
Nuclear Pyruvate Kinase M2 (PKM2) Contributes to Phosphoserine Aminotransferase 1 (PSAT1)-Mediated Cell Migration in EGFR-Activated Lung Cancer Cells

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Reagents and Antibodies
2.2. GST-Pulldown and Mass Spectrometry
2.3. LC/MS Data Collection and Analysis
2.4. Cell Culture
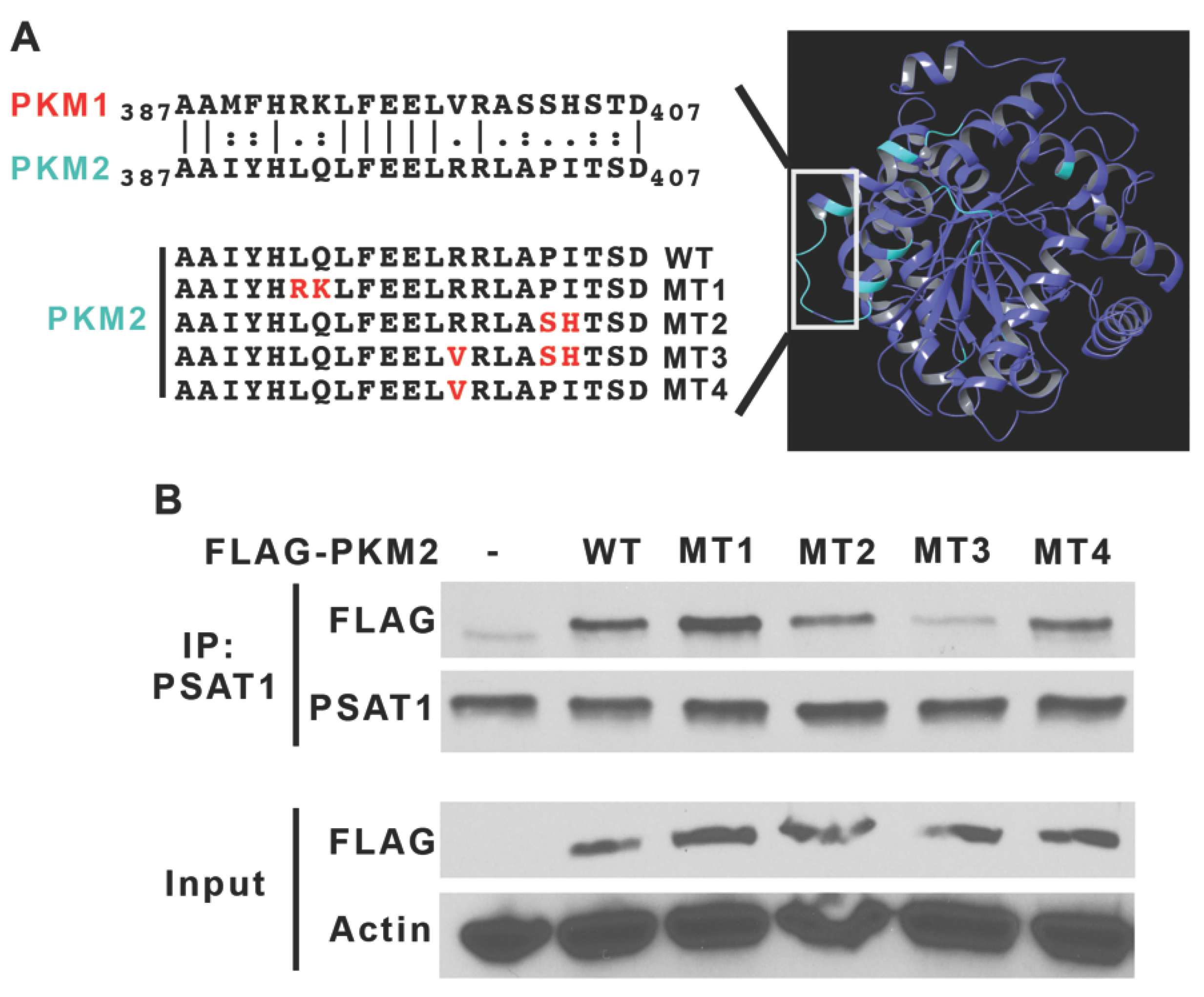
2.5. Plasmid Constructions for Wild-Type and Mutant PKM2
2.6. Transient Transfection
2.7. Generation of Stable Cell Lines
2.8. EGF and Erlotinib Treatment
2.9. Subcellular Fractionation
2.10. Co-Immunoprecipitation (Co-IP)
2.11. Immunoblotting
2.12. Immunofluorescence Staining
2.13. Wound Healing Assay
2.14. Transwell Migration Assay
2.15. Pyruvate Kinase Activity Assay
2.16. Computational Homology Modeling of PKM1 and PKM2
2.17. Bioinformatic Analysis of PSAT1 in EGFR-Mutant Lung Cancers
2.18. Statistical Analysis
3. Results
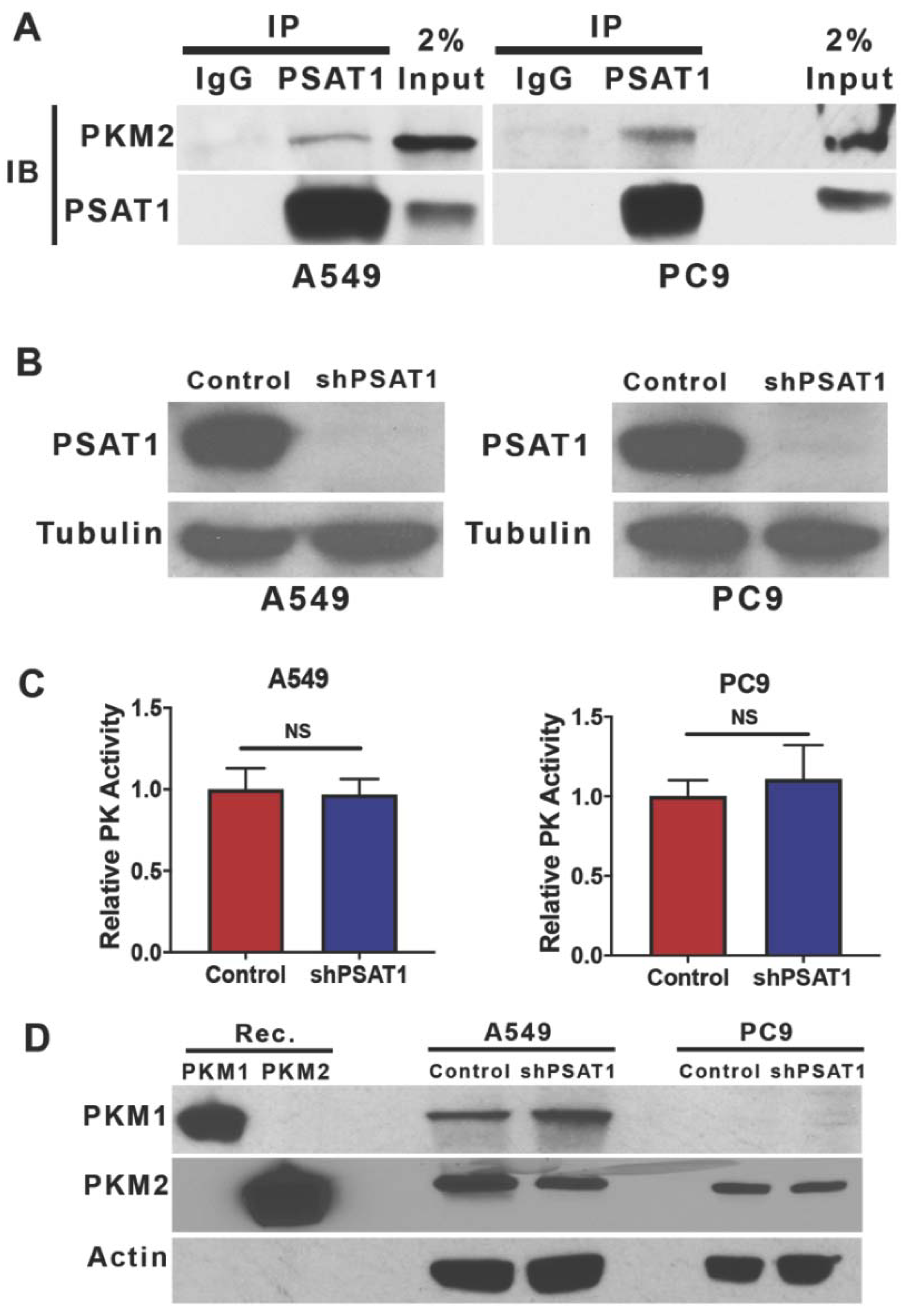
3.1. PKM2 Is a Novel Binding Partner of PSAT1
3.2. Suppression of PSAT1 in NSCLC Cancer Cells Does Not Alter PKM2 Expression or Pyruvate Kinase Activity
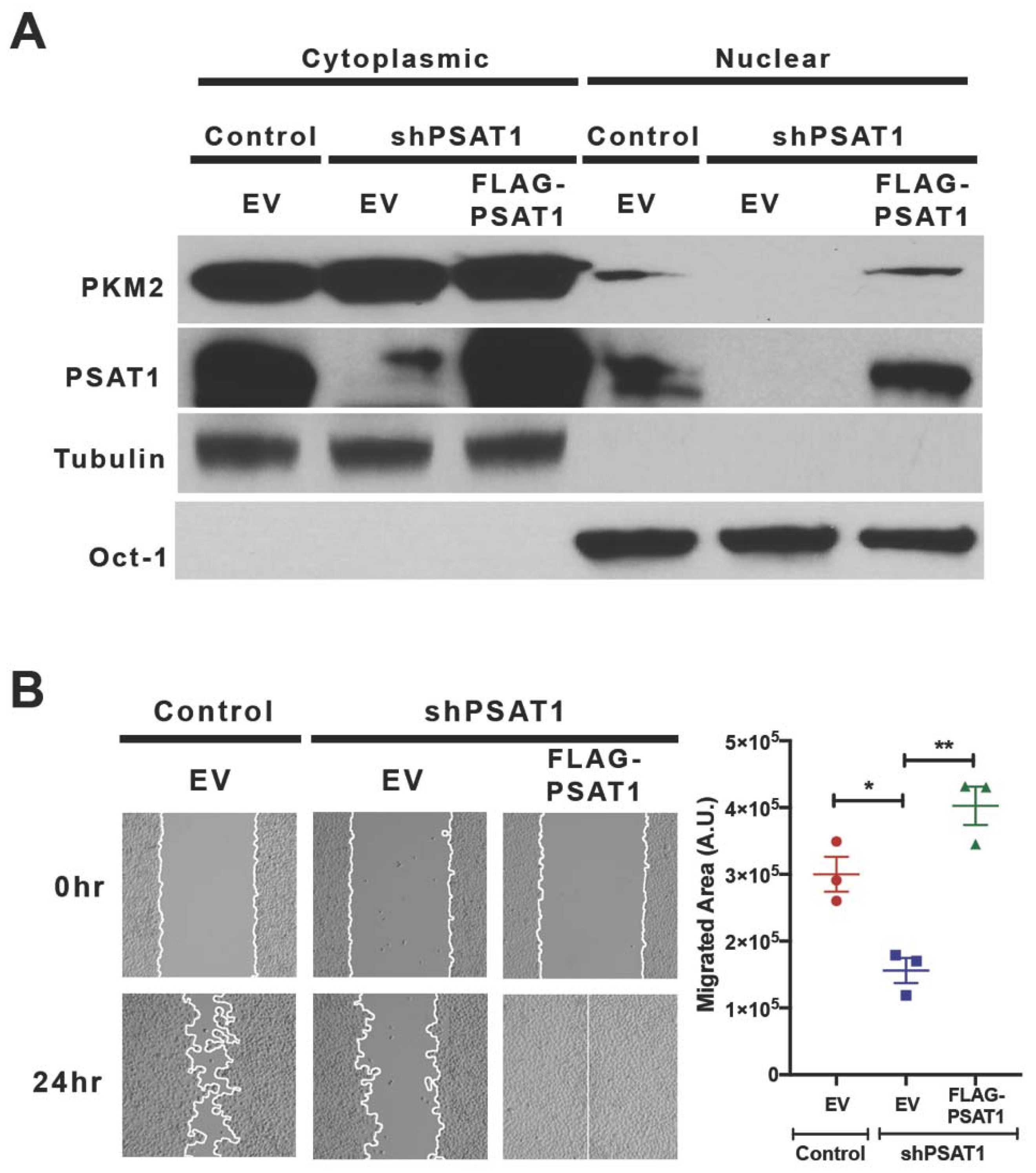
3.3. Silencing of PSAT1 Suppresses the Nuclear Localization of PKM2 in EGFR-Activated NSCLC Cells
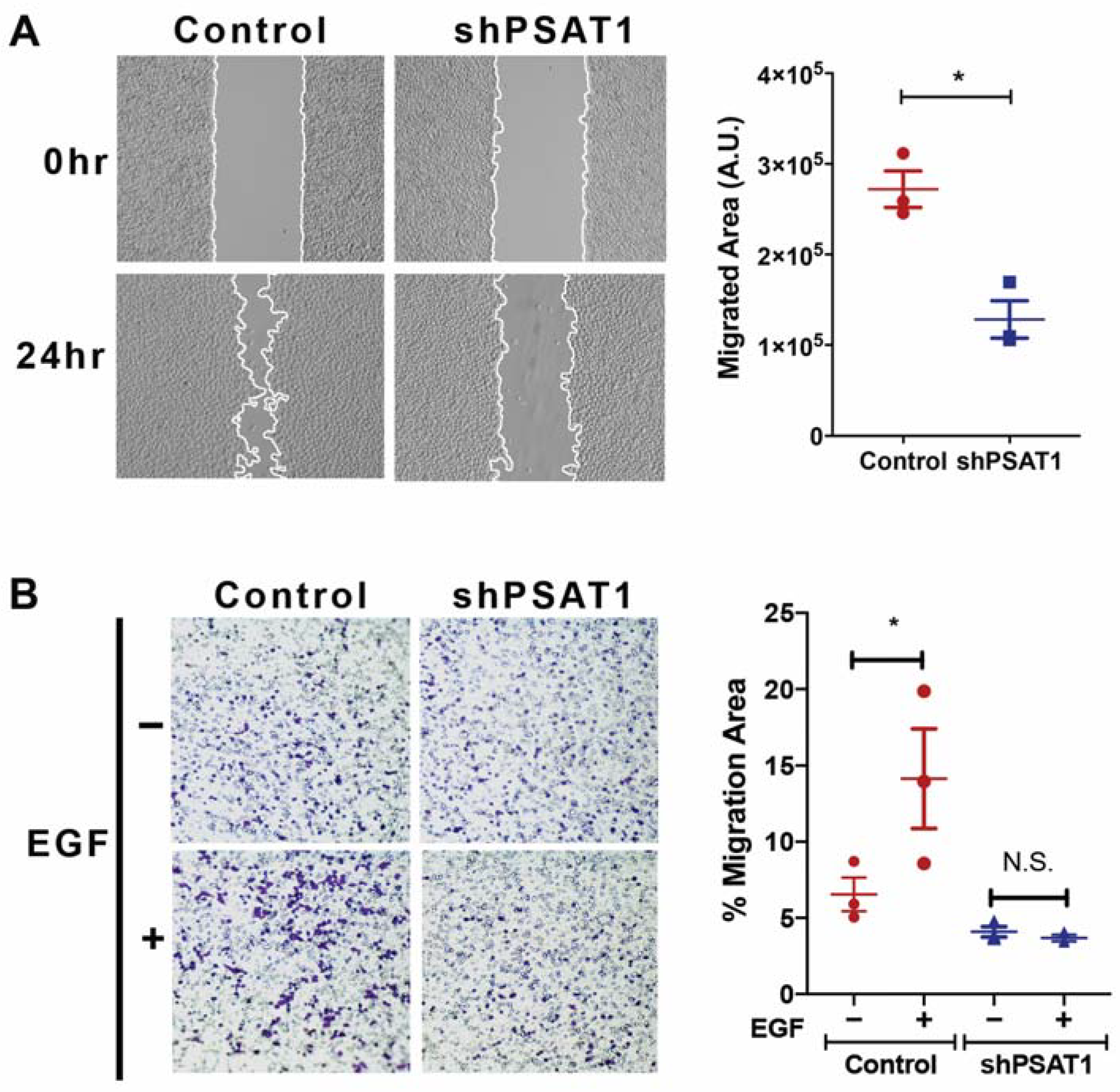
3.4. Loss of PSAT1 Suppresses the Migration of EGFR-Mutant and EGF-Induced EGFR-WT Lung Cancer Cells
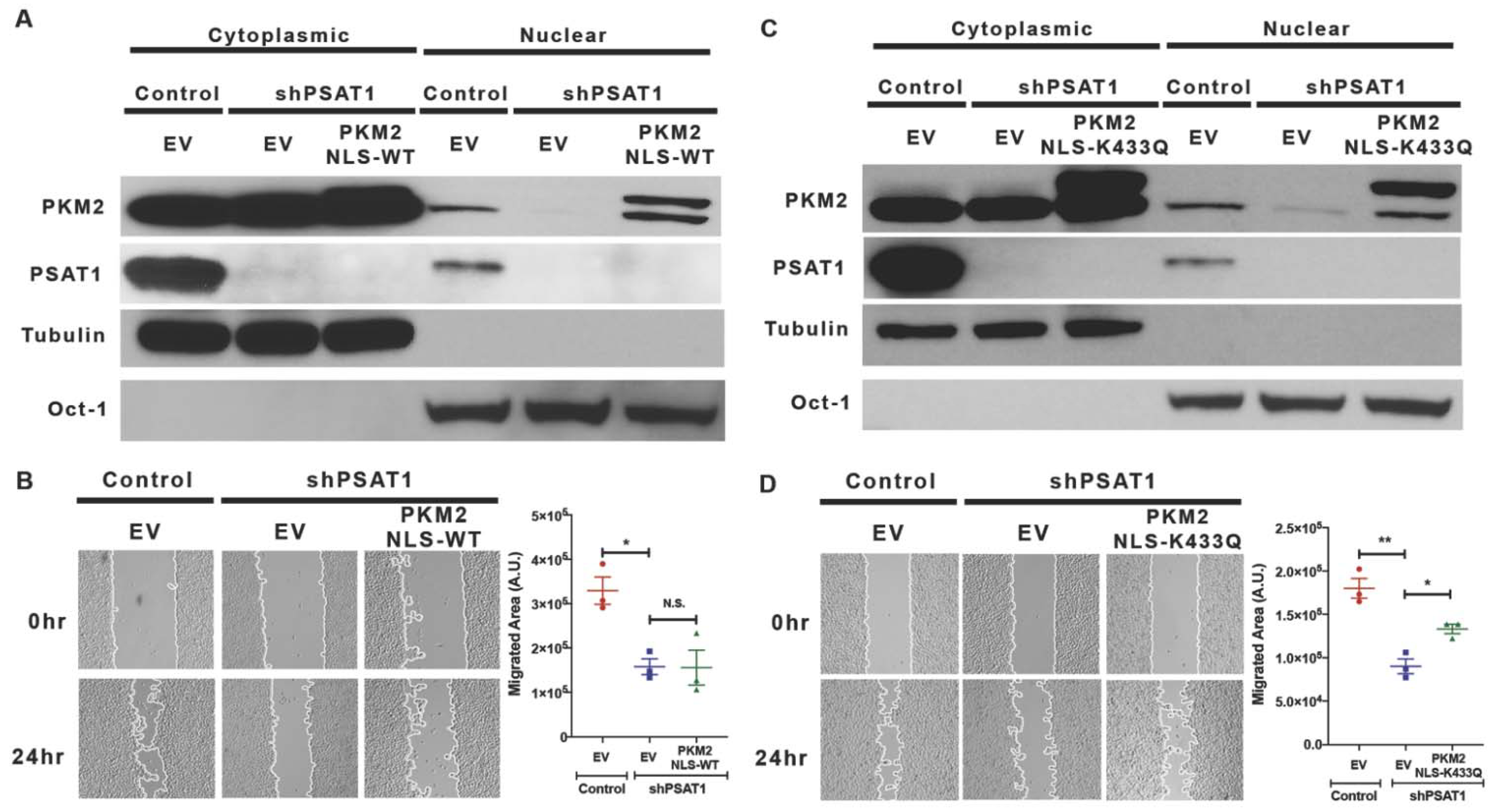
3.5. Re-Expression of Nuclear-Localized Acetyl-Mimetic (K433Q) PKM2 Partially Restores the Migration Defect Due to the Loss of PSAT1 in EGFR-Mutant Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pavlova, N.N.; Thompson, C.B. The Emerging Hallmarks of Cancer Metabolism. Cell Metab. 2016, 23, 27–47. [Google Scholar] [CrossRef] [Green Version]
- Choi, Y.K.; Park, K.G. Targeting Glutamine Metabolism for Cancer Treatment. Biomol. Ther. 2018, 26, 19–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruiz-Perez, M.V.; Sanchez-Jimenez, F.; Alonso, F.J.; Segura, J.A.; Marquez, J.; Medina, M.A. Glutamine, glucose and other fuels for cancer. Curr. Pharm. Des. 2014, 20, 2557–2579. [Google Scholar] [CrossRef] [PubMed]
- Lunt, S.Y.; Vander Heiden, M.G. Aerobic glycolysis: Meeting the metabolic requirements of cell proliferation. Annu. Rev. Cell Dev. Biol. 2011, 27, 441–464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, X.; Li, S. Non-metabolic functions of glycolytic enzymes in tumorigenesis. Oncogene 2017, 36, 2629–2636. [Google Scholar] [CrossRef]
