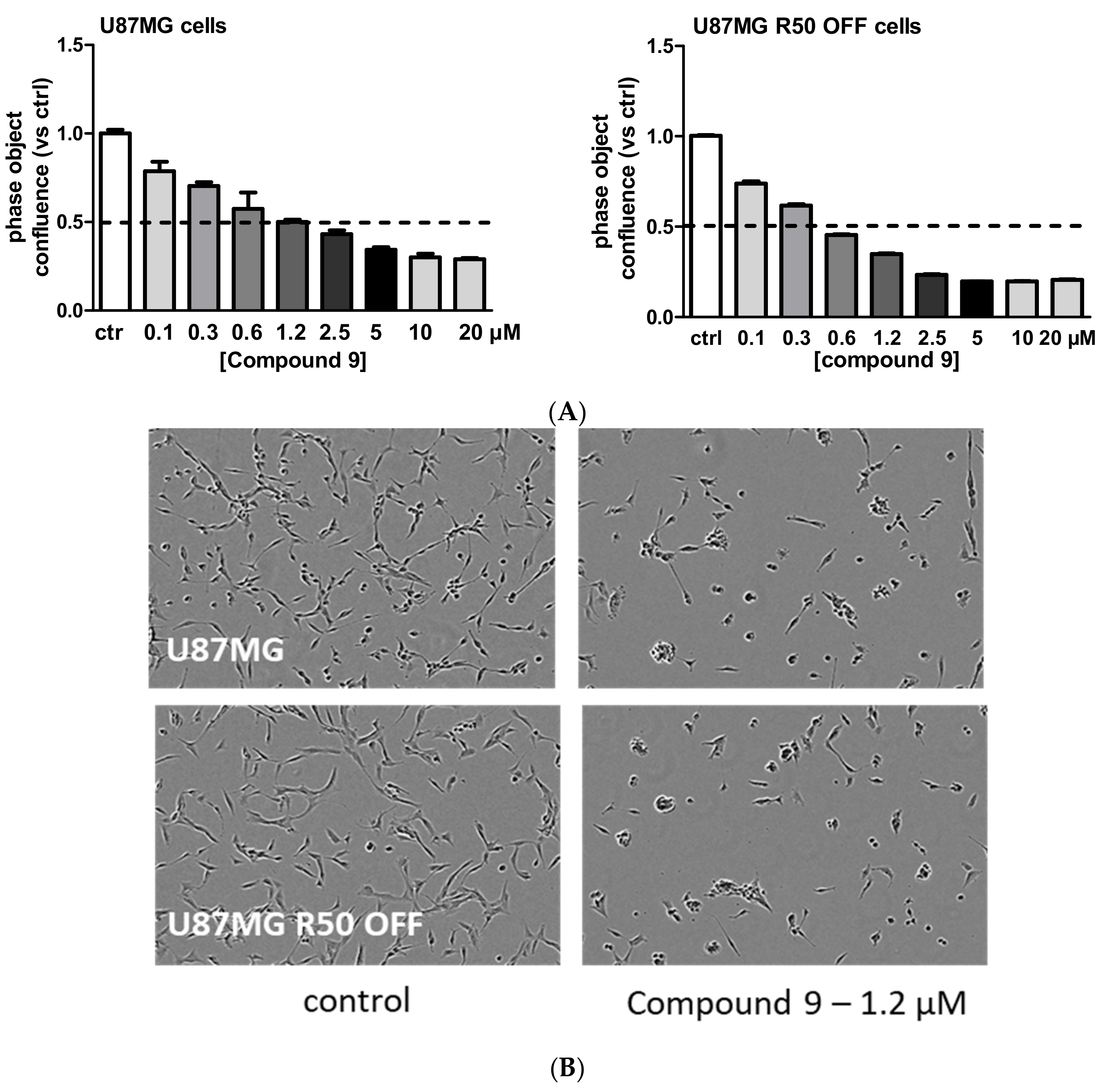
3.4. TMZ Affects Glioma Cell Proliferation and Migration through Modulation of α5β1 Integrin
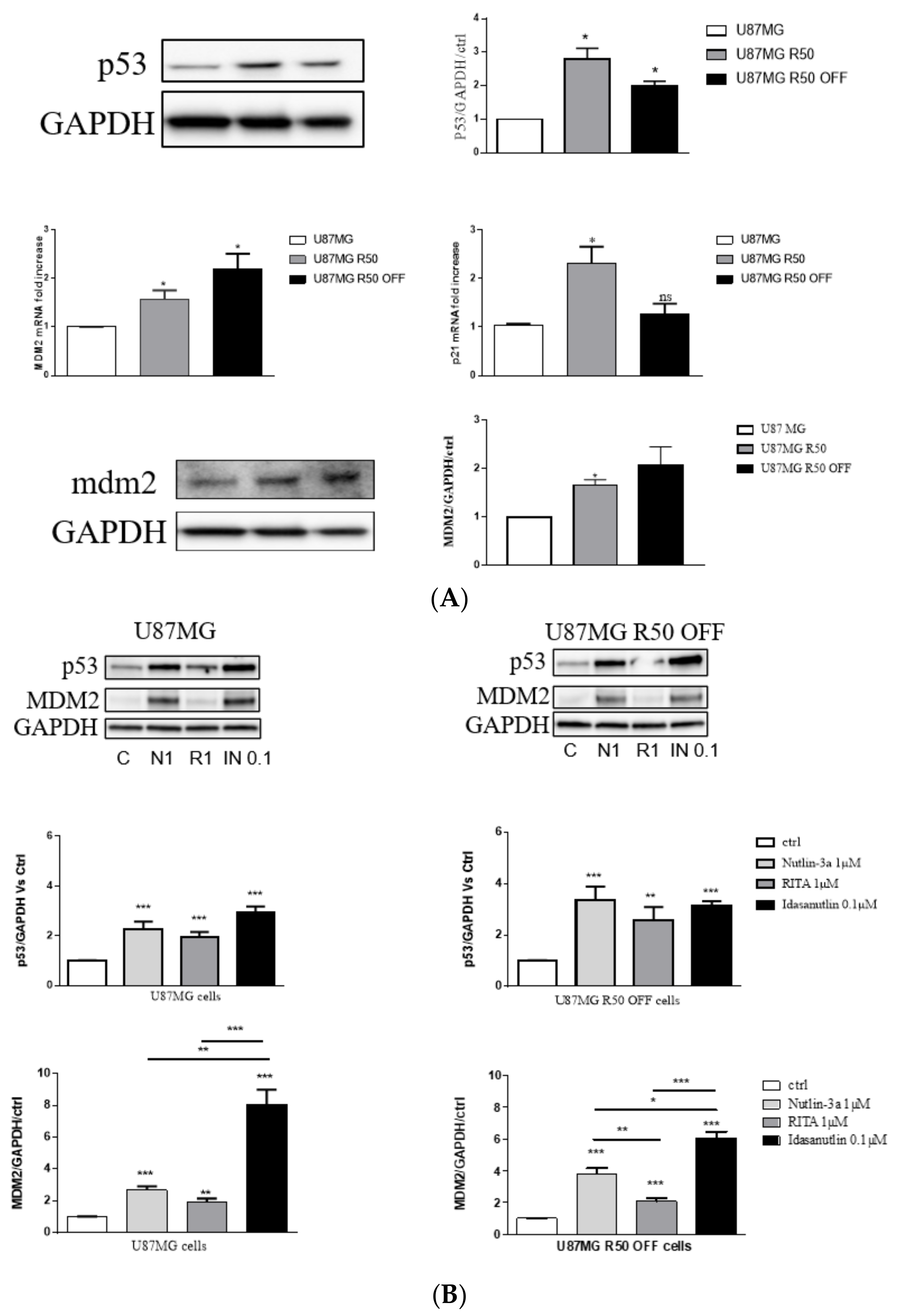
3.5. p53 Signalling Pathway in U87MG and TMZ-Resistant Cells
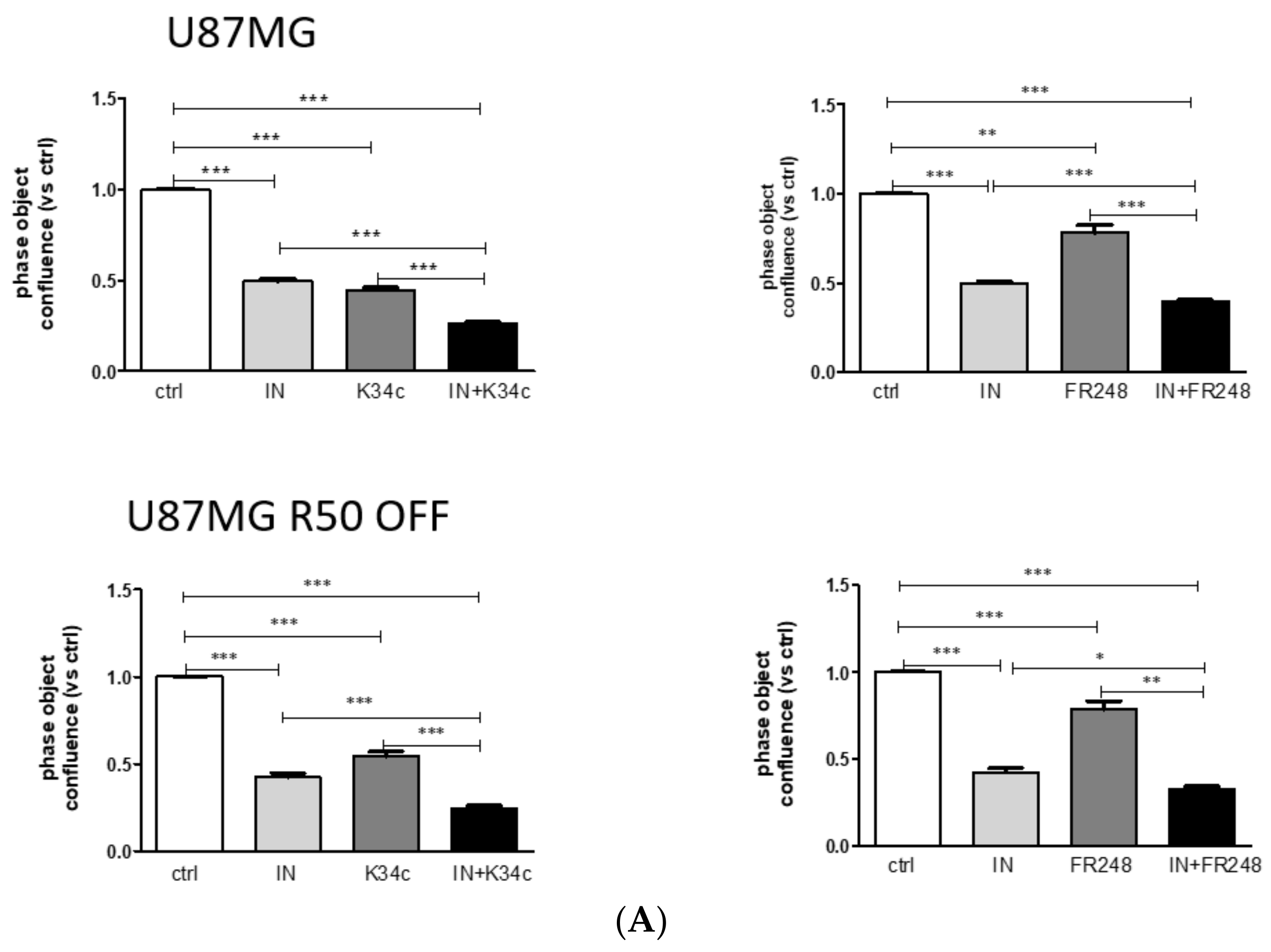
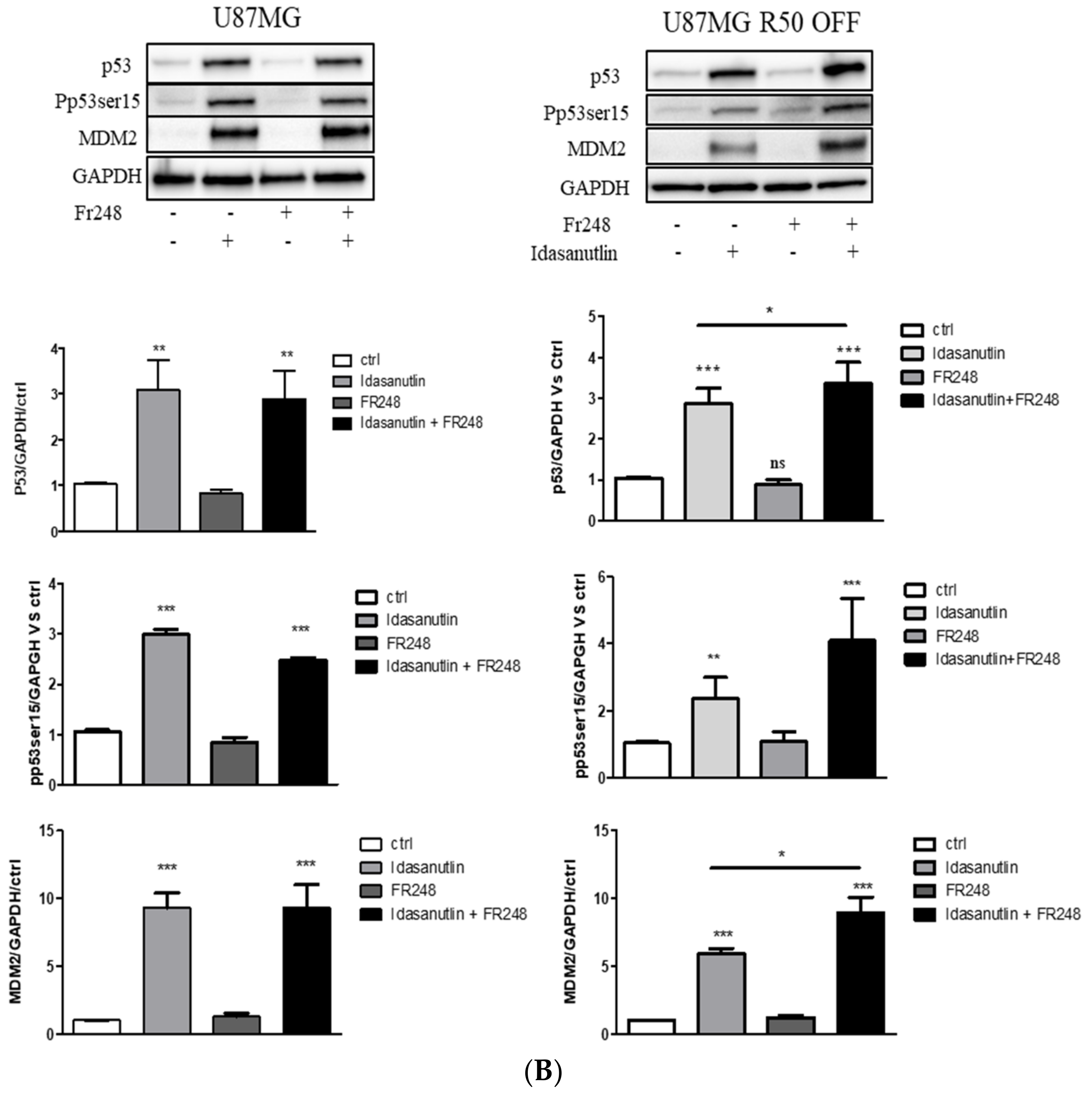
3.6. p53 Activation and Integrin Inhibition as a Therapeutic Option for TMZ-Resistant Cells
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Geraldo, L.H.M.; Garcia, C.; da Fonseca, A.C.C.; Dubois, L.G.F.; de Sampaio e Spohr, T.C.L.; Matias, D.; de Camargo Magalhães, E.S.; do Amaral, R.F.; da Rosa, B.G.; Grimaldi, I.; et al. Glioblastoma Therapy in the Age of Molecular Medicine. Trends Cancer 2019, 5, 46–65. [Google Scholar] [CrossRef] [PubMed]
- Omuro, A. Glioblastoma and Other Malignant Gliomas: A Clinical Review. JAMA 2013, 310, 1842. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Weller, M.; Belanger, K.; Bogdahn, U.; Ludwin, S.K.; Lacombe, D.; Mirimanoff, R.O. Radiotherapy plus Concomitant and Adjuvant Temozolomide for Glioblastoma. N. Engl. J. Med. 2005, 10, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Cruz Da Silva, E.; Mercier, M.-C.; Etienne-Selloum, N.; Dontenwill, M.; Choulier, L. A Systematic Review of Glioblastoma-Targeted Therapies in Phases II, III, IV Clinical Trials. Cancers 2021, 13, 1795. [Google Scholar] [CrossRef]
- Khaddour, K.; Johanns, T.; Ansstas, G. The Landscape of Novel Therapeutics and Challenges in Glioblastoma Multiforme: Contemporary State and Future Directions. Pharmaceuticals 2020, 13, 389. [Google Scholar] [CrossRef]
- Hotchkiss, K.M.; Sampson, J.H. Temozolomide Treatment Outcomes and Immunotherapy Efficacy in Brain Tumor. J. Neurooncol. 2020, 151, 55–62. [Google Scholar] [CrossRef]
- Park, C.-K.; Kim, J.E.; Kim, J.Y.; Song, S.W.; Kim, J.W.; Choi, S.H.; Kim, T.M.; Lee, S.-H.; Kim, I.H.; Park, S.-H. The Changes in MGMT Promoter Methylation Status in Initial and Recurrent Glioblastomas. Transl. Oncol. 2012, 5, 393–397. [Google Scholar] [CrossRef] [Green Version]
- Grombacher, T.; Eichhorn, U.; Kaina, B. P53 Is Involved in Regulation of the DNA Repair Gene O6-Methylguanine-DNA Methyltransferase (MGMT) by DNA Damaging Agents. Oncogene 1998, 17, 845–851. [Google Scholar] [CrossRef] [Green Version]
- Harris, L.C.; Remack, J.S.; Houghton, P.J.; Brent, T.P. Wild-Type P53 Suppresses Transcription of the Human O6-Methylguanine-DNA Methyltransferase Gene. Cancer Res. 1996, 56, 2029–2032. [Google Scholar]
