
Metabolic Reprogramming: A Friend or Foe to Cancer Therapy?

{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
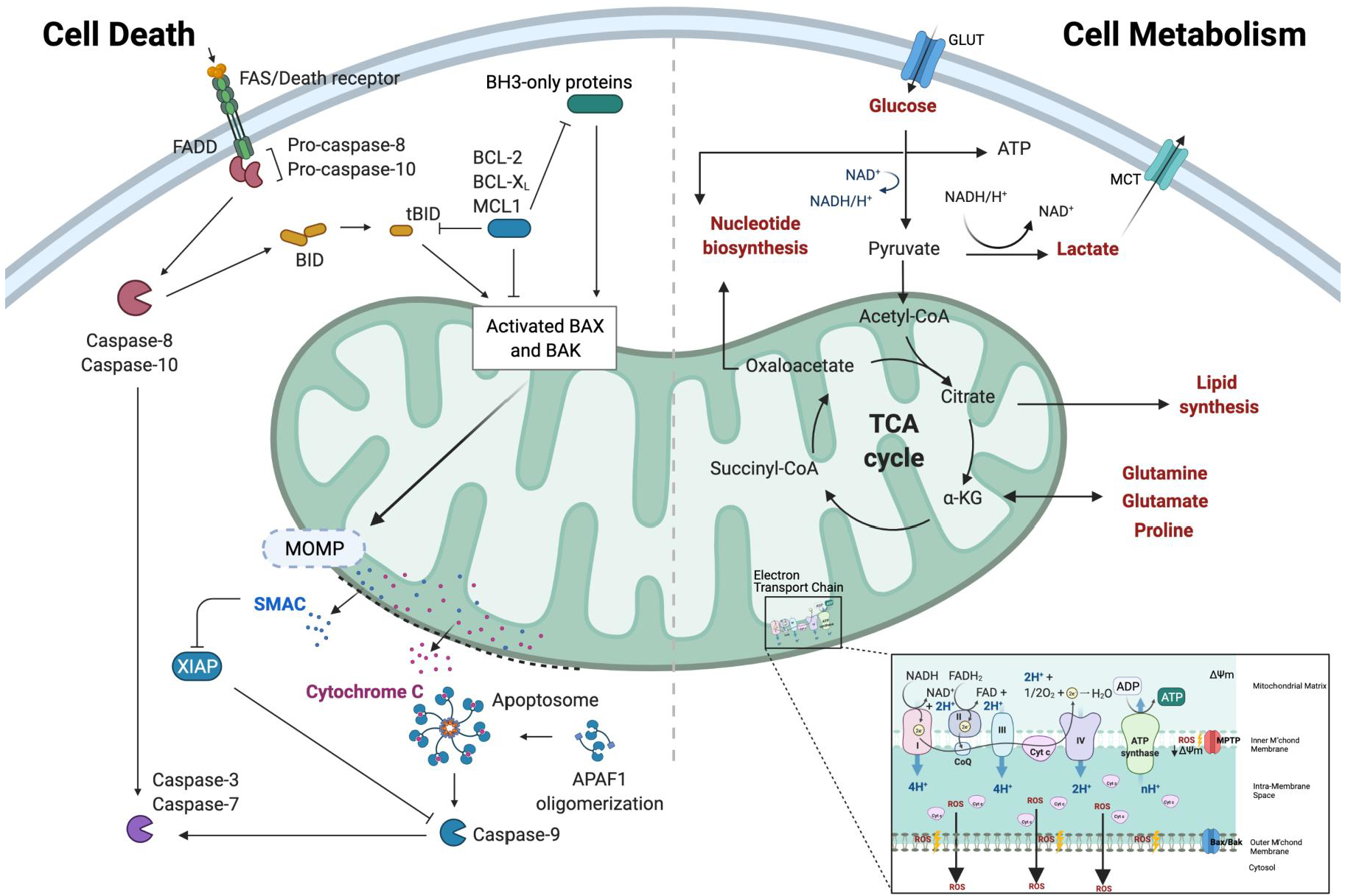
1.1. Mitochondria—A Signalling Hub That Dictates the Balance between Life and Death
1.2. Driving Enhanced Cancer Growth Simultaneously Promotes Resistance to Death
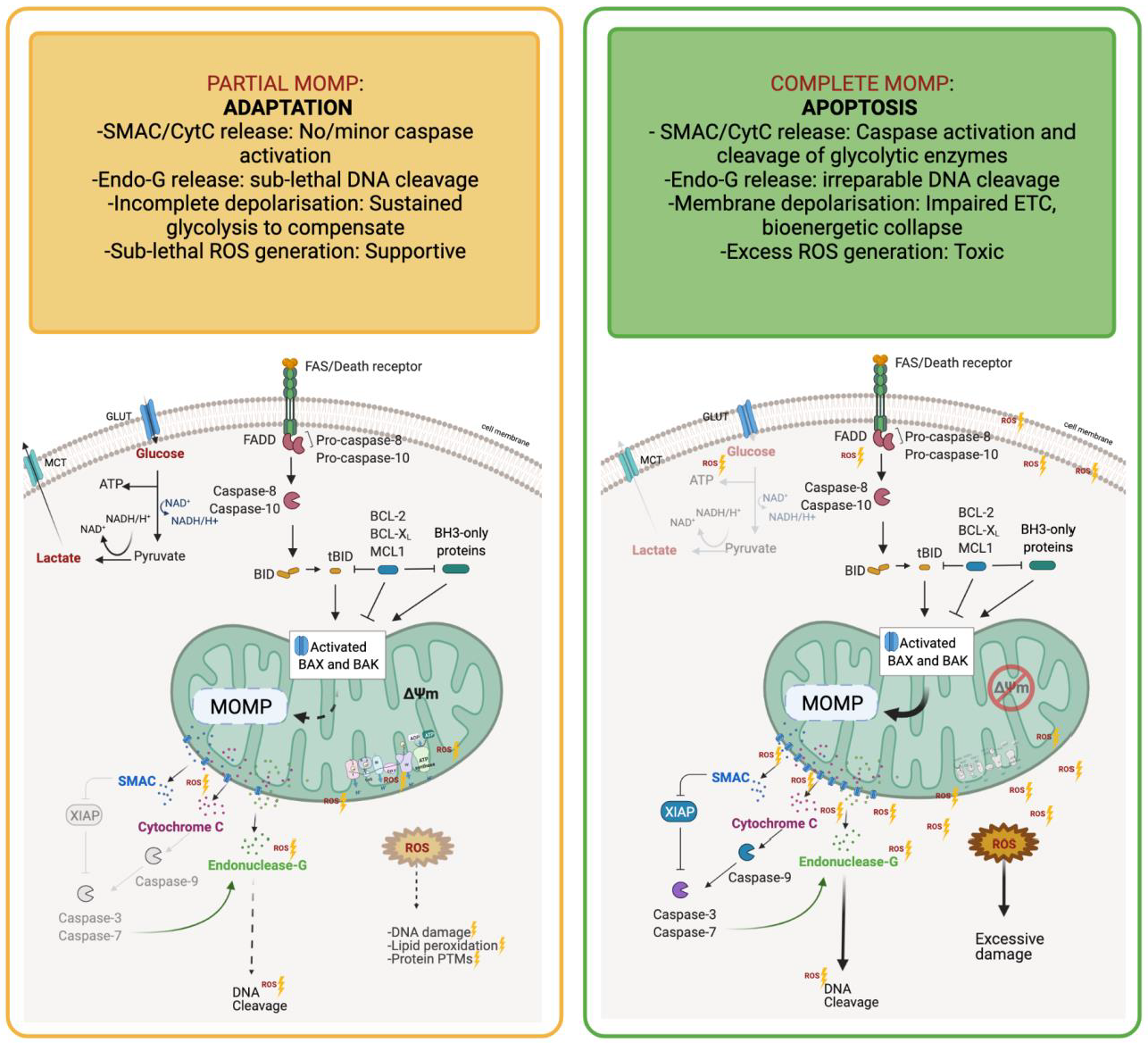
1.3. Adaptations to Cellular Stress Prevent Metabolic Catastrophe and Death
1.4. Metabolites as Direct Mediators of Cancer Cell Death
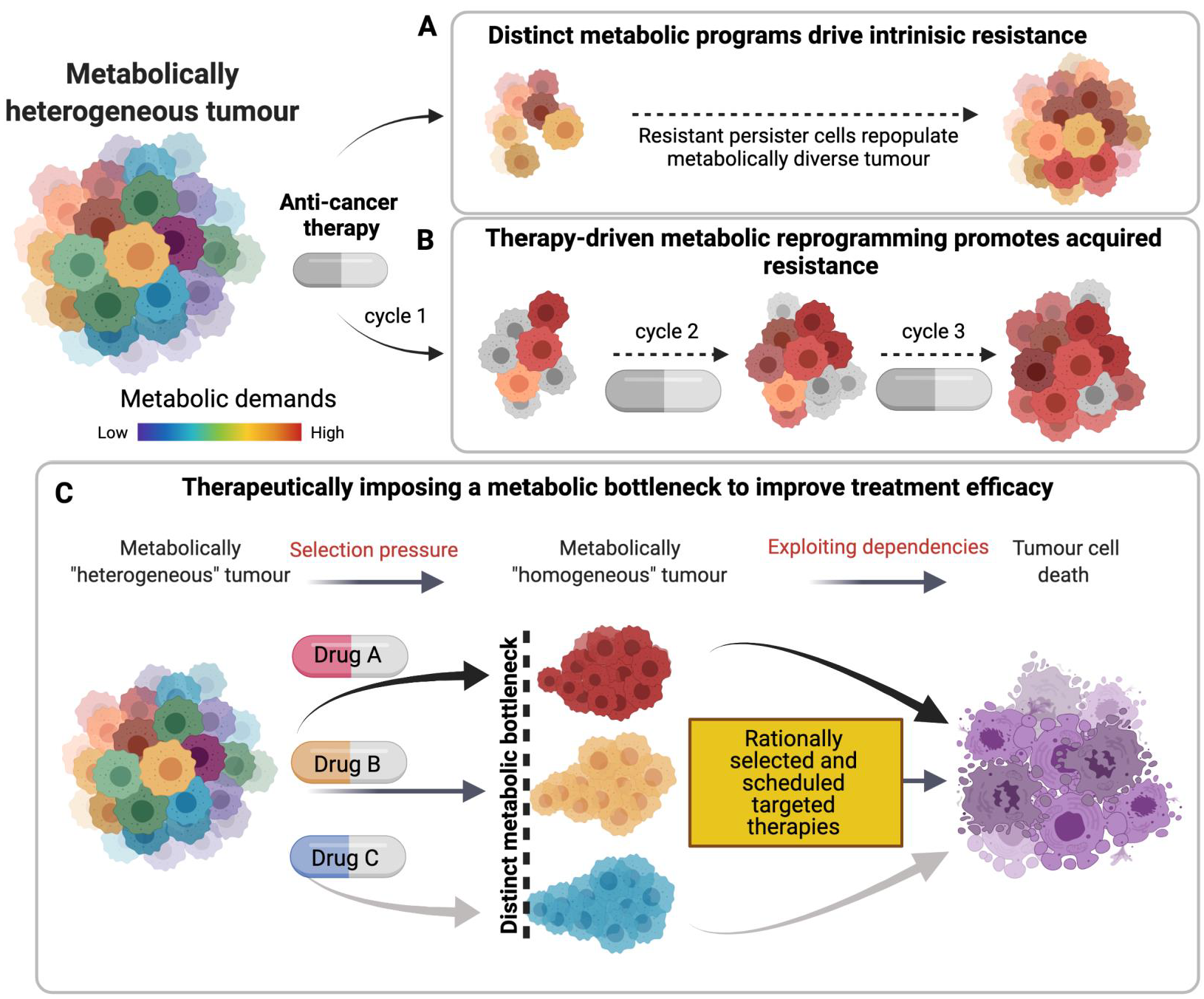
1.5. Metabolism and Therapy Resistance: Exploiting Imposed Vulnerabilities to Improve Response
1.6. Summary: Switching Metabolism from ‘Foe’ to ‘Friend’ of Cancer Therapy
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Warburg, O.; Wind, F.; Negelein, E. The metabolism of tumours. J. Gen. Physiol. 1927, 8, 519–530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Warburg, O. On the Origin of Cancer Cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Ward, S.P.; Thompson, B.C. Metabolic Reprogramming: A Cancer Hallmark Even Warburg Did Not Anticipate. Cancer Cell 2013, 21, 297–308. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Pavlova, N.; Thompson, C.B. The Emerging Hallmarks of Cancer Metabolism. Cell Metab. 2016, 23, 27–47. [Google Scholar] [CrossRef] [Green Version]
- Yuneva, M.O.; Fan, T.W.; Allen, T.; Higashi, R.M.; Ferraris, D.V.; Tsukamoto, T.; Matés, J.M.; Alonso, F.J.; Wang, C.; Seo, Y.; et al. The Metabolic Profile of Tumors Depends on Both the Responsible Genetic Lesion and Tissue Type. Cell Metab. 2012, 15, 157–170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kerr, E.M.; Gaude, E.; Turrell, F.K.; Frezza, C.; Martins, E.M.K. Mutant Kras copy number defines metabolic reprogramming and therapeutic susceptibilities. Nat. Cell Biol. 2016, 531, 110–113. [Google Scholar] [CrossRef] [Green Version]
