
CXCR6-CXCL16 Axis Promotes Breast Cancer by Inducing Oncogenic Signaling

 ,
,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Tissue Specimens
2.2. Immunohistochemical Staining and Evaluation of CXCR6/CXCL16 and ADAM10
2.3. Cell Culture
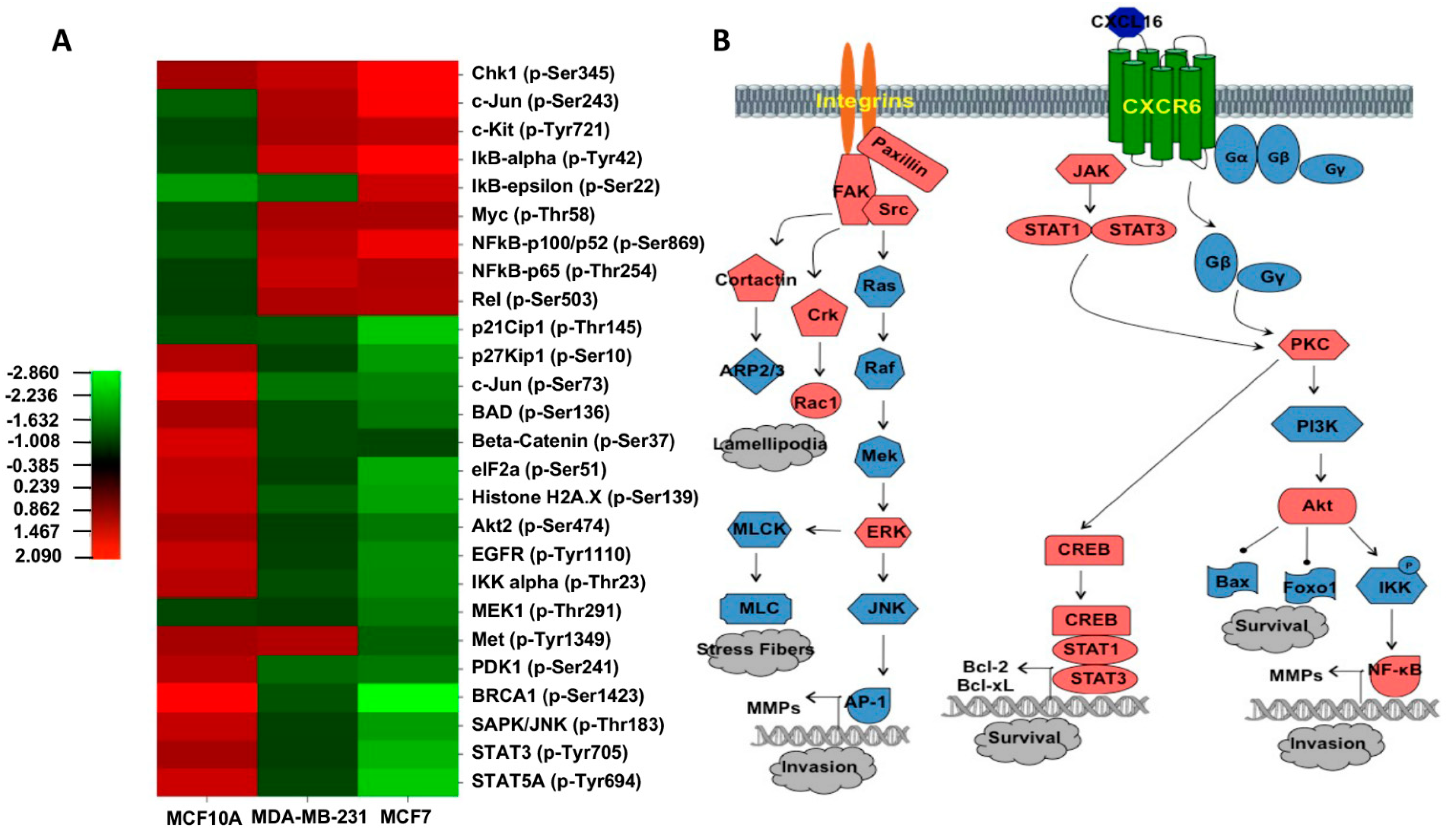
2.4. Cancer Signaling Phosphorylation Antibody Array
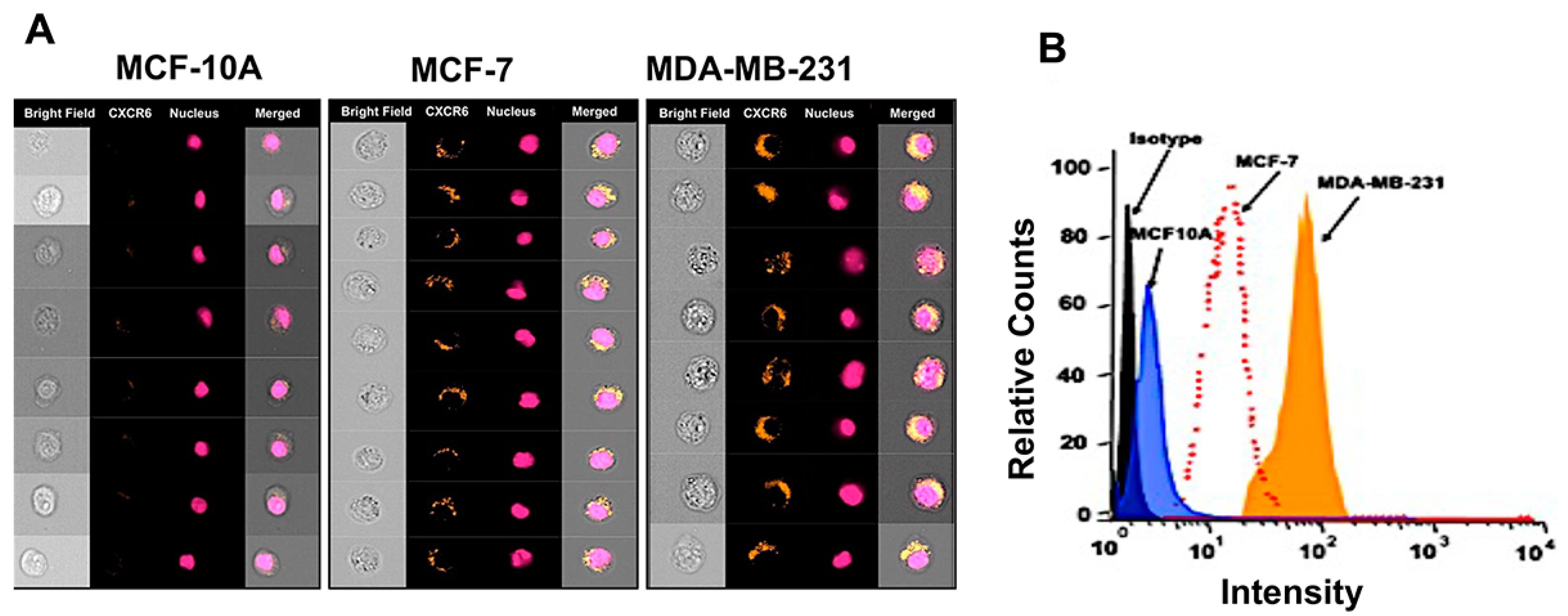
2.5. Image-Based Analysis of CXCR6 Expression
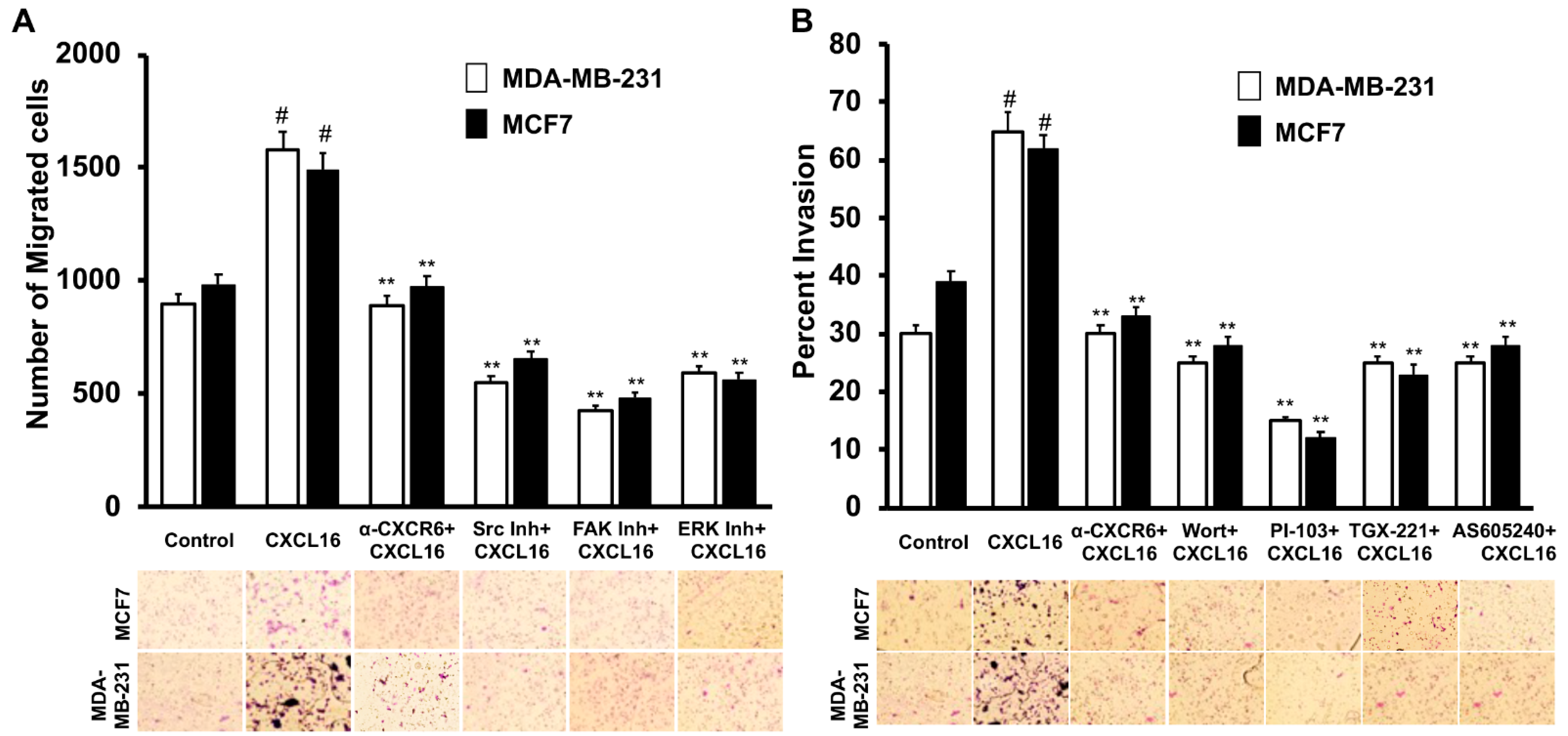
2.6. Cell Migration and Invasion Assay
2.7. Actin Polymerization
2.8. Statistics
3. Results
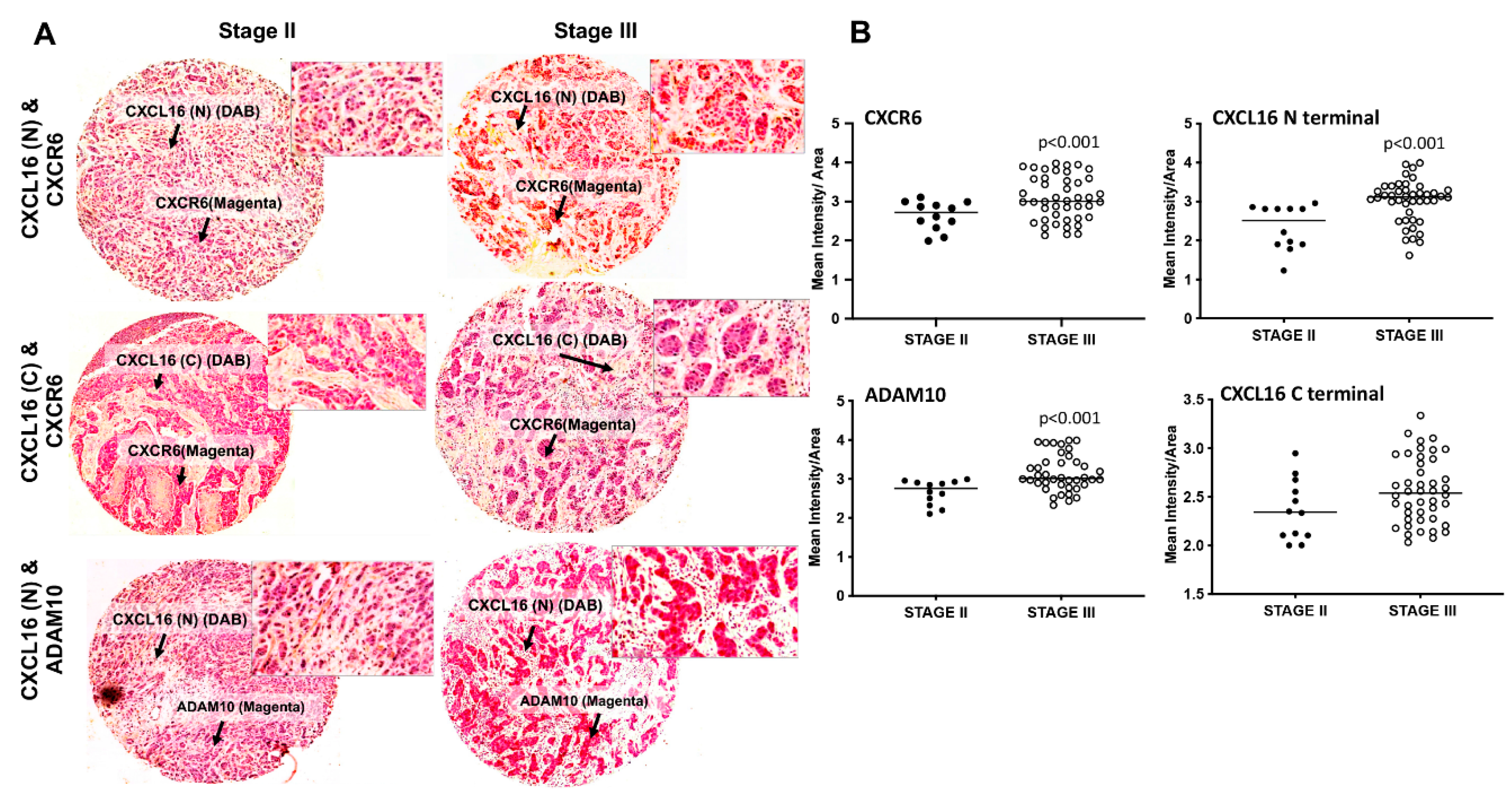
3.1. Expression of ADAM10, CXCL16, and CXCR6 in BrCa Tissues and Cell Lines
3.2. Molecular Pathways Activated by the CXCR6/CXCL16 Axis
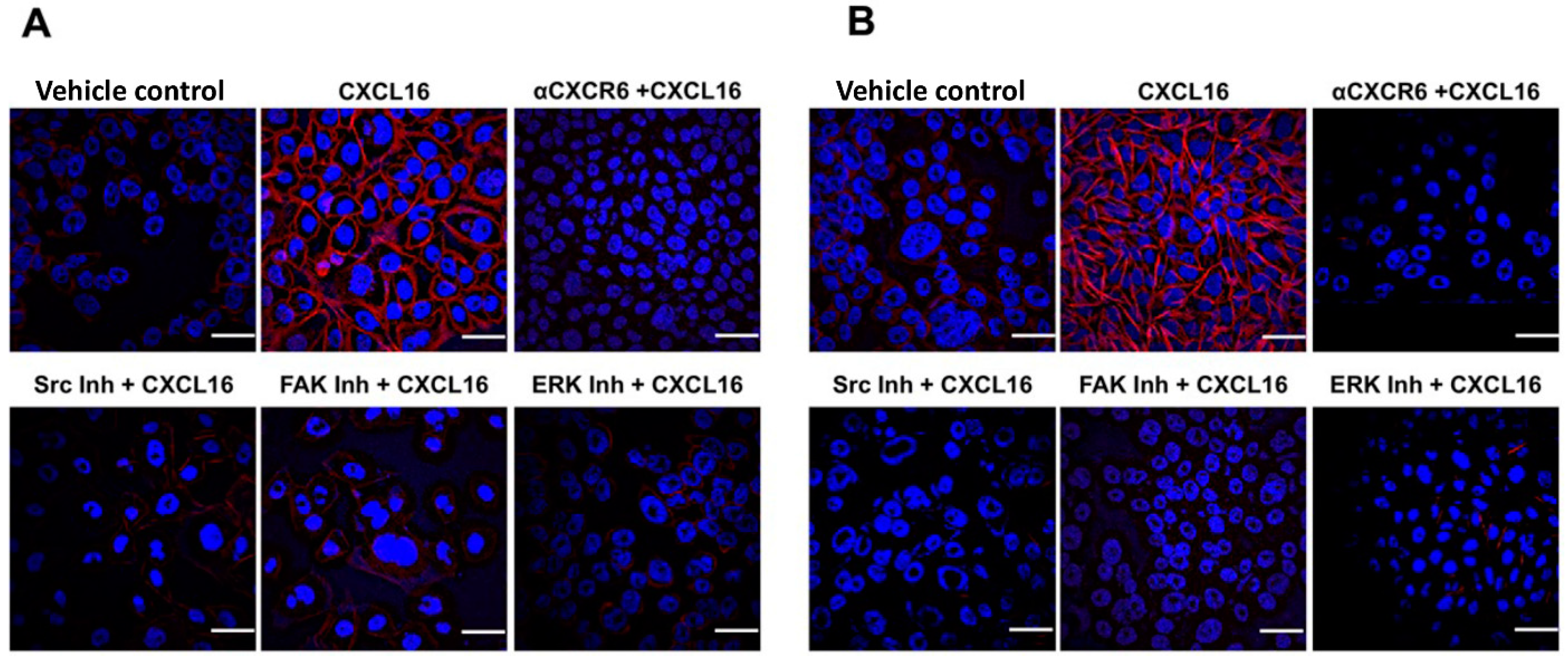
3.3. CXCR6 Signaling Promotes BrCa Cell Migration through Src, FAK, and ERK1/2 Pathways and Invasion through the PI3K Pathway
3.4. CXCR6 Signaling Promotes F-Actin Polymerization in BrCa Cells
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Palacios-Arreola, M.I.; Nava-Castro, K.E.; Castro, J.I.; Garcia-Zepeda, E.; Carrero, J.C.; Morales-Montor, J. The role of chemokines in breast cancer pathology and its possible use as therapeutic targets. J. Immunol. Res. 2014, 2014, 849720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esquivel-Velazquez, M.; Ostoa-Saloma, P.; Palacios-Arreola, M.I.; Nava-Castro, K.E.; Castro, J.I.; Morales-Montor, J. The role of cytokines in breast cancer development and progression. J. Interferon Cytokine Res. 2015, 35, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tao, Z.; Shi, A.; Lu, C.; Song, T.; Zhang, Z.; Zhao, J. Breast Cancer: Epidemiology and Etiology. Cell Biochem. Biophys. 2015, 72, 333–338. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics. CA Cancer J. Clin. 2018, 68, 7–30. [Google Scholar] [CrossRef]