- Brennan, C.W.; Verhaak, R.G.W.; McKenna, A.; Campos, B.; Noushmehr, H.; Salama, S.R.; Zheng, S.; Chakravarty, D.; Sanborn, J.Z.; Berman, S.H.; et al. The Somatic Genomic Landscape of Glioblastoma. Cell 2013, 155, 462–477. [Google Scholar] [CrossRef]
- Verhaak, R.G.W.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated Genomic Analysis Identifies Clinically Relevant Subtypes of Glioblastoma Characterized by Abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef] [Green Version]
- Crespo, I.; Vital, A.L.; Gonzalez-Tablas, M.; del Carmen Patino, M.; Otero, A.; Lopes, M.C.; de Oliveira, C.; Domingues, P.; Orfao, A.; Tabernero, M.D. Molecular and Genomic Alterations in Glioblastoma Multiforme. Am. J. Pathol. 2015, 185, 1820–1833. [Google Scholar] [CrossRef] [Green Version]
- England, B.; Huang, T.; Karsy, M. Current Understanding of the Role and Targeting of Tumor Suppressor P53 in Glioblastoma Multiforme. Tumor Biol. 2013, 34, 2063–2074. [Google Scholar] [CrossRef]
- Vassilev, L.T. In Vivo Activation of the P53 Pathway by Small-Molecule Antagonists of MDM2. Science 2004, 303, 844–848. [Google Scholar] [CrossRef] [Green Version]
- Her, N.-G.; Oh, J.-W.; Oh, Y.J.; Han, S.; Cho, H.J.; Lee, Y.; Ryu, G.H.; Nam, D.-H. Potent Effect of the MDM2 Inhibitor AMG232 on Suppression of Glioblastoma Stem Cells. Cell Death Dis. 2018, 9, 792. [Google Scholar] [CrossRef]
- Reifenberger, G.; Liu, L.; Ichimura, K.; Schmidt, E.E.; Collins, V.P. Amplification and Overexpression of the MDM2 Gene in a Subset of Human Malignant Gliomas without P53 Mutations. Cancer Res. 1993, 53, 2736–2739. [Google Scholar]
- Verreault, M.; Schmitt, C.; Goldwirt, L.; Pelton, K.; Haidar, S.; Levasseur, C.; Guehennec, J.; Knoff, D.; Labussiere, M.; Marie, Y.; et al. Preclinical Efficacy of the MDM2 Inhibitor RG7112 in MDM2-Amplified and TP53 Wild-Type Glioblastomas. Clin. Cancer Res. 2016, 22, 1185–1196. [Google Scholar] [CrossRef] [Green Version]
- Berberich, A.; Kessler, T.; Thomé, C.M.; Pusch, S.; Hielscher, T.; Sahm, F.; Oezen, I.; Schmitt, L.-M.; Ciprut, S.; Hucke, N.; et al. Targeting Resistance against the MDM2 Inhibitor RG7388 in Glioblastoma Cells by the MEK Inhibitor Trametinib. Clin. Cancer Res. 2019, 25, 253–265. [Google Scholar] [CrossRef] [Green Version]
- Fishlock, D.; Diodone, R.; Hildbrand, S.; Kuhn, B.; Mössner, C.; Peters, C.; Rege, P.D.; Rimmler, G.; Schantz, M. Efficient Industrial Synthesis of the MDM2 Antagonist Idasanutlin via a Cu(I)-Catalyzed [3+2] Asymmetric Cycloaddition. Chim. Int. J. Chem. 2018, 72, 492–500. [Google Scholar] [CrossRef]
