
Bone Metastatic Breast Cancer: Advances in Cell Signaling and Autophagy Related Mechanisms


Abstract
:Simple Summary
Abstract
1. Introduction
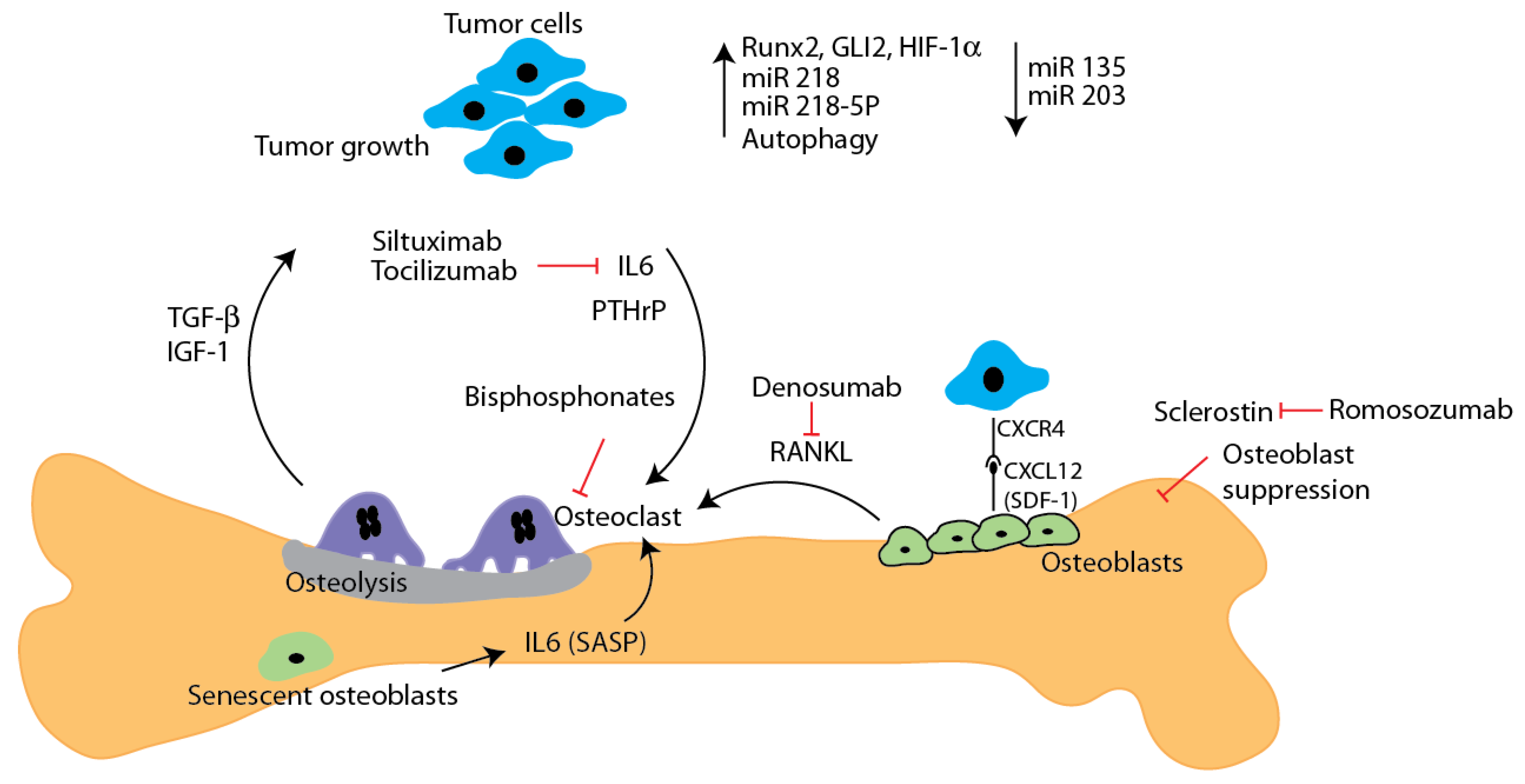
2. Bone Metastasis
2.1. Metastatic Potential and Organ Tropism
2.2. Disease Onset and Progression
3. Regulatory Mechanisms
3.1. Tumor-Derived Factors
3.1.1. Cell Signaling
3.1.2. Tumor Microenvironment Derived Factors
4. Regulatory Pathways
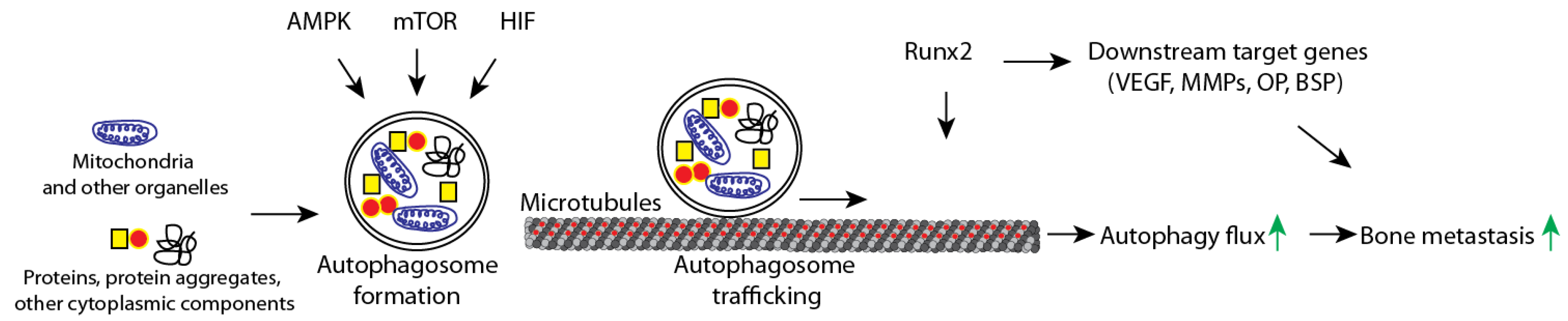
4.1. Autophagy Dysregulation and Metastasis
4.2. Micro RNA and Metastasis

{kind=link}
{kind=link}
| Factor | Functional Description | Reference |
|---|---|---|
| Signaling/Secreted Factors | ||
| ADM | Potentiates osteolytic responses in bone to metastatic breast cancer | [92] |
| ANGPTL2 | Up-regulates CXCR4 expression in tumor cells, enhancing responsiveness of breast cancer cells to bone tissues secreting CXCL12 | [93] |
| DKK1 | Promotes bone metastasis by regulating canonical WNT signaling of osteoblasts | [94] |
| IL-1β | Stimulates breast cancer colonization by inducing NFkB/CREB-Wnt signaling | [95] |
| IL-6 | Inhibition of IL-6 reduces MDA-231 bone metastasis by inhibiting cell proliferation and decreasing expression of P-Stat3, VEGF, and RANK | [96] |
| IL-11 | Promotes bone metastasis through JAK1/STAT3 signaling pathway in BoM-1833 cells | [97] |
| ITGBL1 | Facilitates recruitment, residence, and growth of breast cancer in bone | [98] |
| Jagged1 | Tumor-derived ligand that activates Notch signaling in bone, promoting IL-6 secretion and subsequent osteolytic bone metastasis | [63,99] |
| OPN | Enhances ability of CD44+ breast cancer cells to migrate to the bone, potentially through activation of WINK-1 and PRAS40-related pathways | [100] |
| Sclerostin | Promotes cell migration, invasion, and bone osteolysis | [60] |
| VCAM-1 | Promotes metastasis by interacting and recruiting α4β1-positive osteoclast progenitors | [101] |
| Transcription Factors | ||
| HIF-1α | Promotes metastatic spread by upregulating PTGS2/COX-2 and by increasing expression of CXCL12 in osteoprogenitor cells | [102,103] |
| Runx2 | An essential regulator of skeletal development, Runx2 is highly expressed in breast cancer skeletal metastases, and is associated with tumor-induced osteolysis. Runx2 activity is promoted via the PI3K/AKT signaling pathway. Evidence shows that expression in bone metastasis is regulated by miR-135 and miR-203. Functions also in increasing cell proliferation via disrupting growth-arresting acini structures, and promoting cell survival by enhancing the autophagic process. | [20,42,48,49,50,51,52,54,58,59,85] |
| GLI-2 | Increases secretion of osteolytic factors such as parathyroid hormone-related protein (PTHrP) | [46,47] |
| STAT3 | Contributes to migration, invasion, and angiogenesis | [104] |
| Micro RNA | ||
| miRNA 135 miRNA 203 | Both show diminished expression in bone metastatic MDA-231 cells, and are related to aberrant expression of Runx2 | [88] |
| miRNA 218-5P | Highly expressed in bone metastatic breast cancer cells, functioning to promote WNT signaling and enhance osteolysis | [89] |
| Enzymatic Proteins | ||
| ADAMTS1 MMP1 | Modulate bone microenvironment in favor of osteoclastogenesis and bone metastasis | [105] |
| MMP13 | Upregulation contributes to osteoclast differentiation by activating MMP-9 and promoting cleavage of galectin-3 | [106] |
| MFAP5 | Increases and accelerates bone metastasis, possibly by increasing expression of MMP2, MMP9, and activating the ERK signaling pathway | [107] |
| Receptors and Growth Factors | ||
| βAR | βAR stimulation in osteoblasts may activate bone marrow vessels to favor skeletal engraftment of breast cancer cells | [108] |
| BMPR1a | BMPR1a promotes osteolytic metastasis of breast cancer cells by promoting RANKL production via the p38 pathway | [109] |
| FGFR1/FGF | FGFR1 activation by tumor cell-derived FGF ligands enhance osteoclast function, contributing to metastatic lesions | [110] |
| Notch | Activation of Notch signaling in bone microenvironment via tumor-derived Jagged1 promotes osteoclast differentiation and facilitates metastasis by initiation EMT. Activation via tumor-derived Galectin-3 has been shown to inhibit osteoblast differentiation | [63,99,111] |
| IGF-1R/IGF | IGF-1R activation by bone-derived IGF stimulates bone metastasis via activation of the Akt/NF-κB signaling pathway | [112] |
| C-Met/HGF | C-Met and cognate ligand HGF expression in bone correlates with bone metastasis | [113] |
| RANK/RANKL | RANKL expression and RANK activation is induced by tumor-derived factors, promoting osteoclast activity and increasing bone invasiveness | [114] |
| TGF-β | TGF-β released in bone matrix upon tumor-induced osteolysis further stimulates bone metastatic cells to secrete factors driving osteolytic bone destruction | [115] |
| VEGF | VEGF is expressed strongly in breast cancer metastases, and in the presence of RANKL can stimulate formation of osteoclasts | [116] |
| Autophagy | ||
| ATG7 ATG12 | ATG7 and ATG12 inhibition in MDA-231 cells resulted in decreased colony formation and proliferation | [77] |
| Rab5a | Rab5a expression is elevated in metastasized 1833 cells, and may be related to cell survival by triggering autophagy and autophagosome sealing | [84] |
| Chemokines | ||
| COX-2/CXCR2 | CXCR2 enhances breast cancer metastasis to bone by suppressing AKT1 and activating COX-2 | [117] |