- Mayers, J.R.; Torrence, M.E.; Danai, L.V.; Papagiannakopoulos, T.; Davidson, S.M.; Bauer, M.R.; Lau, A.N.; Ji, B.W.; Dixit, P.D.; Hosios, A.M.; et al. Tissue of origin dictates branched-chain amino acid metabolism in mutant Kras-driven cancers. Science 2016, 353, 1161–1165. [Google Scholar] [CrossRef] [Green Version]
- Heaster, T.M.; Landman, B.A.; Skala, M.C. Quantitative Spatial Analysis of Metabolic Heterogeneity Across In Vivo and In Vitro Tumor Models. Front. Oncol. 2019, 9, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Najumudeen, A.K.; CRUK Rosetta Grand Challenge Consortium; Ceteci, F.; Fey, S.K.; Hamm, G.; Steven, R.T.; Hall, H.; Nikula, C.J.; Dexter, A.; Murta, T.; et al. The amino acid transporter SLC7A5 is required for efficient growth of KRAS-mutant colorectal cancer. Nat. Genet. 2021, 53, 16–26. [Google Scholar] [CrossRef]
- Weissleder, R. Molecular Imaging in Cancer. Science 2006, 312, 1168–1171. [Google Scholar] [CrossRef] [Green Version]
- Valcarcel-Jimenez, L.; Gaude, E.; Torrano, V.; Frezza, C.; Carracedo, A. Mitochondrial Metabolism: Yin and Yang for Tumor Progression. Trends Endocrinol. Metab. 2017, 28, 748–757. [Google Scholar] [CrossRef] [PubMed]
- Vakifahmetoglu-Norberg, H.; Ouchida, A.T.; Norberg, E. The role of mitochondria in metabolism and cell death. Biochem. Biophys. Res. Commun. 2017, 482, 426–431. [Google Scholar] [CrossRef]
- Holohan, C.; Van Schaeybroeck, S.; Longley, D.B.; Johnston, P.G. Cancer drug resistance: An evolving paradigm. Nat. Rev. Cancer 2013, 13, 714–726. [Google Scholar] [CrossRef]
- Vasan, N.; Baselga, J.; Hyman, D.M. A view on drug resistance in cancer. Nature 2019, 575, 299–309. [Google Scholar] [CrossRef] [Green Version]
- Bock, F.J.; Tait, S.W.G. Mitochondria as multifaceted regulators of cell death. Nat. Rev. Mol. Cell Biol. 2020, 21, 85–100. [Google Scholar] [CrossRef] [PubMed]
- Taanman, J.-W. The mitochondrial genome: Structure, transcription, translation and replication. Biochim. Biophys. Acta Bioenerg. 1999, 1410, 103–123. [Google Scholar] [CrossRef] [Green Version]