- Nieberler, M.; Reuning, U.; Reichart, F.; Notni, J.; Wester, H.-J.; Schwaiger, M.; Weinmüller, M.; Räder, A.; Steiger, K.; Kessler, H. Exploring the Role of RGD-Recognizing Integrins in Cancer. Cancers 2017, 9, 116. [Google Scholar] [CrossRef]
- Ludwig, B.S.; Kessler, H.; Kossatz, S.; Reuning, U. RGD-Binding Integrins Revisited: How Recently Discovered Functions and Novel Synthetic Ligands (Re-)Shape an Ever-Evolving Field. Cancers 2021, 13, 1711. [Google Scholar] [CrossRef]
- Su, C.; Li, J.; Zhang, L.; Wang, H.; Wang, F.; Tao, Y.; Wang, Y.; Guo, Q.; Li, J.; Liu, Y.; et al. The Biological Functions and Clinical Applications of Integrins in Cancers. Front. Pharmacol. 2020, 11, 579068. [Google Scholar] [CrossRef]
- Malric, L.; Monferran, S.; Gilhodes, J.; Boyrie, S.; Dahan, P.; Skuli, N.; Sesen, J.; Filleron, T.; Kowalski-Chauvel, A.; Cohen-Jonathan Moyal, E.; et al. Interest of Integrins Targeting in Glioblastoma According to Tumor Heterogeneity and Cancer Stem Cell Paradigm: An Update. Oncotarget 2017, 8, 86947–86968. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blandin, A.-F.; Renner, G.; Lehmann, M.; Lelong-Rebel, I.; Martin, S.; Dontenwill, M. Β1 Integrins as Therapeutic Targets to Disrupt Hallmarks of Cancer. Front. Pharmacol. 2015, 6, 279. [Google Scholar] [CrossRef]
- Mas-Moruno, C.; Rechenmacher, F.; Kessler, H. Cilengitide: The First Anti-Angiogenic Small Molecule Drug Candidate. Design, Synthesis and Clinical Evaluation. Anticancer Agents Med. Chem. 2010, 10, 753–768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stupp, R.; Hegi, M.E.; Gorlia, T.; Erridge, S.C.; Perry, J.; Hong, Y.-K.; Aldape, K.D.; Lhermitte, B.; Pietsch, T.; Grujicic, D.; et al. Cilengitide Combined with Standard Treatment for Patients with Newly Diagnosed Glioblastoma with Methylated MGMT Promoter (CENTRIC EORTC 26071-22072 Study): A Multicentre, Randomised, Open-Label, Phase 3 Trial. Lancet Oncol. 2014, 15, 1100–1108. [Google Scholar] [CrossRef] [Green Version]
- Tucci, M.; Stucci, S.; Silvestris, F. Does Cilengitide Deserve Another Chance? Lancet Oncol. 2014, 15, e584–e585. [Google Scholar] [CrossRef]
- Ellert-Miklaszewska, A.; Poleszak, K.; Pasierbinska, M.; Kaminska, B. Integrin Signaling in Glioma Pathogenesis: From Biology to Therapy. Int. J. Mol. Sci. 2020, 21, 888. [Google Scholar] [CrossRef] [Green Version]