- Lv, L.; Xu, Y.P.; Zhao, D.; Li, F.L.; Wang, W.; Sasaki, N.; Jiang, Y.; Zhou, X.; Li, T.T.; Guan, K.L.; et al. Mitogenic and oncogenic stimulation of K433 acetylation promotes PKM2 protein kinase activity and nuclear localization. Mol. Cell 2013, 52, 340–352. [Google Scholar] [CrossRef] [Green Version]
- Yang, W.; Xia, Y.; Ji, H.; Zheng, Y.; Liang, J.; Huang, W.; Gao, X.; Aldape, K.; Lu, Z. Nuclear PKM2 regulates beta-catenin transactivation upon EGFR activation. Nature 2011, 480, 118–122. [Google Scholar] [CrossRef]
- Yang, W.; Xia, Y.; Hawke, D.; Li, X.; Liang, J.; Xing, D.; Aldape, K.; Hunter, T.; Alfred Yung, W.K.; Lu, Z. PKM2 phosphorylates histone H3 and promotes gene transcription and tumorigenesis. Cell 2012, 150, 685–696. [Google Scholar] [CrossRef] [Green Version]
- Gao, X.; Wang, H.; Yang, J.J.; Liu, X.; Liu, Z.R. Pyruvate kinase M2 regulates gene transcription by acting as a protein kinase. Mol. Cell 2012, 45, 598–609. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.C.; Chien, M.H.; Liu, H.Y.; Chang, Y.C.; Chen, C.K.; Lee, W.J.; Kuo, T.C.; Hsiao, M.; Hua, K.T.; Cheng, T.Y. Nuclear translocation of PKM2/AMPK complex sustains cancer stem cell populations under glucose restriction stress. Cancer Lett. 2018, 421, 28–40. [Google Scholar] [CrossRef]
- Zhang, D.; Jin, N.; Sun, W.; Li, X.; Liu, B.; Xie, Z.; Qu, J.; Xu, J.; Yang, X.; Su, Y.; et al. Phosphoglycerate mutase 1 promotes cancer cell migration independent of its metabolic activity. Oncogene 2017, 36, 2900–2909. [Google Scholar] [CrossRef] [PubMed]
- DeNicola, G.M.; Chen, P.H.; Mullarky, E.; Sudderth, J.A.; Hu, Z.; Wu, D.; Tang, H.; Xie, Y.; Asara, J.M.; Huffman, K.E.; et al. NRF2 regulates serine biosynthesis in non-small cell lung cancer. Nat. Genet. 2015, 47, 1475–1481. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Wu, J.; Cai, J.; He, Z.; Yuan, J.; Zhu, X.; Li, Y.; Li, M.; Guan, H. PSAT1 regulates cyclin D1 degradation and sustains proliferation of non-small cell lung cancer cells. Int. J. Cancer 2015, 136, E39–E50. [Google Scholar] [CrossRef] [PubMed]
