
SHP2 as a Potential Therapeutic Target in Diffuse-Type Gastric Carcinoma Addicted to Receptor Tyrosine Kinase Signaling

 , , ,
, , ,
Abstract
:Simple Summary
Abstract
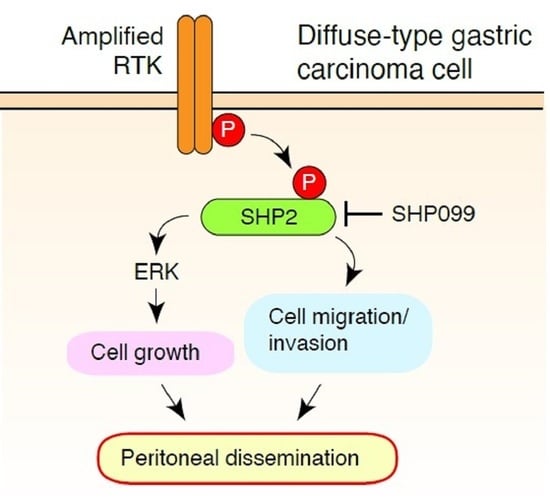
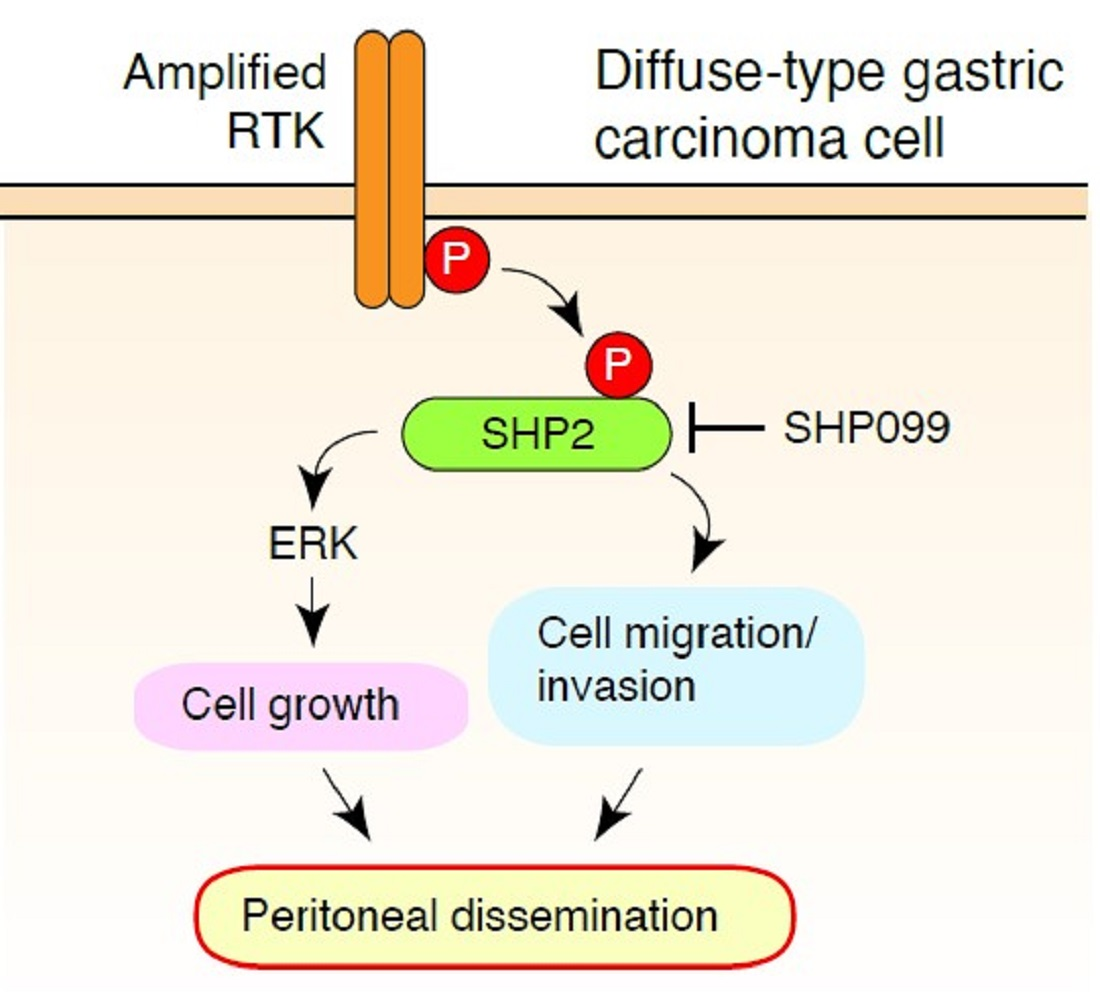{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Antibodies and Reagents
2.3. siRNA Transfection
2.4. Lentiviral shRNA Transduction
2.5. Immunoblotting
2.6. Cell Proliferation Assay
2.7. Quantitative PCR (qPCR)
2.8. Cell Migration and Invasion Assays
2.9. Peritoneal Dissemination Assay
2.10. Statistical Analysis
3. Results
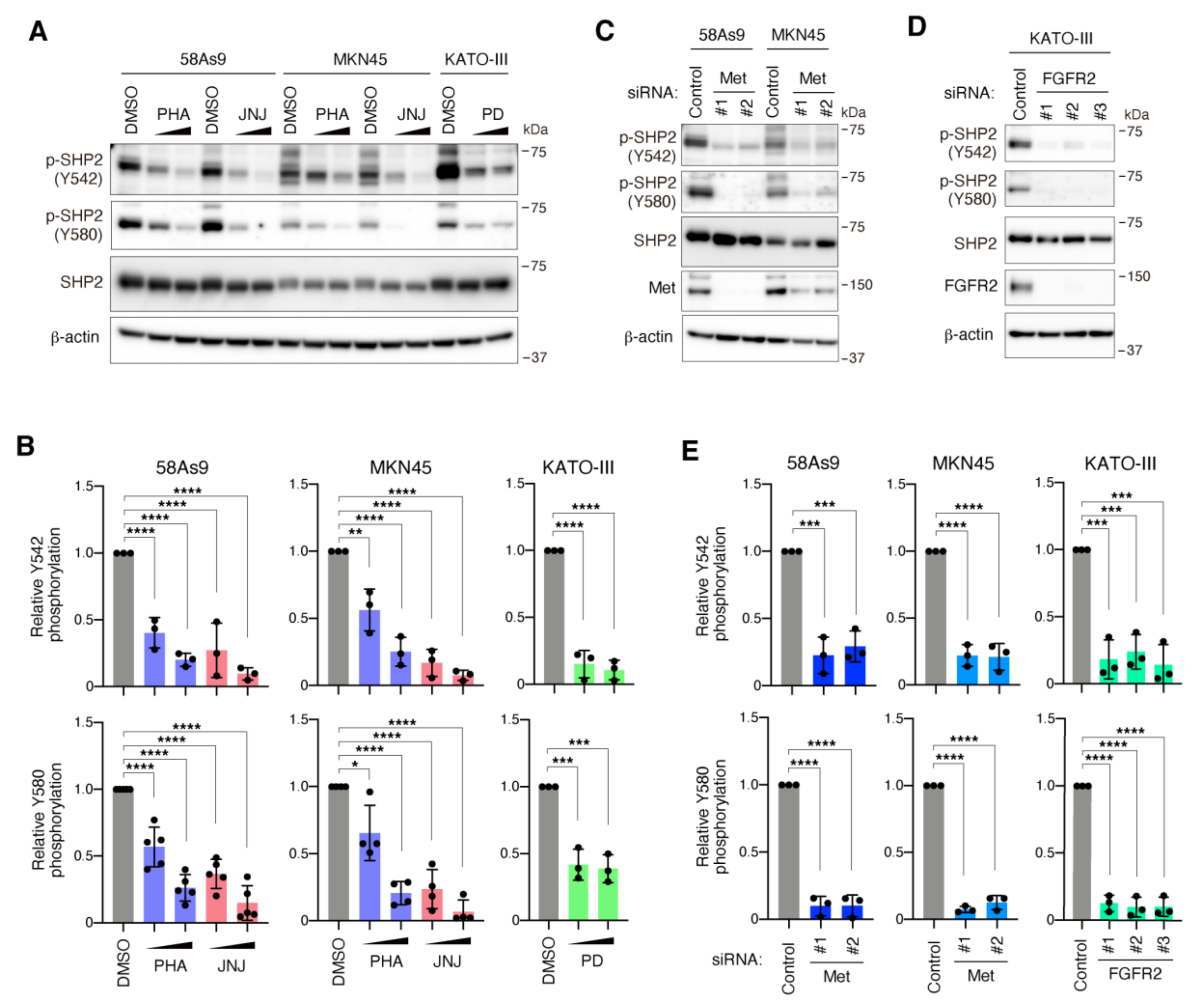
3.1. SHP2 Is Preferentially Tyrosine Phosphorylated in DGC Cell Lines with Met or FGFR2 Gene Amplification
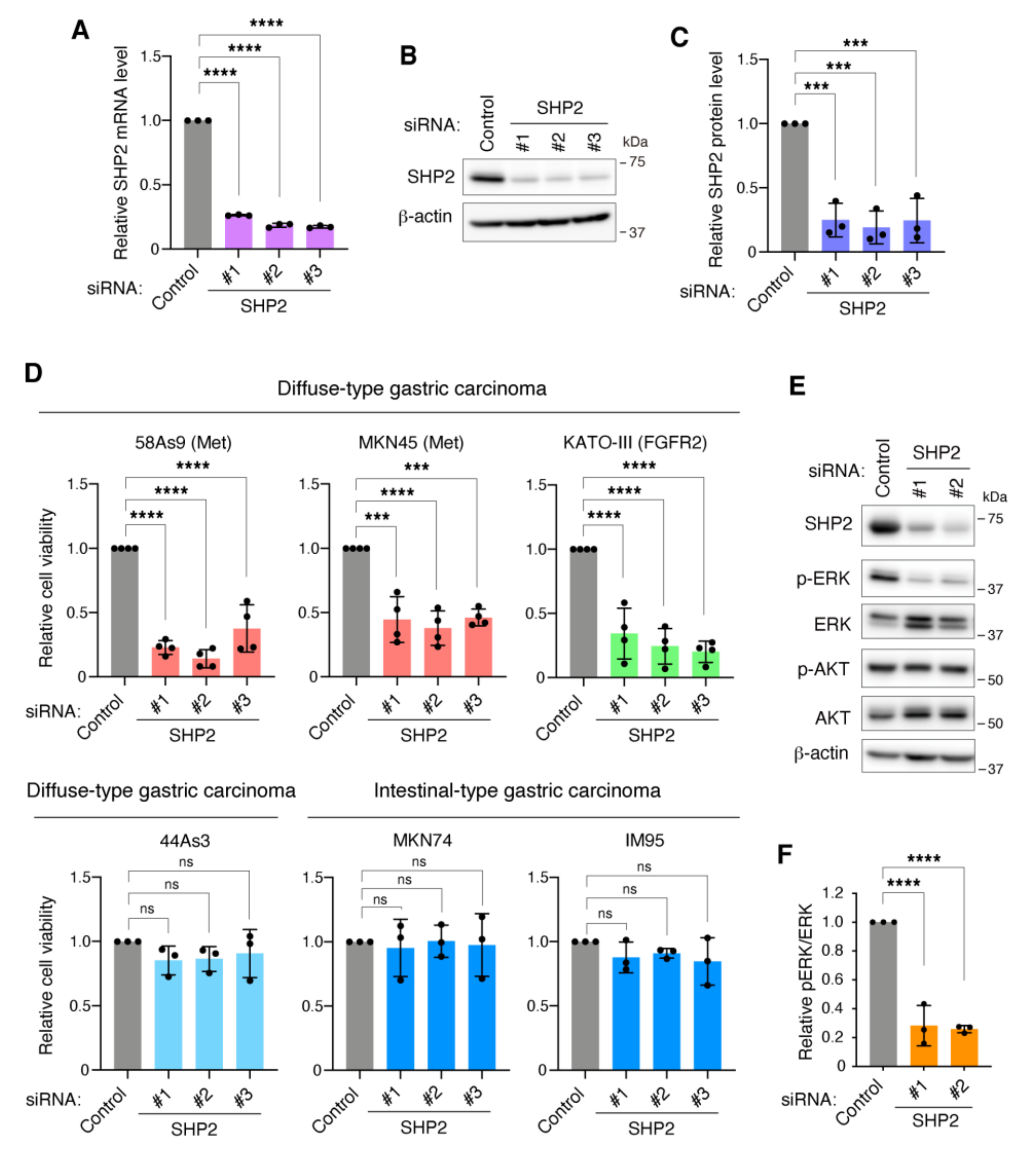
3.2. SHP2 Is Required for the Growth of DGC Cells with Met or FGFR2 Gene Amplification
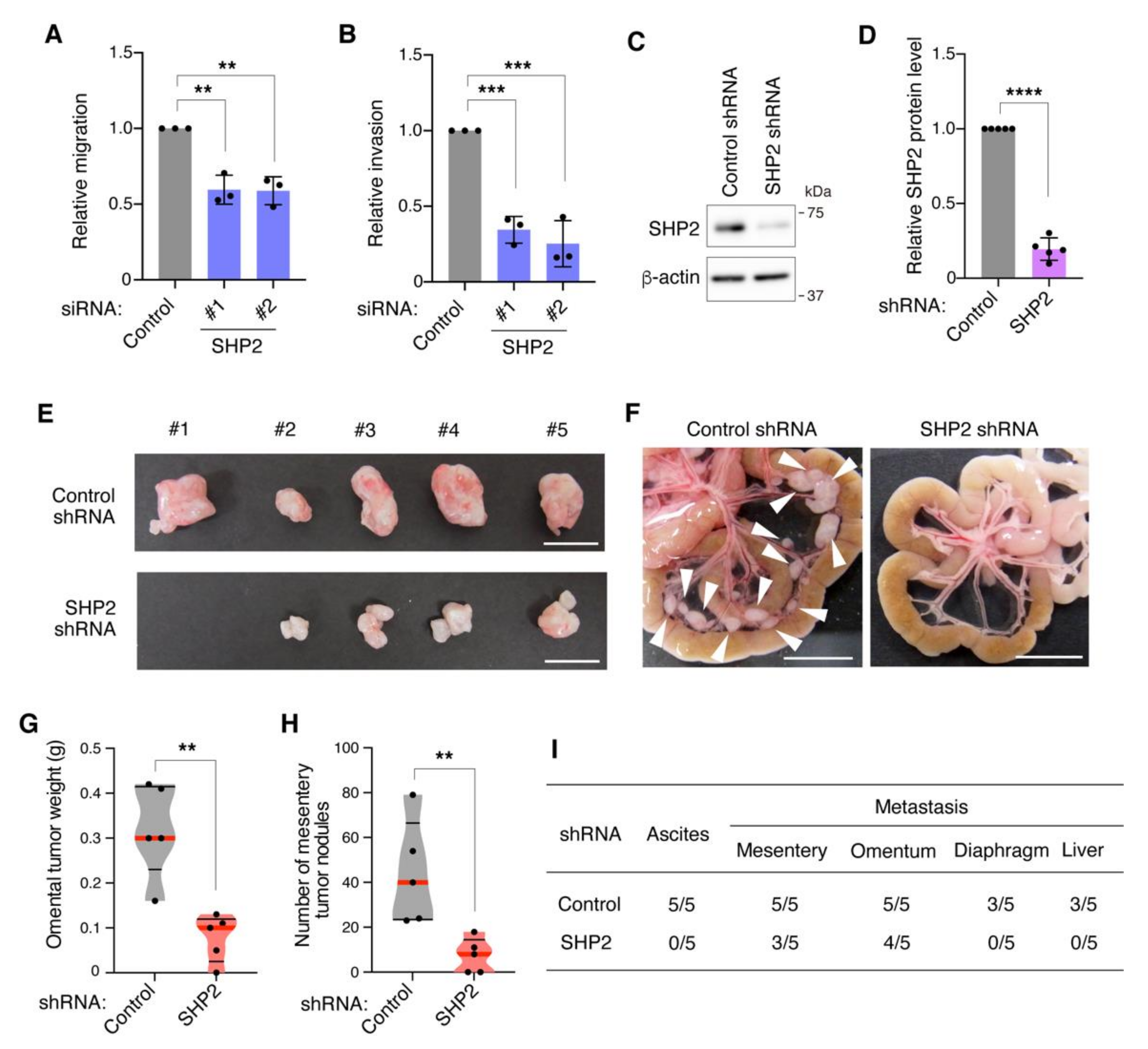
3.3. SHP2 Knockdown Blocks Migration, Invasion, and Peritoneal Dissemination of Met-Addicted DGC Cells
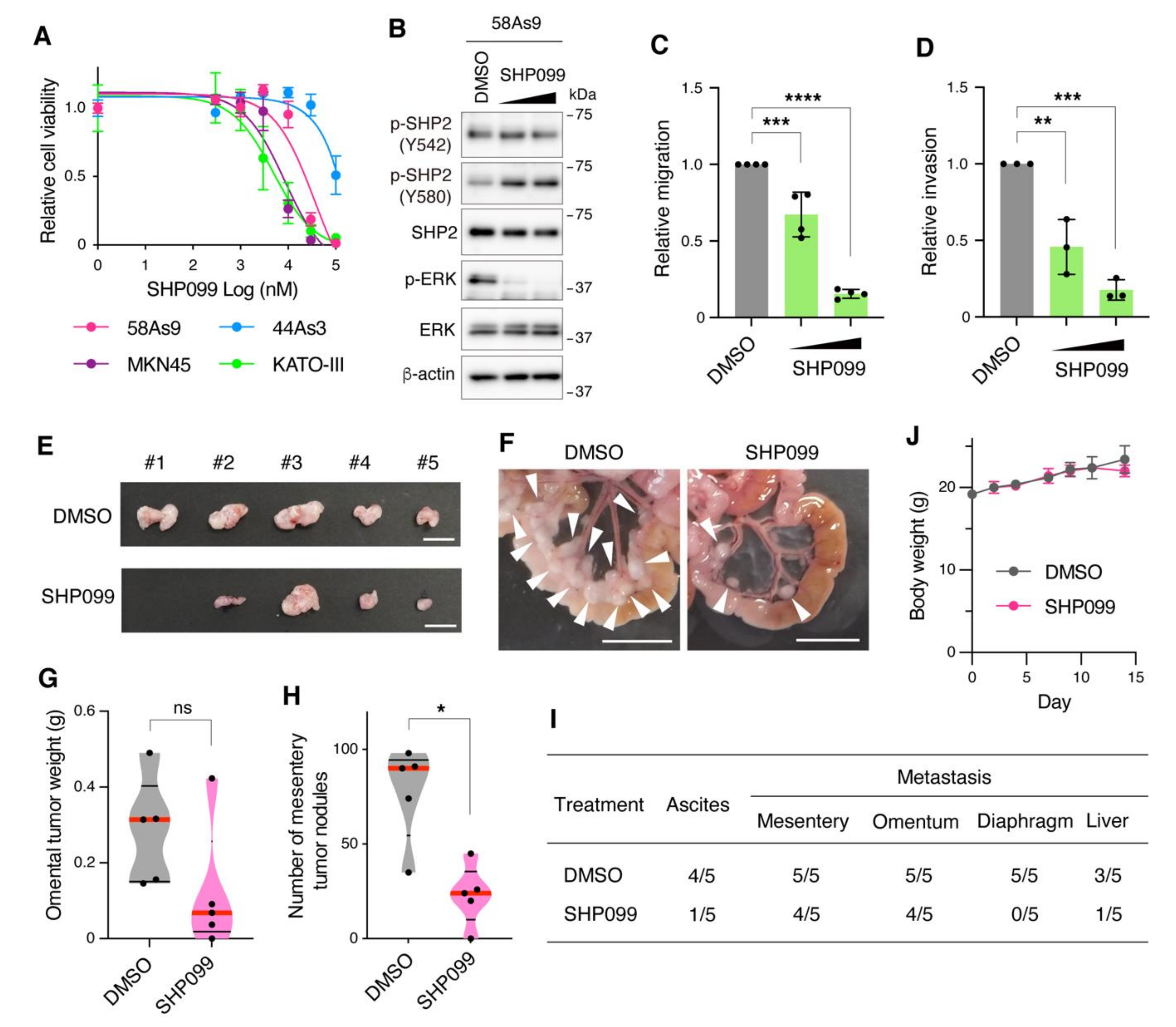
3.4. Pharmacological Inhibition of SHP2 Abrogates Malignant Phenotypes of Met-Addicted DGC Cells
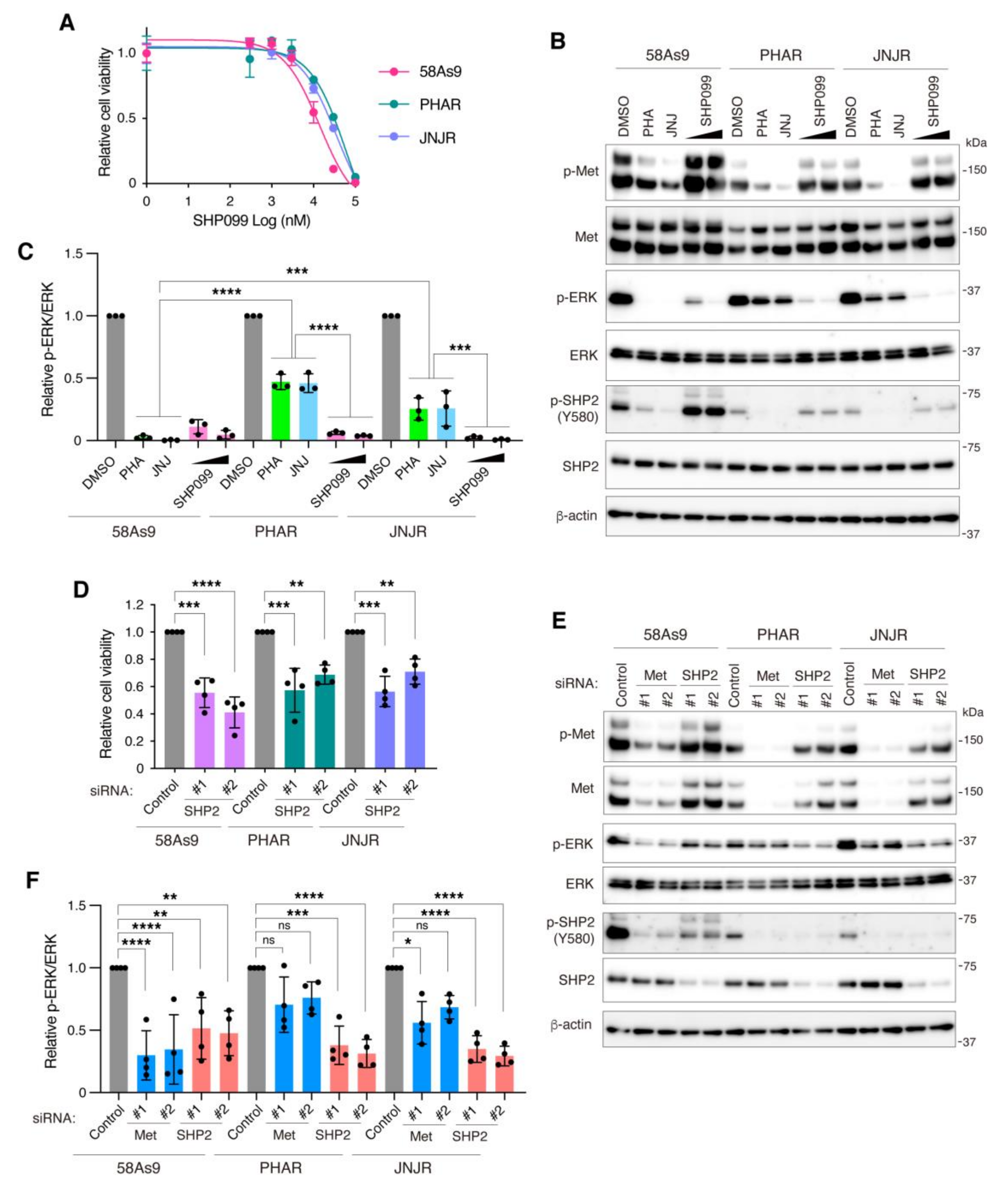
3.5. Inhibition of SHP2 Overcomes Resistance to Met Inhibitors in DGC Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Global Burden of Disease Cancer Collaboration; Fitzmaurice Collaboration; Dicker, D.; Pain, A.; Hamavid, H.; Moradi-Lakeh, M.; MacIntyre, M.F.; Allen, C.; Hansen, G.; Woodbrook, R.; et al. The Global Burden of Cancer 2013. JAMA Oncol. 2015, 1, 505–527. [Google Scholar] [CrossRef]
- Lauren, P. The two histological main types of gastric carcinoma: Diffuse and so-called intestinal-type carcinoma. An attempt at a histo-clinical classification. Acta Pathol. Microbiol. Scand. 1965, 64, 31–49. [Google Scholar] [CrossRef]
- Chen, Y.; Zhou, Q.; Wang, H.; Zhuo, W.; Ding, Y.; Lu, J.; Wu, G.; Xu, N.; Teng, L. Predicting peritoneal dissemination of gastric cancer in the era of precision medicine: Molecular characterization and biomarkers. Cancers 2020, 12, 2236. [Google Scholar] [CrossRef] [PubMed]
- Yashiro, M.; Hirakawa, K. Cancer-stromal interactions in scirrhous gastric carcinoma. Cancer Microenviron. 2010, 3, 127–135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ikeguchi, M.; Miyake, T.; Matsunaga, T.; Yamamoto, M.; Fukumoto, Y.; Yamada, Y.; Fukuda, K.; Saito, H.; Tatebe, S.; Tsujitani, S. Recent results of therapy for scirrhous gastric cancer. Surg. Today 2009, 39, 290–294. [Google Scholar] [CrossRef]