- Holmes, K.M.; Annala, M.; Chua, C.Y.X.; Dunlap, S.M.; Liu, Y.; Hugen, N.; Moore, L.M.; Cogdell, D.; Hu, L.; Nykter, M.; et al. Insulin-like Growth Factor-Binding Protein 2-Driven Glioma Progression Is Prevented by Blocking a Clinically Significant Integrin, Integrin-Linked Kinase, and NF- B Network. Proc. Natl. Acad. Sci. USA 2012, 109, 3475–3480. [Google Scholar] [CrossRef] [Green Version]
- Janouskova, H.; Maglott, A.; Leger, D.Y.; Bossert, C.; Noulet, F.; Guerin, E.; Guenot, D.; Pinel, S.; Chastagner, P.; Plenat, F.; et al. Integrin 5 1 Plays a Critical Role in Resistance to Temozolomide by Interfering with the P53 Pathway in High-Grade Glioma. Cancer Res. 2012, 72, 3463–3470. [Google Scholar] [CrossRef] [Green Version]
- Etienne-Selloum, N.; Prades, J.; Bello-Roufai, D.; Boone, M.; Sevestre, H.; Trudel, S.; Caillet, P.; Coutte, A.; Desenclos, C.; Constans, J.-M.; et al. Expression Analysis of A5 Integrin Subunit Reveals Its Upregulation as a Negative Prognostic Biomarker for Glioblastoma. Pharmaceuticals 2021, 14, 882. [Google Scholar] [CrossRef]
- Schaffner, F.; Ray, A.; Dontenwill, M. Integrin A5β1, the Fibronectin Receptor, as a Pertinent Therapeutic Target in Solid Tumors. Cancers 2013, 5, 27–47. [Google Scholar] [CrossRef] [Green Version]
- Renner, G.; Noulet, F.; Mercier, M.-C.; Choulier, L.; Etienne-Selloum, N.; Gies, J.-P.; Lehmann, M.; Lelong-Rebel, I.; Martin, S.; Dontenwill, M. Expression/Activation of A5β1 Integrin Is Linked to the β-Catenin Signaling Pathway to Drive Migration in Glioma Cells. Oncotarget 2016, 7, 62194–62207. [Google Scholar] [CrossRef] [Green Version]
- Ray, A.-M.; Schaffner, F.; Janouskova, H.; Noulet, F.; Rognan, D.; Lelong-Rebel, I.; Choulier, L.; Blandin, A.-F.; Lehmann, M.; Martin, S.; et al. Single Cell Tracking Assay Reveals an Opposite Effect of Selective Small Non-Peptidic A5β1 or Avβ3/Β5 Integrin Antagonists in U87MG Glioma Cells. Biochim. Biophys. Acta BBA Gen. Subj. 2014, 1840, 2978–2987. [Google Scholar] [CrossRef]
- Dudvarski Stanković, N.; Bicker, F.; Keller, S.; Jones, D.T.; Harter, P.N.; Kienzle, A.; Gillmann, C.; Arnold, P.; Golebiewska, A.; Keunen, O.; et al. EGFL7 Enhances Surface Expression of Integrin α 5 β 1 to Promote Angiogenesis in Malignant Brain Tumors. EMBO Mol. Med. 2018, 10, e8420. [Google Scholar] [CrossRef]
- Malric, L.; Monferran, S.; Delmas, C.; Arnauduc, F.; Dahan, P.; Boyrie, S.; Deshors, P.; Lubrano, V.; Da Mota, D.F.; Gilhodes, J.; et al. Inhibiting Integrin Β8 to Differentiate and Radiosensitize Glioblastoma-Initiating Cells. Mol. Cancer Res. 2019, 17, 384–397. [Google Scholar] [CrossRef] [Green Version]