- Johnson-Holiday, C.; Singh, R.; Johnson, E.L.; Grizzle, W.E.; Lillard, J.W., Jr.; Singh, S. CCR9-CCL25 interactions promote cisplatin resistance in breast cancer cell through Akt activation in a PI3K-dependent and FAK-independent fashion. World J. Surg. Oncol. 2011, 9, 46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Hayre, M.; Salanga, C.L.; Handel, T.M.; Allen, S.J. Chemokines and cancer: Migration, intracellular signalling and intercellular communication in the microenvironment. Biochem. J. 2008, 409, 635–649. [Google Scholar] [CrossRef] [Green Version]
- Singh, R.; Lillard, J.W., Jr.; Singh, S. Chemokines: Key players in cancer progression and metastasis. Front. Biosci. Schol. Ed. 2011, 3, 1569–1582. [Google Scholar] [PubMed] [Green Version]
- Sarvaiya, P.J.; Guo, D.; Ulasov, I.; Gabikian, P.; Lesniak, M.S. Chemokines in tumor progression and metastasis. Oncotarget 2013, 4, 2171–2185. [Google Scholar] [CrossRef] [Green Version]
- King, J.; Mir, H.; Singh, S. Association of Cytokines and Chemokines in Pathogenesis of Breast Cancer. Prog. Mol. Biol. Transl. Sci. 2017, 151, 113–136. [Google Scholar] [CrossRef] [PubMed]
- Xiao, G.; Wang, X.; Wang, J.; Zu, L.; Cheng, G.; Hao, M.; Sun, X.; Xue, Y.; Lu, J.; Wang, J. CXCL16/CXCR6 chemokine signaling mediates breast cancer progression by pERK1/2-dependent mechanisms. Oncotarget 2015, 6, 14165–14178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilbanks, A.; Zondlo, S.C.; Murphy, K.; Mak, S.; Soler, D.; Langdon, P.; Andrew, D.P.; Wu, L.; Briskin, M. Expression cloning of the STRL33/BONZO/TYMSTRligand reveals elements of CC, CXC, and CX3C chemokines. J. Immunol. 2001, 166, 5145–5154. [Google Scholar] [CrossRef] [Green Version]