- Otsuji, E.; Kuriu, Y.; Okamoto, K.; Ochiai, T.; Ichikawa, D.; Hagiwara, A.; Yamagishi, H. Outcome of surgical treatment for patients with scirrhous carcinoma of the stomach. Am. J. Surg. 2004, 188, 327–332. [Google Scholar] [CrossRef]
- Saraon, P.; Pathmanathan, S.; Snider, J.; Lyakisheva, A.; Wong, V.; Stagljar, I. Receptor tyrosine kinases and cancer: Oncogenic mechanisms and therapeutic approaches. Oncogene 2021, 40, 4079–4093. [Google Scholar] [CrossRef] [PubMed]
- Kuniyasu, H.; Yasui, W.; Kitadai, Y.; Yokozaki, H.; Ito, H.; Tahara, E. Frequent amplification of the c-met gene in scirrhous type stomach cancer. Biochem Biophys Res. Commun. 1992, 189, 227–232. [Google Scholar] [CrossRef]
- Hattori, Y.; Itoh, H.; Uchino, S.; Hosokawa, K.; Ochiai, A.; Ino, Y.; Ishii, H.; Sakamoto, H.; Yamaguchi, N.; Yanagihara, K.; et al. Immunohistochemical detection of K-sam protein in stomach cancer. Clin. Cancer Res. 1996, 2, 1373–1381. [Google Scholar] [PubMed]
- Hattori, Y.; Odagiri, H.; Nakatani, H.; Miyagawa, K.; Naito, K.; Sakamoto, H.; Katoh, O.; Yoshida, T.; Sugimura, T.; Terada, M. K-sam, an amplified gene in stomach cancer, is a member of the heparin-binding growth factor receptor genes. Proc. Natl. Acad. Sci. USA 1990, 87, 5983–5987. [Google Scholar] [CrossRef] [Green Version]
- Katoh, M.; Nakagama, H. FGF receptors: Cancer biology and therapeutics. Med. Res. Rev. 2014, 34, 280–300. [Google Scholar] [CrossRef]
- Recondo, G.; Che, J.; Janne, P.A.; Awad, M.M. Targeting MET dysregulation in cancer. Cancer Discov. 2020, 10, 922–934. [Google Scholar] [CrossRef]
- Su, X.; Zhan, P.; Gavine, P.R.; Morgan, S.; Womack, C.; Ni, X.; Shen, D.; Bang, Y.J.; Im, S.A.; Ho Kim, W.; et al. FGFR2 amplification has prognostic significance in gastric cancer: Results from a large international multicentre study. Br. J. Cancer 2014, 110, 967–975. [Google Scholar] [CrossRef]
- Ahn, S.; Lee, J.; Hong, M.; Kim, S.T.; Park, S.H.; Choi, M.G.; Lee, J.H.; Sohn, T.S.; Bae, J.M.; Kim, S.; et al. FGFR2 in gastric cancer: Protein overexpression predicts gene amplification and high H-index predicts poor survival. Mod. Pathol. 2016, 29, 1095–1103. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Seo, J.W.; Jun, H.J.; Ki, C.S.; Park, S.H.; Park, Y.S.; Lim, H.Y.; Choi, M.G.; Bae, J.M.; Sohn, T.S.; et al. Impact of MET amplification on gastric cancer: Possible roles as a novel prognostic marker and a potential therapeutic target. Oncol. Rep. 2011, 25, 1517–1524. [Google Scholar] [CrossRef] [PubMed]