- DeLay, M.; Jahangiri, A.; Carbonell, W.S.; Hu, Y.-L.; Tsao, S.; Tom, M.W.; Paquette, J.; Tokuyasu, T.A.; Aghi, M.K. Microarray Analysis Verifies Two Distinct Phenotypes of Glioblastomas Resistant to Antiangiogenic Therapy. Clin. Cancer Res. 2012, 18, 2930–2942. [Google Scholar] [CrossRef] [Green Version]
- Martin, S.; Janouskova, H.; Dontenwill, M. Integrins and P53 Pathways in Glioblastoma Resistance to Temozolomide. Front. Oncol. 2012, 2, 157. [Google Scholar] [CrossRef] [Green Version]
- Renner, G.; Janouskova, H.; Noulet, F.; Koenig, V.; Guerin, E.; Bär, S.; Nuesch, J.; Rechenmacher, F.; Neubauer, S.; Kessler, H.; et al. Integrin A5β1 and P53 Convergent Pathways in the Control of Anti-Apoptotic Proteins PEA-15 and Survivin in High-Grade Glioma. Cell Death Differ. 2016, 23, 640–653. [Google Scholar] [CrossRef] [Green Version]
- Heckmann, D.; Meyer, A.; Laufer, B.; Zahn, G.; Stragies, R.; Kessler, H. Rational Design of Highly Active and Selective Ligands for the A5β1 Integrin Receptor. ChemBioChem 2008, 9, 1397–1407. [Google Scholar] [CrossRef]
- Rechenmacher, F.; Neubauer, S.; Polleux, J.; Mas-Moruno, C.; De Simone, M.; Cavalcanti-Adam, E.A.; Spatz, J.P.; Fässler, R.; Kessler, H. Functionalizing Avβ3- or A5β1-Selective Integrin Antagonists for Surface Coating: A Method to Discriminate Integrin Subtypes In Vitro. Angew. Chem. Int. Ed. 2013, 52, 1572–1575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merlino, F.; Daniele, S.; La Pietra, V.; Di Maro, S.; Di Leva, F.S.; Brancaccio, D.; Tomassi, S.; Giuntini, S.; Cerofolini, L.; Fragai, M.; et al. Simultaneous Targeting of RGD-Integrins and Dual Murine Double Minute Proteins in Glioblastoma Multiforme. J. Med. Chem. 2018, 61, 4791–4809. [Google Scholar] [CrossRef] [PubMed]
- Blandin, A.-F.; Noulet, F.; Renner, G.; Mercier, M.-C.; Choulier, L.; Vauchelles, R.; Ronde, P.; Carreiras, F.; Etienne-Selloum, N.; Vereb, G.; et al. Glioma Cell Dispersion Is Driven by A5 Integrin-Mediated Cell–Matrix and Cell–Cell Interactions. Cancer Lett. 2016, 376, 328–338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aasland, D.; Götzinger, L.; Hauck, L.; Berte, N.; Meyer, J.; Effenberger, M.; Schneider, S.; Reuber, E.E.; Roos, W.P.; Tomicic, M.T.; et al. Temozolomide Induces Senescence and Repression of DNA Repair Pathways in Glioblastoma Cells via Activation of ATR–CHK1, P21, and NF-ΚB. Cancer Res. 2019, 79, 99–113. [Google Scholar] [CrossRef] [Green Version]