| CXCL10/CXCR3 | CXCL10/CXCR3 axis contributes to breast cancer metastasis and osteoclast activation in 4T1 cells | [118] |
| CXCL12/CXCR4 | CXCL12/CXCR4 axis promotes breast cancer metastasis to tissues expressing high levels of CXCL12 | [119] |
5. Metastatic Bone Disease: Clinical Management and Perspectives
5.1. Therapeutic Approaches to Managing Metastatic Bone Disease
5.2. Current Challenges and Future Directions
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Howlader, N.; Noone, A.M.; Krapcho, M.; Garshell, J.; Miller, D.; Altekruse, S.F.; Kosary, C.L.; Yu, M.; Ruhl, J.; Tatalovich, Z. SEER Cancer Statistics Review, 1975–2012; National Cancer Institute: Bethesda, MD, USA, 2014. [Google Scholar]
- Wang, H.; Zhang, C.; Zhang, J.; Kong, L.; Zhu, H.; Yu, J. The prognosis analysis of different metastasis pattern in patients with different breast cancer subtypes: A SEER based study. Oncotarget 2016, 8, 26368–26379. [Google Scholar] [CrossRef] [PubMed]
- Gong, Y.; Liu, Y.-R.; Ji, P.; Hu, X.; Shao, Z.-M. Impact of molecular subtypes on metastatic breast cancer patients: A SEER population-based study. Sci. Rep. 2017, 7, srep45411. [Google Scholar] [CrossRef] [Green Version]
- Molnár, I.A.; Molnár, B.Á.; Vízkeleti, L.; Fekete, K.; Tamás, J.; Deák, P.; Szundi, C.; Székely, B.; Moldvay, J.; Vári-Kakas, S.; et al. Breast carcinoma subtypes show different patterns of metastatic behavior. Virchows Archiv 2017, 470, 275–283. [Google Scholar] [CrossRef]
- Wu, Q.; Li, J.; Zhu, S.; Wu, J.; Chen, C.; Liu, Q.; Wei, W.; Zhang, Y.; Sun, S. Breast cancer subtypes predict the preferential site of distant metastases: A SEER based study. Oncotarget 2017, 8, 27990–27996. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weilbaecher, K.N.; Guise, T.A.; McCauley, L.K. Cancer to bone: A fatal attraction. Nat. Rev. Cancer 2011, 11, 411–425. [Google Scholar] [CrossRef]
- Roodman, G.D. Biology of Osteoclast Activation in Cancer. J. Clin. Oncol. 2001, 19, 3562–3571. [Google Scholar] [CrossRef]
- Ryan, C.; Stoltzfus, K.C.; Horn, S.; Chen, H.; Louie, A.V.; Lehrer, E.J.; Trifiletti, D.M.; Fox, E.J.; Abraham, J.A.; Zaorsky, N.G. Epidemiology of bone metastases. Bone 2020, 115783. [Google Scholar] [CrossRef] [PubMed]
- O’Shaughnessy, J. Extending Survival with Chemotherapy in Metastatic Breast Cancer. Oncologist 2005, 10, 20–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Meglio, A.; Freedman, R.A.; Lin, N.U.; Barry, W.T.; Metzger-Filho, O.; Keating, N.L.; King, T.A.; Sertoli, M.R.; Boccardo, F.; Winer, E.P.; et al. Time trends in incidence rates and survival of newly diagnosed stage IV breast cancer by tumor histology: A population-based analysis. Breast Cancer Res. Treat. 2016, 157, 587–596. [Google Scholar] [CrossRef]
- Hadji, P.; Coleman, R.E.; Wilson, C.; Powles, T.J.; Clézardin, P.; Aapro, M.; Costa, L.; Body, J.-J.; Markopoulos, C.; Santini, D.; et al. Adjuvant bisphosphonates in early breast cancer: Consensus guidance for clinical practice from a European Panel. Ann. Oncol. 2016, 27, 379–390. [Google Scholar] [CrossRef]
- O’Carrigan, B.; Wong, M.H.; Willson, M.L.; Stockler, M.R.; Pavlakis, N.; Goodwin, A. Bisphosphonates and other bone agents for breast cancer. Cochrane Database Syst. Rev. 2017, 2018, CD003474. [Google Scholar] [CrossRef]
- Coleman, R.; Body, J.J.; Aapro, M.; Hadji, P.; Herrstedt, J. Bone health in cancer patients: ESMO Clinical Practice Guidelines. Ann. Oncol. 2014, 25, iii124–iii137. [Google Scholar] [CrossRef] [PubMed]
- Von Moos, R.; von Moos, R.; Costa, L.; Costa, L.; Ripamonti, C.I.; Ripamonti, C.I.; Niepel, D.; Niepel, D.; Santini, D.; Santini, D.; et al. Improving quality of life in patients with advanced cancer: Targeting metastatic bone pain. Eur. J. Cancer 2017, 71, 80–94. [Google Scholar] [CrossRef] [Green Version]
- Martin, M.; Bell, R.; Bourgeois, H.; Brufsky, A.; Diel, I.; Eniu, A.; Fallowfield, L.; Fujiwara, Y.; Jassem, J.; Paterson, A.H.; et al. Bone-Related Complications and Quality of Life in Advanced Breast Cancer: Results from a Randomized Phase III Trial of Denosumab versus Zoledronic Acid. Clin. Cancer Res. 2012, 18, 4841–4849. [Google Scholar] [CrossRef] [Green Version]
- Schulman, K.L.; Kohles, J. Economic burden of metastatic bone disease in the U.S. Cancer 2007, 109, 2334–2342. [Google Scholar] [CrossRef]
- Coleman, R.E. Bisphosphonates: Clinical Experience. Oncologist 2004, 9, 14–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blank, A.T.; Lerman, D.M.; Patel, N.; Rapp, T.B. Is Prophylactic Intervention More Cost-effective Than the Treatment of Pathologic Fractures in Metastatic Bone Disease? Clin. Orthop. Relat. Res. 2016, 474, 1563–1570. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. The Hallmarks of Cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef] [Green Version]
- Pratap, J.; Lian, J.B.; Javed, A.; Barnes, G.L.; Van Wijnen, A.J.; Stein, J.L.; Stein, G.S. Regulatory roles of Runx2 in metastatic tumor and cancer cell interactions with bone. Cancer Metastasis Rev. 2006, 25, 589–600. [Google Scholar] [CrossRef] [PubMed]
- Gupta, G.P.; Minn, A.J.; Kang, Y.; Siegel, P.M.; Serganova, I.; Cordon-Cardo, C.; Olshen, A.B.; Gerald, W.L.; Massagué, J. Identifying site-specific metastasis genes and functions. Cold Spring Harb Symp Quant Biol. 2005, 70, 149–158. [Google Scholar]