- Kawakami, H.; Okamoto, I.; Arao, T.; Okamoto, W.; Matsumoto, K.; Taniguchi, H.; Kuwata, K.; Yamaguchi, H.; Nishio, K.; Nakagawa, K.; et al. MET amplification as a potential therapeutic target in gastric cancer. Oncotarget 2012, 4, 9–17. [Google Scholar] [CrossRef] [Green Version]
- Hong, L.; Han, Y.; Liu, J.; Brain, L. Fibroblast growth factor receptor 2: A therapeutic target in gastric cancer. Expert Rev. Gastroenterol. Hepatol. 2013, 7, 759–765. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.T.; Lee, I.K.; Rom, E.; Sirkis, R.; Park, S.H.; Park, J.O.; Park, Y.S.; Lim, H.Y.; Kang, W.K.; Kim, K.M.; et al. Neutralizing antibody to FGFR2 can act as a selective biomarker and potential therapeutic agent for gastric cancer with FGFR2 amplification. Am. J. Transl. Res. 2019, 11, 4508–4515. [Google Scholar] [PubMed]
- Nakagawa, T.; Tohyama, O.; Yamaguchi, A.; Matsushima, T.; Takahashi, K.; Funasaka, S.; Shirotori, S.; Asada, M.; Obaishi, H. E7050: A dual c-Met and VEGFR-2 tyrosine kinase inhibitor promotes tumor regression and prolongs survival in mouse xenograft models. Cancer Sci. 2010, 101, 210–215. [Google Scholar] [CrossRef]
- Nakamura, K.; Yashiro, M.; Matsuoka, T.; Tendo, M.; Shimizu, T.; Miwa, A.; Hirakawa, K. A novel molecular targeting compound as K-samII/FGF-R2 phosphorylation inhibitor, Ki23057, for Scirrhous gastric cancer. Gastroenterology 2006, 131, 1530–1541. [Google Scholar] [CrossRef]
- Yamaguchi, H.; Takanashi, M.; Yoshida, N.; Ito, Y.; Kamata, R.; Fukami, K.; Yanagihara, K.; Sakai, R. Saracatinib impairs the peritoneal dissemination of diffuse-type gastric carcinoma cells resistant to Met and fibroblast growth factor receptor inhibitors. Cancer Sci. 2014, 105, 528–536. [Google Scholar] [CrossRef] [Green Version]
- Lau, W.M.; Teng, E.; Huang, K.K.; Tan, J.W.; Das, K.; Zang, Z.; Chia, T.; Teh, M.; Kono, K.; Yong, W.P.; et al. Acquired resistance to FGFR inhibitor in diffuse-type gastric cancer through an AKT-independent PKC-mediated phosphorylation of GSK3beta. Mol. Cancer Ther. 2018, 17, 232–242. [Google Scholar] [CrossRef] [Green Version]