- Lathia, J.D.; Gallagher, J.; Heddleston, J.M.; Wang, J.; Eyler, C.E.; MacSwords, J.; Wu, Q.; Vasanji, A.; McLendon, R.E.; Hjelmeland, A.B.; et al. Integrin Alpha 6 Regulates Glioblastoma Stem Cells. Cell Stem Cell 2010, 6, 421–432. [Google Scholar] [CrossRef] [Green Version]
- Guerrero, P.A.; Tchaicha, J.H.; Chen, Z.; Morales, J.E.; McCarty, N.; Wang, Q.; Sulman, E.P.; Fuller, G.; Lang, F.F.; Rao, G.; et al. Glioblastoma Stem Cells Exploit the Avβ8 Integrin-TGFβ1 Signaling Axis to Drive Tumor Initiation and Progression. Oncogene 2017, 36, 6568–6580. [Google Scholar] [CrossRef] [Green Version]
- Stupp, R.; Hegi, M.E.; Mason, W.P.; van den Bent, M.J.; Taphoorn, M.J.; Janzer, R.C.; Ludwin, S.K.; Allgeier, A.; Fisher, B.; Belanger, K.; et al. Effects of Radiotherapy with Concomitant and Adjuvant Temozolomide versus Radiotherapy Alone on Survival in Glioblastoma in a Randomised Phase III Study: 5-Year Analysis of the EORTC-NCIC Trial. Lancet Oncol. 2009, 10, 459–466. [Google Scholar] [CrossRef]
- Fisher, J.P.; Adamson, D.C. Current FDA-Approved Therapies for High-Grade Malignant Gliomas. Biomedicines 2021, 9, 324. [Google Scholar] [CrossRef]
- Ortiz, R.; Perazzoli, G.; Cabeza, L.; Jiménez-Luna, C.; Luque, R.; Prados, J.; Melguizo, C. Temozolomide: An Updated Overview of Resistance Mechanisms, Nanotechnology Advances and Clinical Applications. Curr. Neuropharmacol. 2021, 19, 513–537. [Google Scholar] [CrossRef]
- Lee, S.Y. Temozolomide Resistance in Glioblastoma Multiforme. Genes Dis. 2016, 3, 198–210. [Google Scholar] [CrossRef] [Green Version]
- Lang, F.; Liu, Y.; Chou, F.-J.; Yang, C. Genotoxic Therapy and Resistance Mechanism in Gliomas. Pharmacol. Ther. 2021, 228, 107922. [Google Scholar] [CrossRef]
- Faget, D.V.; Ren, Q.; Stewart, S.A. Unmasking Senescence: Context-Dependent Effects of SASP in Cancer. Nat. Rev. Cancer 2019, 19, 439–453. [Google Scholar] [CrossRef]
- Putavet, D.A.; de Keizer, P.L.J. Residual Disease in Glioma Recurrence: A Dangerous Liaison with Senescence. Cancers 2021, 13, 1560. [Google Scholar] [CrossRef]
- Martinkova, E.; Maglott, A.; Leger, D.Y.; Bonnet, D.; Stiborova, M.; Takeda, K.; Martin, S.; Dontenwill, M. A5β1 Integrin Antagonists Reduce Chemotherapy-Induced Premature Senescence and Facilitate Apoptosis in Human Glioblastoma Cells. Int. J. Cancer 2010, 127, 1240–1248. [Google Scholar] [CrossRef]
- Li, L.; Hu, Y.; Ylivinkka, I.; Li, H.; Chen, P.; Keski-Oja, J.; Hyytiäinen, M. NETRIN-4 Protects Glioblastoma Cells FROM Temozolomide Induced Senescence. PLoS ONE 2013, 8, e80363. [Google Scholar] [CrossRef] [Green Version]