- Sedlackova, L.; Korolchuk, V.I. Mitochondrial quality control as a key determinant of cell survival. Biochim. Biophys. Acta Bioenerg. 2019, 1866, 575–587. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Reyes, I.; Chandel, N.S. Mitochondrial TCA cycle metabolites control physiology and disease. Nat. Commun. 2020, 11, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herzig, S.; Shaw, R.J. AMPK: Guardian of metabolism and mitochondrial homeostasis. Nat. Rev. Mol. Cell Biol. 2018, 19, 121–135. [Google Scholar] [CrossRef] [Green Version]
- Song, X.; Kim, S.-Y.; Zhang, L.; Tang, D.; Bartlett, D.L.; Kwon, Y.T.; Lee, Y.J. Role of AMP-activated protein kinase in cross-talk between apoptosis and autophagy in human colon cancer. Cell Death Dis. 2014, 5, e1504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reczek, C.R.; Chandel, N.S. The Two Faces of Reactive Oxygen Species in Cancer. Annu. Rev. Cancer Biol. 2017, 1, 79–98. [Google Scholar] [CrossRef]
- Letouzé, E.; Martinelli, C.; Loriot, C.; Burnichon, N.; Abermil, N.; Ottolenghi, C.; Janin, M.; Menara, M.; Nguyen, A.T.; Benit, P.; et al. SDH Mutations Establish a Hypermethylator Phenotype in Paraganglioma. Cancer Cell 2013, 23, 739–752. [Google Scholar] [CrossRef] [Green Version]
- Yang, M.; Pollard, P.J. Succinate: A New Epigenetic Hacker. Cancer Cell 2013, 23, 709–711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sciacovelli, M.; Gonçalves, E.; Johnson, T.I.; Zecchini, V.R.; Da, C.A.S.H.; Gaude, E.; Drubbel, A.V.; Theobald, S.J.; Abbo, S.; Tran, M.; et al. Fumarate is an epigenetic modifier that elicits epithelial-to-mesenchymal transition. Nature 2016, 537, 544–547. [Google Scholar] [CrossRef]
- West, A.P.; Shadel, G.S. Mitochondrial DNA in innate immune responses and inflammatory pathology. Nat. Rev. Immunol. 2017, 17, 363–375. [Google Scholar] [CrossRef]
- Dai, H.; Meng, X.W.; Kaufmann, S.H. Mitochondrial apoptosis and BH3 mimetics. F1000Research 2016, 5, 2804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Del Gaizo Moore, V.; Letai, A. BH3 profiling--measuring integrated function of the mitochondrial apoptotic pathway to predict cell fate decisions. Cancer Lett. 2013, 332, 202–205. [Google Scholar] [CrossRef] [Green Version]