- Gough, P.J.; Garton, K.J.; Wille, P.T.; Rychlewski, M.; Dempsey, P.J.; Raines, E.W. A disintegrin and metalloproteinase 10-mediated cleavage and shedding regulates the cell surface expression of CXC chemokine ligand. J. Immunol. 2004, 172, 3678–3685. [Google Scholar] [CrossRef] [Green Version]
- Matloubian, M.; David, A.; Engel, S.; Ryan, J.E.; Cyster, J.G. A transmembrane CXC chemokine is a ligand for HIV-coreceptor Bonzo. Nat. Immunol. 2000, 1, 298–304. [Google Scholar] [CrossRef]
- Mir, H.; Singh, R.; Kloecker, G.H.; Lillard, J.W., Jr.; Singh, S. CXCR6 expression in non-small cell lung carcinoma supports metastatic process via modulating metalloproteinases. Oncotarget 2015, 6, 9985–9998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.T.; Lee, S.D.; Lee, J.Z.; Chung, M.K.; Ha, H.K. Expression analysis and clinical significance of CXCL16/CXCR6 in patients with bladder cancer. Oncol. Lett. 2013, 5, 229–235. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Kapur, N.; Mir, H.; Singh, N.; Lillard, J.W., Jr.; Singh, S. CXCR6-CXCL16 axis promotes prostate cancer by mediating cytoskeleton rearrangement via Ezrin activation and alphavbeta3 integrin clustering. Oncotarget 2016, 7, 7343–7353. [Google Scholar] [CrossRef] [PubMed]
- Duffy, M.J.; Mullooly, M.; O’Donovan, N.; Sukor, S.; Crown, J.; Pierce, A.; McGowan, P.M. The ADAMs family of proteases: New biomarkers and therapeutic targets for cancer? Clin. Proteom. 2011, 8, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duffy, M.J.; McKiernan, E.; O’Donovan, N.; McGowan, P.M. Role of ADAMs in cancer formation and progression. Clin. Cancer Res. 2009, 15, 1140–1144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mullooly, M.; McGowan, P.M.; Crown, J.; Duffy, M.J. The ADAMs family of proteases as targets for the treatment of cancer. Cancer Biol. 2016, 17, 870–880. [Google Scholar] [CrossRef] [PubMed]