- Ribatti, D.; Mangialardi, G.; Vacca, A. Stephen Paget and the ‘seed and soil’theory of metastatic dissemination. Clin. Exp. Med. 2006, 6, 145–149. [Google Scholar] [CrossRef]
- Fidler, I.J. The pathogenesis of cancer metastasis: The ‘seed and soil’ hypothesis revisited. Nat. Rev. Cancer 2003, 3, 453–458. [Google Scholar] [CrossRef]
- Press, D.; Miller, M.E.; Liederbach, E.; Yao, K.; Huo, D. De novo metastasis in breast cancer: Occurrence and overall survival stratified by molecular subtype. Clin. Exp. Metastasis 2017, 34, 457–465. [Google Scholar] [CrossRef] [PubMed]
- Coleman, R.E. Clinical Features of Metastatic Bone Disease and Risk of Skeletal Morbidity. Clin. Cancer Res. 2006, 12, 6243s–6249s. [Google Scholar] [CrossRef] [Green Version]
- Hage, W.D.; Aboulafia, A.J.; Aboulafia, D.M. Incidence, Location, and Diagnostic Evaluation of Metastatic Bone Disease. Orthop. Clin. N. Am. 2000, 31, 515–528. [Google Scholar] [CrossRef]
- Parkes, A.; Clifton, K.; Al-Awadhi, A.; Oke, O.; Warneke, C.L.; Litton, J.K.; Hortobagyi, G.N. Characterization of bone only metastasis patients with respect to tumor subtypes. NPJ Breast Cancer 2018, 4, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Parkes, A.; Warneke, C.L.; Clifton, K.; Al-Awadhi, A.; Oke, O.; Pestana, R.C.; Alhalabi, O.; Litton, J.K.; Hortobagyi, G.N. Prognostic Factors in Patients with Metastatic Breast Cancer with Bone-Only Metastases. Oncologist 2018, 23, 1282–1288. [Google Scholar] [CrossRef] [Green Version]
- Macedo, F.; Ladeira, K.; Pinho, F.; Saraiva, N.; Bonito, N.; Pinto, L.; Gonçalves, F. Bone metastases: An overview. Oncol. Rev. 2017, 11, 321. [Google Scholar] [CrossRef] [PubMed]
- Sowder, M.E.; Johnson, R.W. Bone as a Preferential Site for Metastasis. JBMR Plus 2019, 3, e10126. [Google Scholar] [CrossRef] [PubMed]
- Li, B.T.; Wong, M.H.; Pavlakis, N. Treatment and Prevention of Bone Metastases from Breast Cancer: A Comprehensive Review of Evidence for Clinical Practice. J. Clin. Med. 2014, 3, 1–24. [Google Scholar] [CrossRef] [Green Version]
- Soki, F.N.; Park, S.I.; McCauley, L.K. The multifaceted actions of PTHrP in skeletal metastasis. Futur. Oncol. 2012, 8, 803–817. [Google Scholar] [CrossRef] [Green Version]
- Chavez, K.J.; Garimella, S.V.; Lipkowitz, S. Triple negative breast cancer cell lines: One tool in the search for better treatment of triple negative breast cancer. Breast Dis. 2010, 32, 35–48. [Google Scholar] [CrossRef] [Green Version]
- Rodler, E.; Korde, L.; Gralow, J. Current treatment options in triple negative breast cancer. Breast Dis. 2011, 32, 99–122. [Google Scholar] [CrossRef]
- Ismail-Khan, R.; Bui, M.M. A Review of Triple-Negative Breast Cancer. Cancer Control 2010, 17, 173–176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richard, S.; Selle, F.; Lotz, J.-P.; Khalil, A.; Gligorov, J.; Soares, D.G. Pertuzumab and trastuzumab: The rationale way to synergy. An. Acad. Bras. Ciências 2016, 88, 565–577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Ruijter, T.C.; Veeck, J.; De Hoon, J.P.J.; Van Engeland, M.; Tjan-Heijnen, V.C. Characteristics of triple-negative breast cancer. J. Cancer Res. Clin. Oncol. 2010, 137, 183–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, S.-G.; Li, H.; Tang, L.-Y.; Sun, J.-Y.; Zhang, W.-W.; Li, F.-Y.; Chen, Y.-X.; He, Z.-Y. The effect of distant metastases sites on survival in de novo stage-IV breast cancer: A SEER database analysis. Tumor Biol. 2017, 39, 1010428317705082. [Google Scholar] [CrossRef] [Green Version]
- Schröder, J.; Fietz, T.; Köhler, A.; Petersen, V.; Tesch, H.; Spring, L.; Fleitz, A.; Jänicke, M.; Marschner, N. Treatment and pattern of bone metastases in 1094 patients with advanced breast cancer–Results from the prospective German Tumour Registry Breast Cancer cohort study. Eur. J. Cancer 2017, 79, 139–148. [Google Scholar] [CrossRef] [Green Version]
- Xiong, Z.; Deng, G.; Huang, X.; Li, X.; Xie, X.; Wang, J.; Shuang, Z.; Wang, X. Bone metastasis pattern in initial metastatic breast cancer: A population-based study. Cancer Manag. Res. 2018, 10, 287–295. [Google Scholar] [CrossRef] [Green Version]
- Hollestelle, A.; Elstrodt, F.; Nagel, J.H.; Kallemeijn, W.W.; Schutte, M. Phosphatidylinositol-3-OH Kinase or RAS Pathway Mutations in Human Breast Cancer Cell Lines. Mol. Cancer Res. 2007, 5, 195–201. [Google Scholar] [CrossRef] [Green Version]
- Tandon, M.; Othman, A.H.; Ashok, V.; Stein, G.S.; Pratap, J. The role of Runx2 in facilitating autophagy in metastatic breast cancer cells. J. Cell. Physiol. 2018, 233, 559–571. [Google Scholar] [CrossRef]
- Strohecker, A.M.; White, E. Targeting Mitochondrial Metabolism by Inhibiting Autophagy in BRAF-Driven Cancers. Cancer Discov. 2014, 4, 766–772. [Google Scholar] [CrossRef] [Green Version]
- Levy, J.M.M.; Thompson, J.C.; Griesinger, A.M.; Amani, V.; Donson, A.M.; Birks, D.K.; Morgan, M.J.; Mirsky, D.M.; Handler, M.H.; Foreman, N.K.; et al. Autophagy Inhibition Improves Chemosensitivity in BRAFV600E Brain Tumors. Cancer Discov. 2014, 4, 773–780. [Google Scholar] [CrossRef] [Green Version]
- Chen, N.; Karantza, V. Autophagy as a therapeutic target in cancer. Cancer Biol. Ther. 2011, 11, 157–168. [Google Scholar] [CrossRef]