- Park, S.M.; Seo, E.H.; Bae, D.H.; Kim, S.S.; Kim, J.; Lin, W.; Kim, K.H.; Park, J.B.; Kim, Y.S.; Yin, J.; et al. Phosphoserine Phosphatase Promotes Lung Cancer Progression through the Dephosphorylation of IRS-1 and a Noncanonical L-Serine-Independent Pathway. Mol. Cells 2019, 42, 604–616. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Guo, S.; Li, Q.; Yang, L.; Xia, Z.; Zhang, L.; Huang, Z.; Zhang, N. Phosphoglycerate dehydrogenase induces glioma cells proliferation and invasion by stabilizing forkhead box M1. J. Neurooncol. 2013, 111, 245–255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mattaini, K.R.; Sullivan, M.R.; Vander Heiden, M.G. The importance of serine metabolism in cancer. J. Cell Biol. 2016, 214, 249–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amelio, I.; Markert, E.K.; Rufini, A.; Antonov, A.V.; Sayan, B.S.; Tucci, P.; Agostini, M.; Mineo, T.C.; Levine, A.J.; Melino, G. p73 regulates serine biosynthesis in cancer. Oncogene 2014, 33, 5039–5046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Metcalf, S.; Dougherty, S.; Kruer, T.; Hasan, N.; Biyik-Sit, R.; Reynolds, L.; Clem, B.F. Selective loss of phosphoserine aminotransferase 1 (PSAT1) suppresses migration, invasion, and experimental metastasis in triple negative breast cancer. Clin. Exp. Metastasis 2019, 37, 187–197. [Google Scholar] [CrossRef]
- Vie, N.; Copois, V.; Bascoul-Mollevi, C.; Denis, V.; Bec, N.; Robert, B.; Fraslon, C.; Conseiller, E.; Molina, F.; Larroque, C.; et al. Overexpression of phosphoserine aminotransferase PSAT1 stimulates cell growth and increases chemoresistance of colon cancer cells. Mol. Cancer 2008, 7, 14. [Google Scholar] [CrossRef] [Green Version]
- Gao, S.; Ge, A.; Xu, S.; You, Z.; Ning, S.; Zhao, Y.; Pang, D. PSAT1 is regulated by ATF4 and enhances cell proliferation via the GSK3beta/beta-catenin/cyclin D1 signaling pathway in ER-negative breast cancer. J. Exp. Clin. Cancer Res. 2017, 36, 179. [Google Scholar] [CrossRef] [Green Version]
- Zheng, M.J.; Li, X.; Hu, Y.X.; Dong, H.; Gou, R.; Nie, X.; Liu, Q.; Ying-Ying, H.; Liu, J.J.; Lin, B. Identification of molecular marker associated with ovarian cancer prognosis using bioinformatics analysis and experiments. J. Cell. Physiol. 2019, 234, 11023–11036. [Google Scholar] [CrossRef]
- Liao, K.M.; Chao, T.B.; Tian, Y.F.; Lin, C.Y.; Lee, S.W.; Chuang, H.Y.; Chan, T.C.; Chen, T.J.; Hsing, C.H.; Sheu, M.J.; et al. Overexpression of the PSAT1 Gene in Nasopharyngeal Carcinoma Is an Indicator of Poor Prognosis. J. Cancer 2016, 7, 1088–1094. [Google Scholar] [CrossRef] [Green Version]