- Miki, Y.; Yashiro, M.; Moyano-Galceran, L.; Sugimoto, A.; Ohira, M.; Lehti, K. Crosstalk between cancer associated fibroblasts and cancer cells in scirrhous type gastric cancer. Front. Oncol. 2020, 10, 568557. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.Y.; Ahn, T.; Bang, H.; Ham, J.S.; Kim, J.; Kim, S.T.; Jang, J.; Shim, M.; Kang, S.Y.; Park, S.H.; et al. Acquired resistance to LY2874455 in FGFR2-amplified gastric cancer through an emergence of novel FGFR2-ACSL5 fusion. Oncotarget 2017, 8, 15014–15022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, K.; Song, X.; Zhu, M.; Ma, H. Overexpression of FGFR2 contributes to inherent resistance to MET inhibitors in MET-amplified patient-derived gastric cancer xenografts. Oncol. Lett. 2015, 10, 2003–2008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matozaki, T.; Murata, Y.; Saito, Y.; Okazawa, H.; Ohnishi, H. Protein tyrosine phosphatase SHP-2: A proto-oncogene product that promotes Ras activation. Cancer Sci. 2009, 100, 1786–1793. [Google Scholar] [CrossRef]
- Tartaglia, M.; Gelb, B.D. Germ-line and somatic PTPN11 mutations in human disease. Eur. J. Med. Genet 2005, 48, 81–96. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zhang, F.; Niu, R. Functions of Shp2 in cancer. J. Cell Mol. Med. 2015, 19, 2075–2083. [Google Scholar] [CrossRef]
- Frankson, R.; Yu, Z.H.; Bai, Y.; Li, Q.; Zhang, R.Y.; Zhang, Z.Y. Therapeutic targeting of oncogenic tyrosine phosphatases. Cancer Res. 2017, 77, 5701–5705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- LaRochelle, J.R.; Fodor, M.; Vemulapalli, V.; Mohseni, M.; Wang, P.; Stams, T.; LaMarche, M.J.; Chopra, R.; Acker, M.G.; Blacklow, S.C. Structural reorganization of SHP2 by oncogenic mutations and implications for oncoprotein resistance to allosteric inhibition. Nat. Commun. 2018, 9, 4508. [Google Scholar] [CrossRef] [Green Version]
- Padua, R.A.P.; Sun, Y.; Marko, I.; Pitsawong, W.; Stiller, J.B.; Otten, R.; Kern, D. Mechanism of activating mutations and allosteric drug inhibition of the phosphatase SHP2. Nat. Commun. 2018, 9, 4507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, W.; Gong, D.; Bar-Sagi, D.; Cole, P.A. Site-specific incorporation of a phosphotyrosine mimetic reveals a role for tyrosine phosphorylation of SHP-2 in cell signaling. Mol. Cell 2001, 8, 759–769. [Google Scholar] [CrossRef]