- Perciavalle, R.M.; Stewart, D.P.; Koss, B.; Lynch, J.; Milasta, S.; Bathina, M. Anti-apoptotic MCL-1 localizes to the mitochondrial matrix and couples mitochondrial fusion to respiration. Nat. Cell Biol. 2012, 14, 575–583. [Google Scholar] [CrossRef] [Green Version]
- Kalkavan, H.; Green, D. MOMP, cell suicide as a BCL-2 family business. Cell Death Differ. 2018, 25, 46–55. [Google Scholar] [CrossRef] [PubMed]
- Bao, X.; Liu, X.; Li, F.; Li, C.-Y. Limited MOMP, ATM, and their roles in carcinogenesis and cancer treatment. Cell Biosci. 2020, 10, 1–10. [Google Scholar] [CrossRef]
- Liu, X.; Li, F.; Huang, Q.; Zhang, Z.; Zhou, L.; Deng, Y.; Zhou, M.; Fleenor, D.E.; Wang, H.; Kastan, M.B.; et al. Self-inflicted DNA double-strand breaks sustain tumorigenicity and stemness of cancer cells. Cell Res. 2017, 27, 764–783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.Y.; Luo, X.; Wang, X. Endonuclease G is an apoptotic DNase when released from mitochondria. Nat. Cell Biol. 2001, 412, 95–99. [Google Scholar] [CrossRef]
- Garrido, C.; Galluzzi, L.; Brunet, M.; Puig, P.E.; Didelot, C.; Kroemer, G. Mechanisms of cytochrome c release from mitochondria. Cell Death Differ. 2006, 13, 1423–1433. [Google Scholar] [CrossRef] [Green Version]
- Scorrano, L.; Ashiya, M.; Buttle, K.; Weiler, S.; Oakes, S.A.; Mannella, C.A.; Korsmeyer, S.J. A Distinct Pathway Remodels Mitochondrial Cristae and Mobilizes Cytochrome c during Apoptosis. Dev. Cell 2002, 2, 55–67. [Google Scholar] [CrossRef] [Green Version]
- Ricci, J.E.; Muñoz-Pinedo, C.; Fitzgerald, P.; Bailly-Maitre, B.; Perkins, G.A.; Yadava, N.; Scheffler, I.E.; Ellisman, M.H.; Green, D.R. Disruption of Mitochondrial Function during Apoptosis Is Mediated by Caspase Cleavage of the p75 Subunit of Complex I of the Electron Transport Chain. Cell 2004, 117, 