- De Marchi, T.; Timmermans, M.A.; Sieuwerts, A.M.; Smid, M.; Look, M.P.; Grebenchtchikov, N.; Sweep, F.; Smits, J.G.; Magdolen, V.; van Deurzen, C.H.M.; et al. Phosphoserine aminotransferase 1 is associated to poor outcome on tamoxifen therapy in recurrent breast cancer. Sci. Rep. 2017, 7, 2099. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Jia, Y.; Cao, Y.; Wu, S.; Jiang, H.; Sun, X.; Ma, J.; Yin, X.; Mao, A.; Shang, M. Overexpression of Phosphoserine Aminotransferase 1 (PSAT1) Predicts Poor Prognosis and Associates with Tumor Progression in Human Esophageal Squamous Cell Carcinoma. Cell. Physiol. Biochem. 2016, 39, 395–406. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.Y.; Chen, L.P.; Zhang, Y.; Hu, S.K.; Dong, Z.J.; Wu, M.; Chen, Q.X.; Zhuang, Z.Z.; Du, X.J. Integrated transcriptomic analysis reveals hub genes involved in diagnosis and prognosis of pancreatic cancer. Mol. Med. 2019, 25, 47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pollari, S.; Kakonen, S.M.; Edgren, H.; Wolf, M.; Kohonen, P.; Sara, H.; Guise, T.; Nees, M.; Kallioniemi, O. Enhanced serine production by bone metastatic breast cancer cells stimulates osteoclastogenesis. Breast Cancer Res. Treat. 2011, 125, 421–430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kottakis, F.; Nicolay, B.N.; Roumane, A.; Karnik, R.; Gu, H.; Nagle, J.M.; Boukhali, M.; Hayward, M.C.; Li, Y.Y.; Chen, T.; et al. LKB1 loss links serine metabolism to DNA methylation and tumorigenesis. Nature 2016, 539, 390–395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, J.; Wei, R.; Zhang, P.; Kong, B. Overexpression of microRNA-195-5p reduces cisplatin resistance and angiogenesis in ovarian cancer by inhibiting the PSAT1-dependent GSK3beta/beta-catenin signaling pathway. J. Transl. Med. 2019, 17, 190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larsen, C.P.; Trivin-Avillach, C.; Coles, P.; Collins, A.B.; Merchant, M.; Ma, H.; Wilkey, D.W.; Ambruzs, J.M.; Messias, N.C.; Cossey, L.N.; et al. LDL Receptor-Related Protein 2 (Megalin) as a Target Antigen in Human Kidney Anti-Brush Border Antibody Disease. J. Am. Soc. Nephrol. 2018, 29, 644–653. [Google Scholar] [CrossRef] [PubMed]