- Feldinger, K.; Generali, D.; Kramer-Marek, G.; Gijsen, M.; Ng, T.B.; Wong, J.H.; Strina, C.; Cappelletti, M.; Andreis, D.; Li, J.L.; et al. ADAM10 mediates trastuzumab resistance and is correlated with survival in HER2 positive breast cancer. Oncotarget 2014, 5, 6633–6646. [Google Scholar] [CrossRef] [Green Version]
- Mir, H.; Kaur, G.; Kapur, N.; Bae, S.; Lillard, J.W., Jr.; Singh, S. Higher CXCL16 exodomain is associated with aggressive ovarian cancer and promotes the disease by CXCR6 activation and MMP modulation. Sci. Rep. 2019, 9, 2527. [Google Scholar] [CrossRef]
- Sharma, P.K.; Singh, R.; Novakovic, K.R.; Eaton, J.W.; Grizzle, W.E.; Singh, S. CCR9 mediates PI3K/AKT-dependent antiapoptotic signals in prostate cancer cells and inhibition of CCR9-CCL25 interaction enhances the cytotoxic effects of etoposide. Int. J. Cancer 2010, 127, 2020–2030. [Google Scholar] [CrossRef] [PubMed]
- Bromann, P.A.; Korkaya, H.; Courtneidge, S.A. The interplay between Src family kinases and receptor tyrosine kinases. Oncogene 2004, 23, 7957–7968. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Goswami, S.; Lapidus, K.; Wells, A.L.; Wyckoff, J.B.; Sahai, E.; Singer, R.H.; Segall, J.E.; Condeelis, J.S. Identification and testing of a gene expression signature of invasive carcinoma cells within primary mammary tumors. Cancer Res. 2004, 64, 8585–8594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martellucci, S.; Clementi, L.; Sabetta, S.; Mattei, V.; Botta, L.; Angelucci, A. Src Family Kinases as Therapeutic Targets in Advanced Solid Tumors: What We Have Learned so Far. Cancers 2020, 12, 1448. [Google Scholar] [CrossRef] [PubMed]
- Rigiracciolo, D.C.; Cirillo, F.; Talia, M.; Muglia, L.; Gutkind, J.S.; Maggiolini, M.; Lappano, R. Focal Adhesion Kinase Fine Tunes Multifaced Signals toward Breast Cancer Progression. Cancers 2021, 13, 645. [Google Scholar] [CrossRef]