- Rabé, M.; Dumont, S.; Álvarez-Arenas, A.; Janati, H.; Belmonte-Beitia, J.; Calvo, G.F.; Thibault-Carpentier, C.; Séry, Q.; Chauvin, C.; Joalland, N.; et al. Identification of a Transient State during the Acquisition of Temozolomide Resistance in Glioblastoma. Cell Death Dis. 2020, 11, 19. [Google Scholar] [CrossRef]
- Serafim, R.B.; da Silva, P.; Cardoso, C.; Di Cristofaro, L.F.M.; Netto, R.P.; de Almeida, R.; Navegante, G.; Storti, C.B.; de Sousa, J.F.; de Souza, F.C.; et al. Expression Profiling of Glioblastoma Cell Lines Reveals Novel Extracellular Matrix-Receptor Genes Correlated with the Responsiveness of Glioma Patients to Ionizing Radiation. Front. Oncol. 2021, 11, 668090. [Google Scholar] [CrossRef]
- Auffinger, B.; Tobias, A.L.; Han, Y.; Lee, G.; Guo, D.; Dey, M.; Lesniak, M.S.; Ahmed, A.U. Conversion of Differentiated Cancer Cells into Cancer Stem-like Cells in a Glioblastoma Model after Primary Chemotherapy. Cell Death Differ. 2014, 21, 1119–1131. [Google Scholar] [CrossRef]
- Dahan, P.; Martinez Gala, J.; Delmas, C.; Monferran, S.; Malric, L.; Zentkowski, D.; Lubrano, V.; Toulas, C.; Cohen-Jonathan Moyal, E.; Lemarie, A. Ionizing Radiations Sustain Glioblastoma Cell Dedifferentiation to a Stem-like Phenotype through Survivin: Possible Involvement in Radioresistance. Cell Death Dis. 2014, 5, e1543. [Google Scholar] [CrossRef] [Green Version]
- Carbonell, W.S.; DeLay, M.; Jahangiri, A.; Park, C.C.; Aghi, M.K. Β1 Integrin Targeting Potentiates Antiangiogenic Therapy and Inhibits the Growth of Bevacizumab-Resistant Glioblastoma. Cancer Res. 2013, 73, 3145–3154. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Cai, S.; Bailey, B.J.; Reza Saadatzadeh, M.; Ding, J.; Tonsing-Carter, E.; Georgiadis, T.M.; Zachary Gunter, T.; Long, E.C.; Minto, R.E.; et al. Combination Therapy in a Xenograft Model of Glioblastoma: Enhancement of the Antitumor Activity of Temozolomide by an MDM2 Antagonist. J. Neurosurg. 2017, 126, 446–459. [Google Scholar] [CrossRef] [PubMed] [Green Version]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antibody | Blocking Solution | Antibody Dilution |
|---|---|---|
| Anti-α5 integrin H104 (Santa Cruz) | 5% milk/1 × TBS/0.1% Tween -20 | 1/1000 |
| Anti-β1 integrin TS2/16 (Millipore) | 5% milk/1 × TBS/0.1% Tween -20 | 1/1000 |
| Anti-αv integrin (Cell signalling) | 5% milk/1 × TBS/0.1% Tween -20 | 1/1000 |
| Anti-β3 integrin (Cell signalling) | 5% milk/1 × TBS/0.1% Tween -20 | 1/1000 |