- Villafuerte, B.C.; Barati, M.T.; Rane, M.J.; Isaacs, S.; Li, M.; Wilkey, D.W.; Merchant, M.L. Over-expression of insulin-response element binding protein-1 (IRE-BP1) in mouse pancreatic islets increases expression of RACK1 and TCTP: Beta cell markers of high glucose sensitivity. Biochim. Biophys. Acta Proteins Proteom. 2017, 1865, 186–194. [Google Scholar] [CrossRef] [Green Version]
- Grada, A.; Otero-Vinas, M.; Prieto-Castrillo, F.; Obagi, Z.; Falanga, V. Research Techniques Made Simple: Analysis of Collective Cell Migration Using the Wound Healing Assay. J. Investig. Dermatol. 2017, 137, e11–e16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baecker, V. ImageJ Macro Tool Sets for Biological Image Analysis. In Proceedings of the ImageJ User and Developer Conference, Luxembourg, 24–26 October 2012; Centre de Recherche Public Henri Tudor: Luxembourg, 2012. [Google Scholar]
- Gallo-Oller, G.; Rey, J.A.; Dotor, J.; Castresana, J.S. Quantitative method for in vitro matrigel invasiveness measurement through image analysis software. Mol. Biol. Rep. 2014, 41, 6335–6341. [Google Scholar] [CrossRef] [PubMed]
- Lumachi, F.; Luisetto, G.; Basso, S.M.; Basso, U.; Brunello, A.; Camozzi, V. Endocrine therapy of breast cancer. Curr. Med. Chem. 2011, 18, 513–522. [Google Scholar] [CrossRef] [PubMed]
- Simon, R.; Lam, A.; Li, M.C.; Ngan, M.; Menenzes, S.; Zhao, Y. Analysis of gene expression data using BRB-ArrayTools. Cancer Inform. 2007, 3, 11–17. [Google Scholar] [CrossRef] [PubMed]
- Snaebjornsson, M.T.; Schulze, A. Non-canonical functions of enzymes facilitate cross-talk between cell metabolic and regulatory pathways. Exp. Mol. Med. 2018, 50, 34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, Z.; Hunter, T. Metabolic Kinases Moonlighting as Protein Kinases. Trends Biochem. Sci. 2018, 43, 301–310. [Google Scholar] [CrossRef] [PubMed]
- Israelsen, W.J.; Vander Heiden, M.G. Pyruvate kinase: Function, regulation and role in cancer. Semin. Cell Dev. Biol. 2015, 43, 43–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaneton, B.; Hillmann, P.; Zheng, L.; Martin, A.C.L.; Maddocks, O.D.K.; Chokkathukalam, A.; Coyle, J.E.; Jankevics, A.; Holding, F.P.; Vousden, K.H.; et al. Serine is a natural ligand and allosteric activator of pyruvate kinase M2. Nature 2012, 491, 458–462. [Google Scholar] [CrossRef] [Green Version]