- McCubrey, J.A.; Steelman, L.S.; Chappell, W.H.; Abrams, S.L.; Wong, E.W.; Chang, F.; Lehmann, B.; Terrian, D.M.; Milella, M.; Tafuri, A.; et al. Roles of the Raf/MEK/ERK pathway in cell growth, malignant transformation and drug resistance. Biochim. Biophys. Acta 2007, 1773, 1263–1284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicholson, K.M.; Anderson, N.G. The protein kinase B/Akt signalling pathway in human malignancy. Cell Signal. 2002, 14, 381–395. [Google Scholar] [CrossRef]
- Rossi, D.; Zlotnik, A. The biology of chemokines and their receptors. Annu. Rev. Immunol. 2000, 18, 217–242. [Google Scholar] [CrossRef]
- Zlotnik, A.; Yoshie, O. Chemokines: A new classification system and their role in immunity. Immunity 2000, 12, 121–127. [Google Scholar] [CrossRef] [Green Version]
- Muller, A.; Homey, B.; Soto, H.; Ge, N.; Catron, D.; Buchanan, M.E.; McClanahan, T.; Murphy, E.; Yuan, W.; Wagner, S.N.; et al. Involvement of chemokine receptors in breast cancer metastasis. Nature 2001, 410, 50–56. [Google Scholar] [CrossRef]
- Scotton, C.J.; Wilson, J.L.; Milliken, D.; Stamp, G.; Balkwill, F.R. Epithelial cancer cell migration: A role for chemokine receptors? Cancer Res. 2001, 61, 4961–4965. [Google Scholar]
- Gutwein, P.; Schramme, A.; Sinke, N.; Abdel-Bakky, M.S.; Voss, B.; Obermuller, N.; Doberstein, K.; Koziolek, M.; Fritzsche, F.; Johannsen, M.; et al. Tumoural CXCL16 expression is a novel prognostic marker of longer survival times in renal cell cancer patients. Eur. J. Cancer 2009, 45, 478–489. [Google Scholar] [CrossRef]
- Wagsater, D.; Dimberg, J. Expression of chemokine receptor CXCR6 in human colorectal adenocarcinomas. Anticancer. Res. 2004, 24, 3711–3714. [Google Scholar]