- Araki, T.; Nawa, H.; Neel, B.G. Tyrosyl phosphorylation of Shp2 is required for normal ERK activation in response to some, but not all, growth factors. J. Biol. Chem. 2003, 278, 41677–41684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.N.; LaMarche, M.J.; Chan, H.M.; Fekkes, P.; Garcia-Fortanet, J.; Acker, M.G.; Antonakos, B.; Chen, C.H.; Chen, Z.; Cooke, V.G.; et al. Allosteric inhibition of SHP2 phosphatase inhibits cancers driven by receptor tyrosine kinases. Nature 2016, 535, 148–152. [Google Scholar] [CrossRef]
- Mainardi, S.; Mulero-Sanchez, A.; Prahallad, A.; Germano, G.; Bosma, A.; Krimpenfort, P.; Lieftink, C.; Steinberg, J.D.; de Wit, N.; Goncalves-Ribeiro, S.; et al. SHP2 is required for growth of KRAS-mutant non-small-cell lung cancer In Vivo. Nat. Med. 2018, 24, 961–967. [Google Scholar] [CrossRef] [PubMed]
- Ruess, D.A.; Heynen, G.J.; Ciecielski, K.J.; Ai, J.; Berninger, A.; Kabacaoglu, D.; Gorgulu, K.; Dantes, Z.; Wormann, S.M.; Diakopoulos, K.N.; et al. Mutant KRAS-driven cancers depend on PTPN11/SHP2 phosphatase. Nat. Med. 2018, 24, 954–960. [Google Scholar] [CrossRef]
- Wong, G.S.; Zhou, J.; Liu, J.B.; Wu, Z.; Xu, X.; Li, T.; Xu, D.; Schumacher, S.E.; Puschhof, J.; McFarland, J.; et al. Targeting wild-type KRAS-amplified gastroesophageal cancer through combined MEK and SHP2 inhibition. Nat. Med. 2018, 24, 968–977. [Google Scholar] [CrossRef] [PubMed]
- Nichols, R.J.; Haderk, F.; Stahlhut, C.; Schulze, C.J.; Hemmati, G.; Wildes, D.; Tzitzilonis, C.; Mordec, K.; Marquez, A.; Romero, J.; et al. RAS nucleotide cycling underlies the SHP2 phosphatase dependence of mutant BRAF-, NF1- and RAS-driven cancers. Nat. Cell Biol. 2018, 20, 1064–1073. [Google Scholar] [CrossRef]
- Nagamura, Y.; Miyazaki, M.; Nagano, Y.; Yuki, M.; Fukami, K.; Yanagihara, K.; Sasaki, K.; Sakai, R.; Yamaguchi, H. PLEKHA5 regulates the survival and peritoneal dissemination of diffuse-type gastric carcinoma cells with Met gene amplification. Oncogenesis 2021, 10, 25. [Google Scholar] [CrossRef]
- Yanagihara, K.; Tanaka, H.; Takigahira, M.; Ino, Y.; Yamaguchi, Y.; Toge, T.; Sugano, K.; Hirohashi, S. Establishment of two cell lines from human gastric scirrhous carcinoma that possess the potential to metastasize spontaneously in nude mice. Cancer Sci. 2004, 95, 575–582. [Google Scholar] [CrossRef] [Green Version]
- Yanagihara, K.; Takigahira, M.; Tanaka, H.; Komatsu, T.; Fukumoto, H.; Koizumi, F.; Nishio, K.; Ochiya, T.; Ino, Y.; Hirohashi, S. Development and biological analysis of peritoneal metastasis mouse models for human scirrhous stomach cancer. Cancer Sci. 2005, 96, 323–332. [Google Scholar] [CrossRef] [PubMed]