| Anti-p53 (BD Bioscience) | 5% milk/1 × TBS/0.1% Tween -20 | 1/1000 |
| Anti-pp53ser15 (Cell signalling) | 5% BSA/1 × TBS/0.1% Tween -20 | 1/1000 |
| Anti-p16 (Cell signalling) | 5% milk/1 × TBS/0.1% Tween -20 | 1/1000 |
| Anti-p21 (Cell signalling) | 5% milk/1 × TBS/0.1% Tween -20 | 1/1000 |
| Anti-MDM2 (Calbiochem) | 5% milk/1 × PBS/0.1% Tween -20 | 1/1000 |
| Anti-GAPDH (Millipore) | 5% milk/1 × TBS/0.1% Tween -20 | 1/5000 |
| Anti-tubulin (Sigma-Aldrich) | 5% milk/1 × TBS/0.1% Tween -20 | 1/3000 |
| Mouse HRP-conjugated secondary antibody | 5% milk/1 × TBS/0.1% Tween -20 | 1/10,000 |
| Rabbit HRP-conjugated secondary antibody | 5% milk/1 × TBS/0.1% Tween -20 | 1/10,000 |
| Treatment | U87MG Cells | U87MG R50 OFF Cells | U87MG R50 |
|---|---|---|---|
| Control | 1 | 1 | 1 |
| TMZ | 0.57 ± 0.01 * | 0.94 ± 0.02 ns | - |
| K34c | 0.44 ± 0.01 * | 0.54 ± 0.03 * | 0.65 ± 0.04 * |
| FR248 | 0.78 ± 0.04 * | 0.78 ± 0.04 * | 0.86 ± 0.09 ns |
| Nutlin-3a | 0.35 ± 0.01 * | 0.42 ± 0.02 * | 0.42 ± 0.01 * |
| Idasanutlin | 0.52 ± 0.05 * | 0.45 ± 0.02 * | _ |
| Nutlin-3a + K34c | 0.21 ± 0.01 * # | 0.24 ± 0.01 * # | 0.25 ± 0.008 * # |
| Nutlin-3a + FR248 | 0.28 ± 0.02 * # | 0.33 ± 0.01 * # | 0.37 ± 0.01 * ns |
| Idasanutlin + K34c | 0.265 ± 0.005 * # | 0.244 ± 0.007 * # | - |
| Idasanutlin + FR248 | 0.399 ± 0.005 * # | 0.319 ± 0.009 * # | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sani, S.; Pallaoro, N.; Messe, M.; Bernhard, C.; Etienne-Selloum, N.; Kessler, H.; Marinelli, L.; Entz-Werle, N.; Foppolo, S.; Martin, S.; et al. Temozolomide-Acquired Resistance Is Associated with Modulation of the Integrin Repertoire in Glioblastoma, Impact of α5β1 Integrin. Cancers 2022, 14, 369. https://doi.org/10.3390/cancers14020369
Sani S, Pallaoro N, Messe M, Bernhard C, Etienne-Selloum N, Kessler H, Marinelli L, Entz-Werle N, Foppolo S, Martin S, et al. Temozolomide-Acquired Resistance Is Associated with Modulation of the Integrin Repertoire in Glioblastoma, Impact of α5β1 Integrin. Cancers. 2022; 14(2):369. https://doi.org/10.3390/cancers14020369
Chicago/Turabian StyleSani, Saidu, Nikita Pallaoro, Mélissa Messe, Chloé Bernhard, Nelly Etienne-Selloum, Horst Kessler, Luciana Marinelli, Natacha Entz-Werle, Sophie Foppolo, Sophie Martin, and et al. 2022. "Temozolomide-Acquired Resistance Is Associated with Modulation of the Integrin Repertoire in Glioblastoma, Impact of α5β1 Integrin" Cancers 14, no. 2: 369. https://doi.org/10.3390/cancers14020369