- Wente, M.N.; Gaida, M.M.; Mayer, C.; Michalski, C.W.; Haag, N.; Giese, T.; Felix, K.; Bergmann, F.; Giese, N.A.; Friess, H. Expression and potential function of the CXC chemokine CXCL16 in pancreatic ductal adenocarcinoma. Int. J. Oncol. 2008, 33, 297–308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, L.; Chen, N.; Li, Y.; Zheng, H.; Lei, Q. CXCR6/CXCL16 functions as a regulator in metastasis and progression of cancer. Biochim. Biophys. Acta 2010, 1806, 42–49. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.; Henderson, F.C., Jr.; Yi, Q.; Lei, Q.; Li, Y.; Chen, N. Chemokine CXCL16 expression suppresses migration and invasiveness and induces apoptosis in breast cancer cells. Mediat. Inflamm. 2014, 2014, 478641. [Google Scholar] [CrossRef] [PubMed]
- You, B.; Shan, Y.; Shi, S.; Li, X.; You, Y. Effects of ADAM10 upregulation on progression, migration, and prognosis of nasopharyngeal carcinoma. Cancer Sci. 2015, 106, 1506–1514. [Google Scholar] [CrossRef] [Green Version]
- Gaida, M.M.; Haag, N.; Gunther, F.; Tschaharganeh, D.F.; Schirmacher, P.; Friess, H.; Giese, N.A.; Schmidt, J.; Wente, M.N. Expression of A disintegrin and metalloprotease 10 in pancreatic carcinoma. Int. J. Mol. Med. 2010, 26, 281–288. [Google Scholar]
- Groot, A.J.; Vooijs, M.A. The role of Adams in Notch signaling. Adv. Exp. Med. Biol. 2012, 727, 15–36. [Google Scholar] [CrossRef] [Green Version]
- Kiefel, H.; Bondong, S.; Hazin, J.; Ridinger, J.; Schirmer, U.; Riedle, S.; Altevogt, P. L1CAM: A major driver for tumor cell invasion and motility. Cell Adh. Migr. 2012, 6, 374–384. [Google Scholar] [CrossRef] [Green Version]
- Thakur, R.; Trivedi, R.; Rastogi, N.; Singh, M.; Mishra, D.P. Inhibition of STAT3, FAK and Src mediated signaling reduces cancer stem cell load, tumorigenic potential and metastasis in breast cancer. Sci. Rep. 2015, 5, 10194. [Google Scholar] [CrossRef]
- Bolos, V.; Gasent, J.M.; Lopez-Tarruella, S.; Grande, E. The dual kinase complex FAK-Src as a promising therapeutic target in cancer. OncoTargets Ther. 2010, 3, 83–97. [Google Scholar] [CrossRef] [Green Version]