- Kung, C.; Hixon, J.; Choe, S.; Marks, K.; Gross, S.; Murphy, E.; DeLaBarre, B.; Cianchetta, G.; Sethumadhavan, S.; Wang, X.; et al. Small molecule activation of PKM2 in cancer cells induces serine auxotrophy. Chem. Biol. 2012, 19, 1187–1198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, J.; Mancuso, A.; Tong, X.; Ward, P.S.; Fan, J.; Rabinowitz, J.D.; Thompson, C.B. Pyruvate kinase M2 promotes de novo serine synthesis to sustain mTORC1 activity and cell proliferation. Proc. Natl. Acad. Sci. USA 2012, 109, 6904–6909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parnell, K.M.; Foulks, J.M.; Nix, R.N.; Clifford, A.; Bullough, J.; Luo, B.; Senina, A.; Vollmer, D.; Liu, J.; McCarthy, V.; et al. Pharmacologic activation of PKM2 slows lung tumor xenograft growth. Mol. Cancer Ther. 2013, 12, 1453–1460. [Google Scholar] [CrossRef] [Green Version]
- Li, N.; Feng, L.; Liu, H.; Wang, J.; Kasembeli, M.; Tran, M.K.; Tweardy, D.J.; Lin, S.H.; Chen, J. PARP Inhibition Suppresses Growth of EGFR-Mutant Cancers by Targeting Nuclear PKM2. Cell Rep. 2016, 15, 843–856. [Google Scholar] [CrossRef] [Green Version]
- Arao, T.; Fukumoto, H.; Takeda, M.; Tamura, T.; Saijo, N.; Nishio, K. Small in-frame deletion in the epidermal growth factor receptor as a target for ZD6474. Cancer Res. 2004, 64, 9101–9104. [Google Scholar] [CrossRef] [Green Version]
- Yamadori, T.; Ishii, Y.; Homma, S.; Morishima, Y.; Kurishima, K.; Itoh, K.; Yamamoto, M.; Minami, Y.; Noguchi, M.; Hizawa, N. Molecular mechanisms for the regulation of Nrf2-mediated cell proliferation in non-small-cell lung cancers. Oncogene 2012, 31, 4768–4777. [Google Scholar] [CrossRef] [Green Version]
- Lauand, C.; Rezende-Teixeira, P.; Cortez, B.A.; Niero, E.L.; Machado-Santelli, G.M. Independent of ErbB1 gene copy number, EGF stimulates migration but is not associated with cell proliferation in non-small cell lung cancer. Cancer Cell Int. 2013, 13, 38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, Y.; Liang, X.; Xu, J.; Cai, X. miR-424 targets AKT3 and PSAT1 and has a tumor-suppressive role in human colorectal cancer. Cancer Manag. Res. 2018, 10, 6537–6547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, S.; Jiang, H.; Fang, S.; Yin, F.; Wang, Z.; Jia, Y.; Sun, X.; Wu, S.; Jiang, T.; Mao, A. MicroRNA-340 Inhibits Esophageal Cancer Cell Growth and Invasion by Targeting Phosphoserine Aminotransferase 1. Cell. Physiol. Biochem. 2015, 37, 375–386. [Google Scholar] [CrossRef] [PubMed]