773–786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Kim, J.; Bae, J.S. ROS homeostasis and metabolism: A critical liaison for cancer therapy. Exp. Mol. Med. 2016, 48, e269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Redza-Dutordoir, M.; Averill-Bates, D.A. Activation of apoptosis signalling pathways by reactive oxygen species. Biochim. Biophys. Acta Bioenerg. 2016, 1863, 2977–2992. [Google Scholar] [CrossRef]
- Giampazolias, E.; Tait, S.W. Mitochondria and the hallmarks of cancer. FEBS J. 2016, 283, 803–814. [Google Scholar] [CrossRef]
- Bailey, M.H.; Tokheim, C.; Porta-Pardo, E.; Sengupta, S.; Bertrand, D.; Weerasinghe, A.; Colaprico, A.; Wendl, M.C.; Kim, J.; Reardon, B.; et al. Comprehensive Characterization of Cancer Driver Genes and Mutations. Cell 2018, 173, 371–385.e18. [Google Scholar] [CrossRef] [Green Version]
- Fridman, J.S.; Lowe, S.W. Control of apoptosis by p53. Oncogene 2003, 22, 9030–9040. [Google Scholar] [CrossRef] [Green Version]
- Berkers, C.R.; Maddocks, O.D.; Cheung, E.C.; Mor, I.; Vousden, K.H. Metabolic Regulation by p53 Family Members. Cell Metab. 2013, 18, 617–633. [Google Scholar] [CrossRef] [Green Version]
- Mirzayans, R.; Andrais, B.; Kumar, P.; Murray, D. Significance of Wild-Type p53 Signaling in Suppressing Apoptosis in Response to Chemical Genotoxic Agents: Impact on Chemotherapy Outcome. Int. J. Mol. Sci. 2017, 18, 928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bensaad, K.; Tsuruta, A.; Selak, M.A.; Vidal, M.N.C.; Nakano, K.; Bartrons, R.; Gottlieb, E.; Vousden, K.H. TIGAR, a p53-Inducible Regulator of Glycolysis and Apoptosis. Cell 2006, 126, 107–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maddocks, O.; Berkers, C.R.; Mason, S.; Zheng, L.; Blyth, K.; Gottlieb, E.; Vousden, K.H. Serine starvation induces stress and p53-dependent metabolic remodelling in cancer cells. Nat. Cell Biol. 2012, 493, 542–546. [Google Scholar] [CrossRef]