- Benzina, S.; Harquail, J.; Guerrette, R.; O’Brien, P.; Jean, S.; Crapoulet, N.; Robichaud, G.A. Breast Cancer Malignant Processes are Regulated by Pax-5 Through the Disruption of FAK Signaling Pathways. J. Cancer 2016, 7, 2035–2044. [Google Scholar] [CrossRef] [Green Version]
- McLean, G.W.; Carragher, N.O.; Avizienyte, E.; Evans, J.; Brunton, V.G.; Frame, M.C. The role of focal-adhesion kinase in cancer—A new therapeutic opportunity. Nat. Rev. Cancer 2005, 5, 505–515. [Google Scholar] [CrossRef]
- Tserga, A.; Chatziandreou, I.; Michalopoulos, N.V.; Patsouris, E.; Saetta, A.A. Mutation of genes of the PI3K/AKT pathway in breast cancer supports their potential importance as biomarker for breast cancer aggressiveness. Virchows Arch. 2016, 469, 35–43. [Google Scholar] [CrossRef]
- Yang, S.X.; Polley, E.; Lipkowitz, S. New insights on PI3K/AKT pathway alterations and clinical outcomes in breast cancer. Cancer Treat. Rev. 2016, 45, 87–96. [Google Scholar] [CrossRef] [Green Version]
- Gavert, N.; Ben-Shmuel, A.; Lemmon, V.; Brabletz, T.; Ben-Ze’ev, A. Nuclear factor-kappaB signaling and ezrin are essential for L1-mediated metastasis of colon cancer cells. J. Cell Sci. 2010, 123, 2135–2143. [Google Scholar] [CrossRef] [Green Version]
- Morton, S.; Davis, R.J.; McLaren, A.; Cohen, P. A reinvestigation of the multisite phosphorylation of the transcription factor c-Jun. EMBO J. 2003, 22, 3876–3886. [Google Scholar] [CrossRef] [Green Version]
- Davis, R.J. Signal transduction by the JNK group of MAP kinases. Cell 2000, 103, 239–252. [Google Scholar] [CrossRef] [Green Version]
- Olson, M.F.; Sahai, E. The actin cytoskeleton in cancer cell motility. Clin. Exp. Metastasis 2009, 26, 273–287. [Google Scholar] [CrossRef] [Green Version]