- Yanagihara, K.; Takigahira, M.; Takeshita, F.; Komatsu, T.; Nishio, K.; Hasegawa, F.; Ochiya, T. A photon counting technique for quantitatively evaluating progression of peritoneal tumor dissemination. Cancer Res. 2006, 66, 7532–7539. [Google Scholar] [CrossRef] [Green Version]
- Yanagihara, K.; Seyama, T.; Tsumuraya, M.; Kamada, N.; Yokoro, K. Establishment and characterization of human signet ring cell gastric carcinoma cell lines with amplification of the c-myc oncogene. Cancer Res. 1991, 51, 381–386. [Google Scholar] [PubMed]
- Yanagihara, K.; Kamada, N.; Tsumuraya, M.; Amano, F. Establishment and characterization of a human gastric scirrhous carcinoma cell line in serum-free chemically defined medium. Int. J. Cancer 1993, 54, 200–207. [Google Scholar] [CrossRef] [PubMed]
- Stewart, S.A.; Dykxhoorn, D.M.; Palliser, D.; Mizuno, H.; Yu, E.Y.; An, D.S.; Sabatini, D.M.; Chen, I.S.; Hahn, W.C.; Sharp, P.A.; et al. Lentivirus-delivered stable gene silencing by RNAi in primary cells. RNA 2003, 9, 493–501. [Google Scholar] [CrossRef] [Green Version]
- Smolen, G.A.; Sordella, R.; Muir, B.; Mohapatra, G.; Barmettler, A.; Archibald, H.; Kim, W.J.; Okimoto, R.A.; Bell, D.W.; Sgroi, D.C.; et al. Amplification of MET may identify a subset of cancers with extreme sensitivity to the selective tyrosine kinase inhibitor PHA-665752. Proc. Natl. Acad. Sci. USA 2006, 103, 2316–2321. [Google Scholar] [CrossRef] [Green Version]
- Okamoto, W.; Okamoto, I.; Yoshida, T.; Okamoto, K.; Takezawa, K.; Hatashita, E.; Yamada, Y.; Kuwata, K.; Arao, T.; Yanagihara, K.; et al. Identification of c-Src as a potential therapeutic target for gastric cancer and of MET activation as a cause of resistance to c-Src inhibition. Mol. Cancer Ther. 2010, 9, 1188–1197. [Google Scholar] [CrossRef] [Green Version]
- Ueda, T.; Sasaki, H.; Kuwahara, Y.; Nezu, M.; Shibuya, T.; Sakamoto, H.; Ishii, H.; Yanagihara, K.; Mafune, K.; Makuuchi, M.; et al. Deletion of the carboxyl-terminal exons of K-sam/FGFR2 by short homology-mediated recombination, generating preferential expression of specific messenger RNAs. Cancer Res. 1999, 59, 6080–6086. [Google Scholar]
- Chen, H.; Libring, S.; Ruddraraju, K.V.; Miao, J.; Solorio, L.; Zhang, Z.Y.; Wendt, M.K. SHP2 is a multifunctional therapeutic target in drug resistant metastatic breast cancer. Oncogene 2020, 39, 7166–7180. [Google Scholar] [CrossRef] [PubMed]