- Johnson, R.W.; Nguyen, M.P.; Padalecki, S.S.; Grubbs, B.G.; Merkel, A.; Oyajobi, B.O.; Matrisian, L.M.; Mundy, G.R.; Sterling, J.A. TGF-β Promotion of Gli2-Induced Expression of Parathyroid Hormone-Related Protein, an Important Osteolytic Factor in Bone Metastasis, Is Independent of Canonical Hedgehog Signaling. Cancer Res. 2011, 71, 822–831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Javelaud, D.; Alexaki, V.I.; Dennler, S.; Mohammad, K.S.; Guise, T.A.; Mauviel, A. TGF-β/SMAD/GLI2 Signaling Axis in Cancer Progression and Metastasis. Cancer Res. 2011, 71, 5606–5610. [Google Scholar] [CrossRef] [Green Version]
- Bendinelli, P.; Maroni, P.; Matteucci, E.; Desiderio, M.A. Cell and Signal Components of the Microenvironment of Bone Metastasis Are Affected by Hypoxia. Int. J. Mol. Sci. 2016, 17, 706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teng, Y.; Ross, J.L.; Cowell, J.K. The involvement of JAK-STAT3 in cell motility, invasion, and metastasis. JAK-STAT 2014, 3, e28086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rankin, E.; Giaccia, A.J. Hypoxic control of metastasis. Science 2016, 352, 175–180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tandon, M.; Chen, Z.; Pratap, J. Runx2 activates PI3K/Akt signaling via mTORC2 regulation in invasive breast cancer cells. Breast Cancer Res. 2014, 16, R16. [Google Scholar] [CrossRef] [Green Version]
- McDonald, L.; Ferrari, N.; Terry, A.; Bell, M.; Mohammed, Z.M.; Orange, C.; Jenkins, A.; Muller, W.J.; Gusterson, B.A.; Neil, J.C.; et al. RUNX2 in subtype specific breast cancer and mammary gland differentiation. Dis. Model. Mech. 2014, 7, 525–534. [Google Scholar] [CrossRef] [Green Version]
- Tandon, M.; Chen, Z.; Othman, A.H.; Pratap, J. Role of Runx2 in IGF-1Rβ/Akt- and AMPK/Erk-dependent growth, survival and sensitivity towards metformin in breast cancer bone metastasis. Oncogene 2016, 35, 4730–4740. [Google Scholar] [CrossRef]
- Pratap, J.; Imbalzano, K.M.; Underwood, J.M.; Cohet, N.; Gokul, K.D.; Akech, J.; Van Wijnen, A.J.; Stein, J.L.; Imbalzano, A.N.; Nickerson, J.A.; et al. Ectopic Runx2 Expression in Mammary Epithelial Cells Disrupts Formation of Normal Acini Structure: Implications for Breast Cancer Progression. Cancer Res. 2009, 69, 6807–6814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barnes, G.L. Fidelity of Runx2 Activity in Breast Cancer Cells Is Required for the Generation of Metastases-Associated Osteolytic Disease. Cancer Res. 2004, 64, 4506–4513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hesse, E.; Schröder, S.; Brandt, D.; Pamperin, J.; Saito, H.; Taipaleenmäki, H. Sclerostin inhibition alleviates breast cancer–induced bone metastases and muscle weakness. JCI Insight 2019, 4, 5. [Google Scholar] [CrossRef]
- Mendoza-Villanueva, D.; Zeef, L.; Shore, P. Metastatic breast cancer cells inhibit osteoblast differentiation through the Runx2/CBFβ-dependent expression of the Wnt antagonist, sclerostin. Breast Cancer Res. 2011, 13, R106. [Google Scholar] [CrossRef] [Green Version]
- McDonald, M.M.; Delgado-Calle, J. Sclerostin: An Emerging Target for the Treatment of Cancer-Induced Bone Disease. Curr. Osteoporos. Rep. 2017, 15, 532–541. [Google Scholar] [CrossRef]
- Sousa, S.; Clézardin, P. Bone-Targeted Therapies in Cancer-Induced Bone Disease. Calcif. Tissue Int. 2017, 102, 227–250. [Google Scholar] [CrossRef]
- Zhu, M.; Liu, C.; Li, S.; Zhang, S.; Yao, Q.; Song, Q. Sclerostin induced tumor growth, bone metastasis and osteolysis in breast cancer. Sci. Rep. 2017, 7, 11399. [Google Scholar] [CrossRef] [PubMed]
- Cohen-Solal, K.A.; Boregowda, R.K.; Lasfar, A. RUNX2 and the PI3K/AKT axis reciprocal activation as a driving force for tumor progression. Mol. Cancer 2015, 14, 137. [Google Scholar] [CrossRef] [Green Version]
- Tan, C.-C.; Li, G.-X.; Tan, L.-D.; Du, X.; Li, X.-Q.; He, R.; Wang, Q.-S.; Feng, Y.-M. Breast cancer cells obtain an osteomimetic feature via epithelial-mesenchymal transition that have undergone BMP2/RUNX2 signaling pathway induction. Oncotarget 2016, 7, 79688–79705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalez, D.M.; Medici, D. Signaling mechanisms of the epithelial-mesenchymal transition. Sci. Signal. 2014, 7, re8. [Google Scholar] [CrossRef] [Green Version]
- Akech, J.; Wixted, J.J.; Bedard, K.; Van Der Deen, M.; Hussain, S.; A Guise, T.; Van Wijnen, A.J.; Stein, J.L.; Languino, L.; Altieri, D.C.; et al. Runx2 association with progression of prostate cancer in patients: Mechanisms mediating bone osteolysis and osteoblastic metastatic lesions. Oncogene 2009, 29, 811–821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roodman, G.D. Mechanisms of Bone Metastasis. N. Engl. J. Med. 2004, 350, 1655–1664. [Google Scholar] [CrossRef] [PubMed]
- Sethi, N.; Dai, X.; Winter, C.G.; Kang, Y. Tumor-Derived Jagged1 Promotes Osteolytic Bone Metastasis of Breast Cancer by Engaging Notch Signaling in Bone Cells. Cancer Cell 2011, 19, 192–205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chirgwin, J.M.; Mohammad, K.S.; Guise, T.A. Tumor-bone cellular interactions in skeletal metastases. J. Musculoskelet. Neuronal Interact. 2004, 4, 308–318. [Google Scholar] [PubMed]
- Mundy, G.R. Metastasis to bone: Causes, consequences and therapeutic opportunities. Nat. Rev. Cancer 2002, 2, 584–593. [Google Scholar] [CrossRef]
- Kang, Y.; Siegel, P.M.; Shu, W.; Drobnjak, M.; Kakonen, S.M.; Cordón-Cardo, C.; Guise, T.A.; Massagué, J. A multigenic program mediating breast cancer metastasis to bone. Cancer Cell 2003, 3, 537–549. [Google Scholar] [CrossRef] [Green Version]