- Friedl, P.; Wolf, K. Tumour-cell invasion and migration: Diversity and escape mechanisms. Nat. Rev. Cancer 2003, 3, 362–374. [Google Scholar] [CrossRef]
- Webb, D.J.; Donais, K.; Whitmore, L.A.; Thomas, S.M.; Turner, C.E.; Parsons, J.T.; Horwitz, A.F. FAK-Src signalling through paxillin, ERK and MLCK regulates adhesion disassembly. Nat. Cell. Biol. 2004, 6, 154–161. [Google Scholar] [CrossRef]
- Dhillon, A.S.; Hagan, S.; Rath, O.; Kolch, W. MAP kinase signalling pathways in cancer. Oncogene 2007, 26, 3279–3290. [Google Scholar] [CrossRef] [Green Version]
- Mendoza, F.A.; Piera-Velazquez, S.; Farber, J.L.; Feghali-Bostwick, C.; Jimenez, S.A. Endothelial Cells Expressing Endothelial and Mesenchymal Cell Gene Products in Lung Tissue From Patients With Systemic Sclerosis-Associated Interstitial Lung Disease. Arthritis Rheumatol. 2016, 68, 210–217. [Google Scholar] [CrossRef]
- Tanaka, T.; Bai, Z.; Srinoulprasert, Y.; Yang, B.G.; Hayasaka, H.; Miyasaka, M. Chemokines in tumor progression and metastasis. Cancer Sci. 2005, 96, 317–322. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mir, H.; Kapur, N.; Gales, D.N.; Sharma, P.K.; Oprea-Ilies, G.; Johnson, A.T.; Singh, R.; Singh, S. CXCR6-CXCL16 Axis Promotes Breast Cancer by Inducing Oncogenic Signaling. Cancers 2021, 13, 3568. https://doi.org/10.3390/cancers13143568
Mir H, Kapur N, Gales DN, Sharma PK, Oprea-Ilies G, Johnson AT, Singh R, Singh S. CXCR6-CXCL16 Axis Promotes Breast Cancer by Inducing Oncogenic Signaling. Cancers. 2021; 13(14):3568. https://doi.org/10.3390/cancers13143568
Chicago/Turabian StyleMir, Hina, Neeraj Kapur, Dominique N. Gales, Praveen K. Sharma, Gabriela Oprea-Ilies, Anita T. Johnson, Rajesh Singh, and Shailesh Singh. 2021. "CXCR6-CXCL16 Axis Promotes Breast Cancer by Inducing Oncogenic Signaling" Cancers 13, no. 14: 3568. https://doi.org/10.3390/cancers13143568