- Dong, S.; Li, F.Q.; Zhang, Q.; Lv, K.Z.; Yang, H.L.; Gao, Y.; Yu, J.R. Expression and clinical significance of SHP2 in gastric cancer. J. Int. Med. Res. 2012, 40, 2083–2089. [Google Scholar] [CrossRef]
- Degirmenci, U.; Wang, M.; Hu, J. Targeting aberrant RAS/RAF/MEK/ERK signaling for cancer therapy. Cells 2020, 9, 198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aceto, N.; Sausgruber, N.; Brinkhaus, H.; Gaidatzis, D.; Martiny-Baron, G.; Mazzarol, G.; Confalonieri, S.; Quarto, M.; Hu, G.; Balwierz, P.J.; et al. Tyrosine phosphatase SHP2 promotes breast cancer progression and maintains tumor-initiating cells via activation of key transcription factors and a positive feedback signaling loop. Nat. Med. 2012, 18, 529–537. [Google Scholar] [CrossRef]
- Zhang, K.; Zhao, H.; Ji, Z.; Zhang, C.; Zhou, P.; Wang, L.; Chen, Q.; Wang, J.; Zhang, P.; Chen, Z.; et al. Shp2 promotes metastasis of prostate cancer by attenuating the PAR3/PAR6/aPKC polarity protein complex and enhancing epithelial-to-mesenchymal transition. Oncogene 2016, 35, 1271–1282. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Li, J.; Gao, Q.; Wei, S.; Yang, B. SHP2 overexpression enhances the invasion and metastasis of ovarian cancer In Vitro and In Vivo. Onco. Targets Ther. 2017, 10, 3881–3891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanda, M.; Kodera, Y. Molecular mechanisms of peritoneal dissemination in gastric cancer. World J. Gastroenterol. 2016, 22, 6829–6840. [Google Scholar] [CrossRef] [PubMed]
- Sun, F.; Feng, M.; Guan, W. Mechanisms of peritoneal dissemination in gastric cancer. Oncol. Lett. 2017, 14, 6991–6998. [Google Scholar] [CrossRef] [Green Version]
- Zhao, M.; Guo, W.; Wu, Y.; Yang, C.; Zhong, L.; Deng, G.; Zhu, Y.; Liu, W.; Gu, Y.; Lu, Y.; et al. SHP2 inhibition triggers anti-tumor immunity and synergizes with PD-1 blockade. Acta Pharm Sin. B 2019, 9, 304–315. [Google Scholar] [CrossRef]
- Duncan, J.S.; Whittle, M.C.; Nakamura, K.; Abell, A.N.; Midland, A.A.; Zawistowski, J.S.; Johnson, N.L.; Granger, D.A.; Jordan, N.V.; Darr, D.B.; et al. Dynamic reprogramming of the kinome in response to targeted MEK inhibition in triple-negative breast cancer. Cell 2012, 149, 307–321. [Google Scholar] [CrossRef] [Green Version]