- Humpton, T.J.; Hock, A.K.; Maddocks, O.D.K.; Vousden, K.H. p53-mediated adaptation to serine starvation is retained by a common tumour-derived mutant. Cancer Metab. 2018, 6, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaude, E.; Frezza, C. Tissue-specific and convergent metabolic transformation of cancer correlates with metastatic potential and patient survival. Nat. Commun. 2016, 7, 13041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muller, P.A.; Vousden, K.H. Mutant p53 in Cancer: New Functions and Therapeutic Opportunities. Cancer Cell 2014, 25, 304–317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, X.; Zhang, W.; Li, S.; Yang, H. The role of cholesterol metabolism in cancer Xiao. Am. J. Cancer Res. 2019, 9, 219–227. [Google Scholar]
- Marino, N.; German, R.; Rao, X.; Simpson, E.; Liu, S.; Wan, J.; Liu, Y.; Sandusky, G.; Jacobsen, M.; Stoval, M.; et al. Upregulation of lipid metabolism genes in the breast prior to cancer diagnosis. NPJ Breast Cancer 2020, 6, 1–13. [Google Scholar] [CrossRef]
- Turrell, F.K.; Kerr, E.M.; Gao, M.; Thorpe, H.; Doherty, G.J.; Cridge, J.; Shorthouse, D.; Speed, A.; Samarajiwa, S.; Hall, B.A.; et al. Lung tumors with distinct p53 mutations respond similarly to p53 targeted therapy but exhibit genotype-specific statin sensitivity. Genes Dev. 2017, 31, 1339–1353. [Google Scholar] [CrossRef] [Green Version]
- Ying, H.; Kimmelman, A.C.; Lyssiotis, C.; Hua, S.; Chu, G.C.; Fletcher-Sananikone, E.; Locasale, J.; Son, J.; Zhang, H.; Coloff, J.L.; et al. Oncogenic Kras Maintains Pancreatic Tumors through Regulation of Anabolic Glucose Metabolism. Cell 2012, 149, 656–670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Son, J.; Lyssiotis, C.; Ying, H.; Wang, X.; Hua, S.; Ligorio, M.; Perera, R.M.; Ferrone, C.R.; Mullarky, E.; Shyh-Chang, N.; et al. Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nat. Cell Biol. 2013, 496, 101–105. [Google Scholar] [CrossRef]