- Pantovic, A.; Krstic, J.; Janjetovic, K.; Kocic, J.; Harhaji-Trajkovic, L.; Bugarski, D.; Trajkovic, V. Coordinated time-dependent modulation of AMPK/Akt/mTOR signaling and autophagy controls osteogenic differentiation of human mesenchymal stem cells. Bone 2013, 52, 524–531. [Google Scholar] [CrossRef]
- Fung, C.; Lock, R.; Gao, S.; Salas, E.; Debnath, J. Induction of Autophagy during Extracellular Matrix Detachment Promotes Cell Survival. Mol. Biol. Cell 2008, 19, 797–806. [Google Scholar] [CrossRef] [Green Version]
- Yu, C.; Wang, H.; Muscarella, A.; Goldstein, A.; Zeng, H.-C.; Bae, Y.; Lee, B.H.I.; Zhang, X.H.-F. Intra-iliac Artery Injection for Efficient and Selective Modeling of Microscopic Bone Metastasis. J. Vis. Exp. 2016, 115, e53982. [Google Scholar] [CrossRef]
- Wright, L.; Ottewell, P.; Rucci, N.; Peyruchaud, O.; Pagnotti, G.M.; Chiechi, A.; Buijs, J.; Sterling, J.A. Murine models of breast cancer bone metastasis. BoneKEy Rep. 2016, 5, 804. [Google Scholar] [CrossRef] [Green Version]
- Kuchimaru, T.; Kataoka, N.; Nakagawa, K.; Isozaki, T.; Miyabara, H.; Minegishi, M.; Kadonosono, T.; Kizaka-Kondoh, S. A reliable murine model of bone metastasis by injecting cancer cells through caudal arteries. Nat. Commun. 2018, 9, 2981. [Google Scholar] [CrossRef] [Green Version]
- Martin, J.W.; Zielenska, M.; Stein, G.S.; Van Wijnen, A.J.; Squire, J.A. The Role of RUNX2 in Osteosarcoma Oncogenesis. Sarcoma 2010, 2011, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Kenific, C.M.; Thorburn, A.; Debnath, J. Autophagy and metastasis: Another double-edged sword. Curr. Opin. Cell Biol. 2010, 22, 241–245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lock, R.; Roy, S.; Kenific, C.M.; Su, J.S.; Salas, E.; Ronen, S.M.; Debnath, J. Autophagy facilitates glycolysis during Ras-mediated oncogenic transformation. Mol. Biol. Cell 2011, 22, 165–178. [Google Scholar] [CrossRef]
- Mori, S.; Chang, J.T.; Andrechek, E.; Matsumura, N.; Baba, T.; Yao, G.; Kim, J.W.; Gatza, M.; Murphy, S.; Nevins, J.R. Anchorage-independent cell growth signature identifies tumors with metastatic potential. Oncogene 2009, 28, 2796–2805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lock, R.; Kenific, C.M.; Leidal, A.M.; Salas, E.; Debnath, J. Autophagy-Dependent Production of Secreted Factors Facilitates Oncogenic RAS-Driven Invasion. Cancer Discov. 2014, 4, 466–479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, X.; Fu, Y.; Loza, A.J.; Murali, B.; Leahy, K.M.; Ruhland, M.K.; Gang, M.; Su, X.; Zamani, A.; Shi, Y.; et al. Stromal-Initiated Changes in the Bone Promote Metastatic Niche Development. Cell Rep. 2016, 14, 82–92. [Google Scholar] [CrossRef] [Green Version]
- Harmer, D.; Falank, C.; Reagan, M.R. Interleukin-6 Interweaves the Bone Marrow Microenvironment, Bone Loss, and Multiple Myeloma. Front. Endocrinol. 2019, 9, 788. [Google Scholar] [CrossRef] [Green Version]
- Tandon, M.; Gokul, K.; A Ali, S.; Chen, Z.; Lian, J.; Stein, G.S.; Pratap, J. Runx2 mediates epigenetic silencing of the bone morphogenetic protein-3B (BMP-3B/GDF10) in lung cancer cells. Mol. Cancer 2012, 11, 27. [Google Scholar] [CrossRef] [Green Version]
- Leong, D.T.; Lim, J.; Goh, X.; Pratap, J.; Pereira, B.P.; Kwok, H.S.; Nathan, S.S.; Dobson, J.R.; Lian, J.B.; Ito, Y.; et al. Cancer-related ectopic expression of the bone-related transcription factor RUNX2 in non-osseous metastatic tumor cells is linked to cell proliferation and motility. Breast Cancer Res. 2010, 12, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Maroni, P.; Bendinelli, P.; Resnati, M.; Matteucci, E.; Milan, E.; Desiderio, M.A. The Autophagic Process Occurs in Human Bone Metastasis and Implicates Molecular Mechanisms Differently Affected by Rab5a in the Early and Late Stages. Int. J. Mol. Sci. 2016, 17, 443. [Google Scholar] [CrossRef] [Green Version]
- Nnah, I.C.; Wang, B.; Saqcena, C.; Weber, G.F.; Bonder, E.M.; Bagley, D.; De Cegli, R.; Napolitano, G.; Medina, D.L.; Ballabio, A.; et al. TFEB-driven endocytosis coordinates MTORC1 signaling and autophagy. Autophagy 2018, 15, 151–164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, R.; Wang, M.; Zhao, C.; Shen, M.; Yu, Y.; He, L.; Zhao, Y.; Chen, H.; Shi, X.; Zhou, M.; et al. TFEB-driven autophagy potentiates TGF-β induced migration in pancreatic cancer cells. J. Exp. Clin. Cancer Res. 2019, 38, 281. [Google Scholar] [CrossRef]
- Hesse, E.; Taipaleenmäki, H. MicroRNAs in Bone Metastasis. Curr. Osteoporos. Rep. 2019, 17, 122–128. [Google Scholar] [CrossRef]
- Taipaleenmäki, H.; Browne, G.; Akech, J.; Zustin, J.; Van Wijnen, A.J.; Stein, J.L.; Hesse, E.; Stein, G.S.; Lian, J.B. Targeting of Runx2 by miR-135 and miR-203 Impairs Progression of Breast Cancer and Metastatic Bone Disease. Cancer Res. 2015, 75, 1433–1444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taipaleenmäki, H.; Farina, N.H.; Van Wijnen, A.J.; Stein, J.L.; Hesse, E.; Stein, G.S.; Lian, J.B. Antagonizing miR-218-5p attenuates Wnt signaling and reduces metastatic bone disease of triple negative breast cancer cells. Oncotarget 2016, 7, 79032–79046. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Cao, M.; Palomares, M.; Wu, X.; Li, A.; Yan, W.; Fong, M.Y.; Chan, W.-C.; Wang, S.E. Metastatic breast cancer cells overexpress and secrete miR-218 to regulate type I collagen deposition by osteoblasts. Breast Cancer Res. 2018, 20, 1–12. [Google Scholar] [CrossRef]