- Zawistowski, J.S.; Bevill, S.M.; Goulet, D.R.; Stuhlmiller, T.J.; Beltran, A.S.; Olivares-Quintero, J.F.; Singh, D.; Sciaky, N.; Parker, J.S.; Rashid, N.U.; et al. Enhancer remodeling during adaptive bypass to MEK inhibition is attenuated by pharmacologic targeting of the P-TEFb complex. Cancer Discov. 2017, 7, 302–321. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Meyers, B.A.; Czako, B.; Leonard, P.; Mseeh, F.; Harris, A.L.; Wu, Q.; Johnson, S.; Parker, C.A.; Cross, J.B.; et al. Allosteric SHP2 inhibitor, IACS-13909, overcomes EGFR-dependent and EGFR-independent resistance mechanisms toward Osimertinib. Cancer Res. 2020, 80, 4840–4853. [Google Scholar] [CrossRef]
- Fedele, C.; Ran, H.; Diskin, B.; Wei, W.; Jen, J.; Geer, M.J.; Araki, K.; Ozerdem, U.; Simeone, D.M.; Miller, G.; et al. SHP2 inhibition prevents adaptive resistance to MEK inhibitors in multiple cancer models. Cancer Discov. 2018, 8, 1237–1249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dardaei, L.; Wang, H.Q.; Singh, M.; Fordjour, P.; Shaw, K.X.; Yoda, S.; Kerr, G.; Yu, K.; Liang, J.; Cao, Y.; et al. SHP2 inhibition restores sensitivity in ALK-rearranged non-small-cell lung cancer resistant to ALK inhibitors. Nat. Med. 2018, 24, 512–517. [Google Scholar] [CrossRef] [PubMed]
- Pudelko, L.; Jaehrling, F.; Reusch, C.; Vitri, S.; Stroh, C.; Linde, N.; Sanderson, M.P.; Musch, D.; Lebrun, C.J.; Keil, M.; et al. SHP2 inhibition influences therapeutic response to tepotinib in tumors with MET alterations. iScience 2020, 23, 101832. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nagamura, Y.; Miyazaki, M.; Nagano, Y.; Tomiyama, A.; Ohki, R.; Yanagihara, K.; Sakai, R.; Yamaguchi, H. SHP2 as a Potential Therapeutic Target in Diffuse-Type Gastric Carcinoma Addicted to Receptor Tyrosine Kinase Signaling. Cancers 2021, 13, 4309. https://doi.org/10.3390/cancers13174309
Nagamura Y, Miyazaki M, Nagano Y, Tomiyama A, Ohki R, Yanagihara K, Sakai R, Yamaguchi H. SHP2 as a Potential Therapeutic Target in Diffuse-Type Gastric Carcinoma Addicted to Receptor Tyrosine Kinase Signaling. Cancers. 2021; 13(17):4309. https://doi.org/10.3390/cancers13174309
Chicago/Turabian StyleNagamura, Yuko, Makoto Miyazaki, Yoshiko Nagano, Arata Tomiyama, Rieko Ohki, Kazuyoshi Yanagihara, Ryuichi Sakai, and Hideki Yamaguchi. 2021. "SHP2 as a Potential Therapeutic Target in Diffuse-Type Gastric Carcinoma Addicted to Receptor Tyrosine Kinase Signaling" Cancers 13, no. 17: 4309. https://doi.org/10.3390/cancers13174309