- Bhardwaj, A.; Das, S. SIRT6 deacetylates PKM2 to suppress its nuclear localization and oncogenic functions. Proc. Natl. Acad. Sci. USA 2016, 113, E538–E547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, B.; Zheng, A.; Hydbring, P.; Ambroise, G.; Ouchida, A.T.; Goiny, M.; Vakifahmetoglu-Norberg, H.; Norberg, E. PHGDH Defines a Metabolic Subtype in Lung Adenocarcinomas with Poor Prognosis. Cell Rep. 2017, 19, 2289–2303. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.C.; Shyh-Chang, N.; Yang, H.; Rai, A.; Umashankar, S.; Ma, S.; Soh, B.S.; Sun, L.L.; Tai, B.C.; Nga, M.E.; et al. Glycine decarboxylase activity drives non-small cell lung cancer tumor-initiating cells and tumorigenesis. Cell 2012, 148, 259–272. [Google Scholar] [CrossRef] [Green Version]
- Huangyang, P.; Simon, M.C. Hidden features: Exploring the non-canonical functions of metabolic enzymes. Dis. Model. Mech. 2018, 11, dmm033365. [Google Scholar] [CrossRef] [Green Version]
- Tsao, A.S.; Scagliotti, G.V.; Bunn, P.A., Jr.; Carbone, D.P.; Warren, G.W.; Bai, C.; de Koning, H.J.; Yousaf-Khan, A.U.; McWilliams, A.; Tsao, M.S.; et al. Scientific Advances in Lung Cancer 2015. J. Thorac. Oncol. 2016, 11, 613–638. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.C.; Cheng, T.Y.; Huang, S.M.; Su, C.Y.; Yang, P.W.; Lee, J.M.; Chen, C.K.; Hsiao, M.; Hua, K.T.; Kuo, M.L. Cytosolic PKM2 stabilizes mutant EGFR protein expression through regulating HSP90-EGFR association. Oncogene 2016, 35, 3387–3398. [Google Scholar] [CrossRef] [PubMed]
- Fan, F.T.; Shen, C.S.; Tao, L.; Tian, C.; Liu, Z.G.; Zhu, Z.J.; Liu, Y.P.; Pei, C.S.; Wu, H.Y.; Zhang, L.; et al. PKM2 regulates hepatocellular carcinoma cell epithelial-mesenchymal transition and migration upon EGFR activation. Asian Pac. J. Cancer Prev. 2014, 15, 1961–1970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, J.; Cao, R.; Zhang, Y.; Xia, Y.; Zheng, Y.; Li, X.; Wang, L.; Yang, W.; Lu, Z. PKM2 dephosphorylation by Cdc25A promotes the Warburg effect and tumorigenesis. Nat. Commun. 2016, 7, 12431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.; Youhong, T.; Tan, Y.; He, Y.; Ban, Y.; Cai, J.; Li, X.; Xiong, W.; Zeng, Z.; Li, G.; et al. EGFR-PKM2 signaling promotes the metastatic potential of nasopharyngeal carcinoma through induction of FOSL1 and ANTXR2. Carcinogenesis 2020, 41, 723–733. [Google Scholar] [CrossRef] [Green Version]