- Browne, G.; Taipaleenmäki, H.; Stein, G.S.; Stein, J.L.; Lian, J.B. MicroRNAs in the control of metastatic bone disease. Trends Endocrinol. Metab. 2014, 25, 320–327. [Google Scholar] [CrossRef] [Green Version]
- Siclari, V.A.; Mohammad, K.S.; Tompkins, D.R.; Davis, H.; McKenna, C.R.; Peng, X.; Wessner, L.L.; Niewolna, M.; Guise, T.A.; Suvannasankha, A.; et al. Tumor-expressed adrenomedullin accelerates breast cancer bone metastasis. Breast Cancer Res. 2014, 16, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Masuda, T.; Endo, M.; Yamamoto, Y.; Odagiri, H.; Kadomatsu, T.; Nakamura, T.; Tanoue, H.; Ito, H.; Yugami, M.; Miyata, K.; et al. ANGPTL2 increases bone metastasis of breast cancer cells through enhancing CXCR4 signaling. Sci. Rep. 2015, 5, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Zhuang, X.; Zhang, H.; Li, X.; Li, X.; Cong, M.; Peng, F.; Yu, J.; Zhang, X.; Yang, Q.; Hu, G. Differential effects on lung and bone metastasis of breast cancer by Wnt signalling inhibitor DKK1. Nat. Cell Biol. 2017, 19, 1274–1285. [Google Scholar] [CrossRef]
- Eyre, R.; Alférez, D.G.; Santiago-Gómez, A.; Spence, K.; McConnell, J.C.; Hart, C.; Simões, B.M.; Lefley, D.; Tulotta, C.; Storer, J.; et al. Microenvironmental IL1β promotes breast cancer metastatic colonisation in the bone via activation of Wnt signalling. Nat. Commun. 2019, 10, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Wakabayashi, H.; Hamaguchi, T.; Nagao, N.; Kato, S.; Iino, T.; Nakamura, T.; Sudo, A. Interleukin-6 receptor inhibitor suppresses bone metastases in a breast cancer cell line. Breast Cancer 2018, 25, 566–574. [Google Scholar] [CrossRef] [PubMed]
- Liang, M.; Ma, Q.; Ding, N.; Luo, F.; Bai, Y.; Kang, F.; Gong, X.; Dong, R.; Dai, J.; Dai, Q.; et al. IL-11 is essential in promoting osteolysis in breast cancer bone metastasis via RANKL-independent activation of osteoclastogenesis. Cell Death Dis. 2019, 10, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.-Q.; Du, X.; Li, D.-M.; Kong, P.-Z.; Sun, Y.; Liu, P.-F.; Wang, Q.-S.; Feng, Y.-M. ITGBL1 Is a Runx2 Transcriptional Target and Promotes Breast Cancer Bone Metastasis by Activating the TGFβ Signaling Pathway. Cancer Res. 2015, 75, 3302–3313. [Google Scholar] [CrossRef] [Green Version]
- Leong, K.G.; Niessen, K.; Kulic, I.; Raouf, A.; Eaves, C.; Pollet, I.; Karsan, A. Jagged1-mediated Notch activation induces epithelial-to-mesenchymal transition through Slug-induced repression of E-cadherin. J. Exp. Med. 2007, 204, 2935–2948. [Google Scholar] [CrossRef] [Green Version]
- Pio, G.M.; Xia, Y.; Piaseczny, M.M.; Chu, J.; Allan, A.L. Soluble bone-derived osteopontin promotes migration and stem-like behavior of breast cancer cells. PLoS ONE 2017, 12, e0177640. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Mu, E.; Wei, Y.; Riethdorf, S.; Yang, Q.; Yuan, M.; Yan, J.; Hua, Y.; Tiede, B.J.; Lu, X.; et al. VCAM-1 Promotes Osteolytic Expansion of Indolent Bone Micrometastasis of Breast Cancer by Engaging α4β1-Positive Osteoclast Progenitors. Cancer Cell 2011, 20, 701–714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Devignes, C.-S.; Aslan, Y.; Brenot, A.; Devillers, A.; Schepers, K.; Fabre, S.; Chou, J.; Casbon, A.-J.; Werb, Z.; Provot, S. HIF signaling in osteoblast-lineage cells promotes systemic breast cancer growth and metastasis in mice. Proc. Natl. Acad. Sci. USA 2018, 115, E992–E1001. [Google Scholar] [CrossRef] [Green Version]
- Maroni, P.; Bendinelli, P.; Matteucci, E.; Desiderio, M.A. The therapeutic effect of miR-125b is enhanced by the prostaglandin endoperoxide synthase 2/cyclooxygenase 2 blockade and hampers ETS1 in the context of the microenvironment of bone metastasis. Cell Death Dis. 2018, 9, 472. [Google Scholar] [CrossRef] [Green Version]
- Qin, J.-J.; Yan, L.; Zhang, J.; Zhang, W.-D. STAT3 as a potential therapeutic target in triple negative breast cancer: A systematic review. J. Exp. Clin. Cancer Res. 2019, 38, 1–16. [Google Scholar] [CrossRef]
- Lu, X.; Wang, Q.; Hu, G.; Van Poznak, C.; Fleisher, M.; Reiss, M.; Massagué, J.; Kang, Y. ADAMTS1 and MMP1 proteolytically engage EGF-like ligands in an osteolytic signaling cascade for bone metastasis. Genes Dev. 2009, 23, 1882–1894. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pivetta, E.; Scapolan, M.; Pecolo, M.; Wassermann, B.; Abu-Rumeileh, I.; Balestreri, L.; Borsatti, E.; Tripodo, C.; Colombatti, A.; Spessotto, P. MMP-13 stimulates osteoclast differentiation and activation in tumour breast bone metastases. Breast Cancer Res. 2011, 13, R105. [Google Scholar] [CrossRef] [Green Version]
- Wu, Z.; Wang, T.; Fang, M.; Huang, W.; Sun, Z.; Xiao, J.; Yan, W. MFAP5 promotes tumor progression and bone metastasis by regulating ERK/MMP signaling pathways in breast cancer. Biochem. Biophys. Res. Commun. 2018, 498, 495–501. [Google Scholar] [CrossRef]
- Clément-Demange, L.; Mulcrone, P.L.; Tabarestani, T.Q.; Sterling, J.A.; Elefteriou, F. β2ARs stimulation in osteoblasts promotes breast cancer cell adhesion to bone marrow endothelial cells in an IL-1β and selectin-dependent manner. J. Bone Oncol. 2018, 13, 1–10. [Google Scholar] [CrossRef]