- Yang, W.; Zheng, Y.; Xia, Y.; Ji, H.; Chen, X.; Guo, F.; Lyssiotis, C.A.; Aldape, K.; Cantley, L.C.; Lu, Z. ERK1/2-dependent phosphorylation and nuclear translocation of PKM2 promotes the Warburg effect. Nat. Cell Biol. 2012, 14, 1295–1304. [Google Scholar] [CrossRef] [Green Version]
- Hamabe, A.; Konno, M.; Tanuma, N.; Shima, H.; Tsunekuni, K.; Kawamoto, K.; Nishida, N.; Koseki, J.; Mimori, K.; Gotoh, N.; et al. Role of pyruvate kinase M2 in transcriptional regulation leading to epithelial-mesenchymal transition. Proc. Natl. Acad. Sci. USA 2014, 111, 15526–15531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, F.; Yoshimoto, S.; Okamura, K.; Ikebe, T.; Hashimoto, S. Nuclear PKM2 promotes the progression of oral squamous cell carcinoma by inducing EMT and post-translationally repressing TGIF2. Oncotarget 2018, 9, 33745–33761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Acuna-Hidalgo, R.; Schanze, D.; Kariminejad, A.; Nordgren, A.; Kariminejad, M.H.; Conner, P.; Grigelioniene, G.; Nilsson, D.; Nordenskjold, M.; Wedell, A.; et al. Neu-Laxova syndrome is a heterogeneous metabolic disorder caused by defects in enzymes of the L-serine biosynthesis pathway. Am. J. Hum. Genet. 2014, 95, 285–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Biyik-Sit, R.; Kruer, T.; Dougherty, S.; Bradley, J.A.; Wilkey, D.W.; Merchant, M.L.; Trent, J.O.; Clem, B.F. Nuclear Pyruvate Kinase M2 (PKM2) Contributes to Phosphoserine Aminotransferase 1 (PSAT1)-Mediated Cell Migration in EGFR-Activated Lung Cancer Cells. Cancers 2021, 13, 3938. https://doi.org/10.3390/cancers13163938
Biyik-Sit R, Kruer T, Dougherty S, Bradley JA, Wilkey DW, Merchant ML, Trent JO, Clem BF. Nuclear Pyruvate Kinase M2 (PKM2) Contributes to Phosphoserine Aminotransferase 1 (PSAT1)-Mediated Cell Migration in EGFR-Activated Lung Cancer Cells. Cancers. 2021; 13(16):3938. https://doi.org/10.3390/cancers13163938
Chicago/Turabian StyleBiyik-Sit, Rumeysa, Traci Kruer, Susan Dougherty, James A. Bradley, Daniel W. Wilkey, Michael L. Merchant, John O. Trent, and Brian F. Clem. 2021. "Nuclear Pyruvate Kinase M2 (PKM2) Contributes to Phosphoserine Aminotransferase 1 (PSAT1)-Mediated Cell Migration in EGFR-Activated Lung Cancer Cells" Cancers 13, no. 16: 3938. https://doi.org/10.3390/cancers13163938