- Liu, Y.; Zhang, R.-X.; Yuan, W.; Chen, H.-Q.; Tian, D.-D.; Li, H.; Jiang, X.; Deng, Z.-L.; Wang, Y. Knockdown of Bone Morphogenetic Proteins Type 1a Receptor (BMPR1a) in Breast Cancer Cells Protects Bone from Breast Cancer-Induced Osteolysis by Suppressing RANKL Expression. Cell. Physiol. Biochem. 2018, 45, 1759–1771. [Google Scholar] [CrossRef] [Green Version]
- Aukes, K.; Forsman, C.; Brady, N.J.; Astleford, K.; Blixt, N.; Sachdev, D.; Jensen, E.D.; Mansky, K.C.; Schwertfeger, K.L. Breast cancer cell-derived fibroblast growth factors enhance osteoclast activity and contribute to the formation of metastatic lesions. PLoS ONE 2017, 12, e0185736. [Google Scholar] [CrossRef] [Green Version]
- Nakajima, K.; Kho, D.H.; Yanagawa, T.; Harazono, Y.; Gao, X.; Hogan, V.; Raz, A. Galectin-3 Inhibits Osteoblast Differentiation through Notch Signaling. Neoplasia 2014, 16, 939–949. [Google Scholar] [CrossRef] [Green Version]
- Hiraga, T.; Myoui, A.; Hashimoto, N.; Sasaki, A.; Hata, K.; Morita, Y.; Yoshikawa, H.; Rosen, C.J.; Mundy, G.R.; Yoneda, T. Bone-Derived IGF Mediates Crosstalk between Bone and Breast Cancer Cells in Bony Metastases. Cancer Res. 2012, 72, 4238–4249. [Google Scholar] [CrossRef] [Green Version]
- Whang, Y.M.; Jung, S.P.; Kim, M.-K.; Chang, I.H.; Park, S.I. Targeting the Hepatocyte Growth Factor and c-Met Signaling Axis in Bone Metastases. Int. J. Mol. Sci. 2019, 20, 384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casimiro, S.; Mohammad, K.S.; Pires, R.; Tato-Costa, J.; Alho, I.; Teixeira, R.L.; Carvalho, A.; Ribeiro, S.; Lipton, A.; Guise, T.A.; et al. RANKL/RANK/MMP-1 Molecular Triad Contributes to the Metastatic Phenotype of Breast and Prostate Cancer Cells In Vitro. PLoS ONE 2013, 8, e63153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiechi, A.; Waning, D.L.; Stayrook, K.R.; Buijs, J.T.; Guise, T.A.; Mohammad, K.S. Role of TGF-β in breast cancer bone metastases. Adv. Biosci. Biotechnol. 2013, 4, 15–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aldridge, S.E.; Lennard, T.W.J.; Williams, J.R.; Birch, M.A. Vascular endothelial growth factor acts as an osteolytic factor in breast cancer metastases to bone. Br. J. Cancer 2005, 92, 1531–1537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, H.; Lin, F.; Wang, Z.; Yang, L.; Meng, J.; Ou, Z.; Shao, Z.-M.; Di, G.; Yang, G. CXCR2 promotes breast cancer metastasis and chemoresistance via suppression of AKT1 and activation of COX2. Cancer Lett. 2018, 412, 69–80. [Google Scholar] [CrossRef] [PubMed]
- Jin, W.; Kim, B.; Kim, D.; Choo, H.-Y.P.; Kim, H.-H.; Ha, H.; Lee, Z.H. NF-κB signaling regulates cell-autonomous regulation of CXCL10 in breast cancer 4T1 cells. Exp. Mol. Med. 2017, 49, e295. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Mao, X.; Fan, C.; Liu, C.; Guo, A.; Guan, S.; Jin, Q.; Li, B.; Yao, F.; Jin, F. CXCL12-CXCR4 axis promotes the natural selection of breast cancer cell metastasis. Tumor Biol. 2014, 35, 7765–7773. [Google Scholar] [CrossRef] [Green Version]
- Jairam, V.; Lee, V.; Yu, J.B.; Park, H.S. Nationwide Patterns of Pathologic Fractures Among Patients Hospitalized With Bone Metastases. Am. J. Clin. Oncol. 2020, 43, 720–726. [Google Scholar] [CrossRef]
- Zhang, H.; Zhu, W.; Biskup, E.; Yang, W.; Yang, Z.; Wang, H.; Qiu, X.; Zhang, C.; Hu, G.; Hu, G. Incidence, risk factors and prognostic characteristics of bone metastases and skeletal-related events (SREs) in breast cancer patients: A systematic review of the real world data. J. Bone Oncol. 2018, 11, 38–50. [Google Scholar] [CrossRef]
- Mirels, H. Metastatic disease in long bones. A proposed scoring system for diagnosing impending pathologic fractures. Clin. Orthop. Relat. Res. 1989, 249, 256–264. [Google Scholar] [CrossRef]
- Damron, T.A.; Mann, K.A. Fracture risk assessment and clinical decision making for patients with metastatic bone disease. J. Orthop. Res. 2020, 38, 1175–1190. [Google Scholar] [CrossRef]
- Behnke, N.K.; Baker, D.K.; Xu, S.; Niemeier, T.E.; Watson, S.L.; Ponce, B.A. Risk factors for same-admission mortality after pathologic fracture secondary to metastatic cancer. Support. Care Cancer 2017, 25, 513–521. [Google Scholar] [CrossRef]
- Patchell, R.A.; Tibbs, P.A.; Regine, W.F.; Payne, R.; Saris, S.; Kryscio, R.J.; Mohiuddin, M.; Young, B. Direct decompressive surgical resection in the treatment of spinal cord compression caused by metastatic cancer: A randomised trial. Lancet 2005, 366, 643–648. [Google Scholar] [CrossRef] [Green Version]
- Piccioli, A.; Maccauro, G.; Rossi, B.; Scaramuzzo, L.; Frenos, F.; Capanna, R. Surgical treatment of pathologic fractures of humerus. Injury 2010, 41, 1112–1116. [Google Scholar] [CrossRef]
- Arvinius, C.; Parra, J.L.C.; Mateo, L.S.; Maroto, R.G.; Borrego, A.F.; Stern, L.L.-D. Benefits of early intramedullary nailing in femoral metastases. Int. Orthop. 2014, 38, 129–132. [Google Scholar] [CrossRef] [Green Version]
- Philipp, T.C.; Mikula, J.; Doung, Y.-C.; Gundle, K.R. Is There an Association Between Prophylactic Femur Stabilization and Survival in Patients with Metastatic Bone Disease? Clin. Orthop. Relat. Res. 2020, 478, 540–546. [Google Scholar] [CrossRef]
- Kotian, R.N.; Puvanesarajah, V.; Rao, S.; El Abiad, J.M.; Morris, C.D.; Levin, A.S. Predictors of survival after intramedullary nail fixation of completed or impending pathologic femur fractures from metastatic disease. Surg. Oncol. 2018, 27, 462–467. [Google Scholar] [CrossRef]
- Johnson, J.D.; Wyles, C.C.; Perry, K.I.; Yuan, B.J.; Rose, P.S.; Houdek, M.T. Outcomes of knee arthroplasty for primary treatment of pathologic peri-articular fractures of the distal femur and proximal tibia. Int. Orthop. 2020, 44, 187–193. [Google Scholar] [CrossRef] [PubMed]
- Alvi, H.; Damron, T.A. Prophylactic Stabilization for Bone Metastases, Myeloma, or Lymphoma: Do We Need to Protect the Entire Bone? Clin. Orthop. Relat. Res. 2013, 471, 706–714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nooh, A.; Goulding, K.; Isler, M.H.; Mottard, S.; Arteau, A.; Dion, N.; Turcotte, R. Early Improvement in Pain and Functional Outcome but Not Quality of Life After Surgery for Metastatic Long Bone Disease. Clin. Orthop. Relat. Res. 2018, 476, 535–545. [Google Scholar] [CrossRef]
- McLynn, R.P.; Ondeck, N.T.; Grauer, J.N.; Lindskog, D.M. What Is the Adverse Event Profile After Prophylactic Treatment of Femoral Shaft or Distal Femur Metastases? Clin. Orthop. Relat. Res. 2018, 476, 2381–2388. [Google Scholar] [CrossRef]
- Mosher, Z.A.; Patel, H.; Ewing, M.A.; Niemeier, T.E.; Hess, M.C.; Wilkinson, E.B.; McGwinJr, G.; Ponce, B.A.; Patt, J.C. Early Clinical and Economic Outcomes of Prophylactic and Acute Pathologic Fracture Treatment. J. Oncol. Pr. 2019, 15, e132–e140. [Google Scholar] [CrossRef]
- Ward, W.G.; Holsenbeck, S.; Dorey, F.J.; Spang, J.; Howe, D. Metastatic Disease of the Femur: Surgical Treatment. Clin. Orthop. Relat. Res. 2003, 415, S230–S244. [Google Scholar] [CrossRef]
- Ristevski, B.; Jenkinson, R.J.; Stephen, D.J.; Finkelstein, J.; Schemitsch, E.H.; McKee, M.D.; Kreder, H.J. Mortality and complications following stabilization of femoral metastatic lesions: A population-based study of regional variation and outcome. Can. J. Surg. 2009, 52, 302–308. [Google Scholar]
- Ampil, F.L.; Sadasivan, K.K. Prophylactic and therapeutic fixation of weight-bearing long bones with metastatic cancer. South. Med. J. 2001, 94, 394–396. [Google Scholar] [CrossRef] [PubMed]
- Johnson, N.A.; Uzoigwe, C.; Venkatesan, M.; Burgula, V.; Kulkarni, A.; Davison, J.N.; Ashford, R.U. Risk factors for intramedullary nail breakage in proximal femoral fractures: A 10-year retrospective review. Ann. R. Coll. Surg. Engl. 2017, 99, 145–150. [Google Scholar] [CrossRef]
- Willeumier, J.J.; Kaynak, M.; van der Zwaal, P.; Meylaerts, S.A.G.; Mathijssen, N.M.C.; Jutte, P.C.; Tsagozis, P.; Wedin, R.; van de Sande, M.A.J.; Fiocco, M.; et al. What Factors Are Associated With Implant Breakage and Revision After Intramedullary Nailing for Femoral Metastases? Clin. Orthop. Relat. Res. 2018, 476, 1823–1833. [Google Scholar] [CrossRef]
- Jacofsky, D.J.; Haidukewych, G.J.; Zhang, H.; Sim, F.H. Complications and Results of Arthroplasty for Salvage of Failed Treatment of Malignant Pathologic Fractures of the Hip. Clin. Orthop. Relat. Res. 2004, 427, 52–56. [Google Scholar] [CrossRef] [PubMed]
- Wong, M.; Pavlakis, N. Optimal management of bone metastases in breast cancer patients. Breast Cancer: Targets Ther. 2011, 3, 35–60. [Google Scholar] [CrossRef] [Green Version]
- Stopeck, A.T.; Lipton, A.; Body, J.-J.; Steger, G.G.; Tonkin, K.; De Boer, R.H.; Lichinitser, M.; Fujiwara, Y.; Yardley, D.A.; Viniegra, M.; et al. Denosumab Compared With Zoledronic Acid for the Treatment of Bone Metastases in Patients With Advanced Breast Cancer: A Randomized, Double-Blind Study. J. Clin. Oncol. 2010, 28, 5132–5139. [Google Scholar] [CrossRef] [Green Version]
- White, E.; DiPaola, R.S. The Double-Edged Sword of Autophagy Modulation in Cancer. Clin. Cancer Res. 2009, 15, 5308–5316. [Google Scholar] [CrossRef] [Green Version]
- Rubinsztein, D.C.; Codogno, P.; Levine, B. Autophagy modulation as a potential therapeutic target for diverse diseases. Nat. Rev. Drug Discov. 2012, 11, 709–730. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.S.; Gernapudi, R.; Choi, E.Y.; Lapidus, R.G.; Passaniti, A. Characterization of CADD522, a small molecule that inhibits RUNX2-DNA binding and exhibits antitumor activity. Oncotarget 2017, 8, 70916–70940. [Google Scholar] [CrossRef] [Green Version]
- Buenrostro, D.; Kwakwa, K.A.; Putnam, N.E.; Merkel, A.; Johnson, J.R.; Cassat, J.E.; Sterling, J.A. Early TGF-β inhibition in mice reduces the incidence of breast cancer induced bone disease in a myeloid dependent manner. Bone 2018, 113, 77–88. [Google Scholar] [CrossRef]
- Ganapathy, V.; Ge, R.; Grazioli, A.; Xie, W.; Banach-Petrosky, W.; Kang, Y.; Lonning, S.; McPherson, J.; Yingling, J.M.; Biswas, S.; et al. Targeting the Transforming Growth Factor-β pathway inhibits human basal-like breast cancer metastasis. Mol. Cancer 2010, 9, 1–16. [Google Scholar] [CrossRef] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Othman, A.; Winogradzki, M.; Lee, L.; Tandon, M.; Blank, A.; Pratap, J. Bone Metastatic Breast Cancer: Advances in Cell Signaling and Autophagy Related Mechanisms. Cancers 2021, 13, 4310. https://doi.org/10.3390/cancers13174310
Othman A, Winogradzki M, Lee L, Tandon M, Blank A, Pratap J. Bone Metastatic Breast Cancer: Advances in Cell Signaling and Autophagy Related Mechanisms. Cancers. 2021; 13(17):4310. https://doi.org/10.3390/cancers13174310
Chicago/Turabian StyleOthman, Ahmad, Marcus Winogradzki, Linus Lee, Manish Tandon, Alan Blank, and Jitesh Pratap. 2021. "Bone Metastatic Breast Cancer: Advances in Cell Signaling and Autophagy Related Mechanisms" Cancers 13, no. 17: 4310. https://doi.org/10.3390/cancers13174310