- Yang, H.; Xiang, S.; Kazi, A.; Sebti, S.M. The GTPase KRAS suppresses the p53 tumor suppressor by activating the NRF2-regulated antioxidant defense system in cancer cells. J. Biol. Chem. 2020, 295, 3055–3063. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.Y.; Chen, H.-Y.; Mathew, R.; Fan, J.; Strohecker, A.M.; Karsli-Uzunbas, G.; Kamphorst, J.; Chen, G.; Lemons, J.M.; Karantza, V.; et al. Activated Ras requires autophagy to maintain oxidative metabolism and tumorigenesis. Genes Dev. 2011, 25, 460–470. [Google Scholar] [CrossRef] [Green Version]
- Kamphorst, J.J.; Cross, J.R.; Fan, J.; De Stanchina, E.; Mathew, R.; White, E.P.; Thompson, C.B.; Rabinowitz, J.D. Hypoxic and Ras-transformed cells support growth by scavenging unsaturated fatty acids from lysophospholipids. Proc. Natl. Acad. Sci. USA 2013, 110, 8882–8887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, J.; Kamphorst, J.J.; Mathew, R.; Chung, M.K.; White, E.; Shlomi, T.; Rabinowitz, J.D. Glutamine-driven oxidative phosphorylation is a major ATP source in transformed mammalian cells in both normoxia and hypoxia. Mol. Syst. Biol. 2013, 9, 712. [Google Scholar] [CrossRef] [PubMed]
- Davidson, S.M.; Papagiannakopoulos, T.; Olenchock, B.A.; Heyman, J.E.; Keibler, M.A.; Luengo, A.; Bauer, M.R.; Jha, A.K.; O’Brien, J.P.; Pierce, K.A.; et al. Environment Impacts the Metabolic Dependencies of Ras-Driven Non-Small Cell Lung Cancer. Cell Metab. 2016, 23, 517–528. [Google Scholar] [CrossRef] [Green Version]
- Zaanan, A.; Okamoto, K.; Kawakami, H.; Khazaie, K.; Huang, S.; Sinicrope, F.A. The Mutant KRAS Gene Up-regulates BCL-XL Protein via STAT3 to Confer Apoptosis Resistance That Is Reversed by BIM Protein Induction and BCL-XL Antagonism. J. Biol. Chem. 2015, 290, 23838–23849. [Google Scholar] [CrossRef] [Green Version]
- Miller, K.A.; Yeager, N.; Baker, K.; Liao, X.-H.; Refetoff, S.; Di Cristofano, A. Oncogenic Kras Requires Simultaneous PI3K Signaling to Induce ERK Activation and Transform Thyroid Epithelial Cells In Vivo. Cancer Res. 2009, 69, 3689–3694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castellano, E.; Downward, J. RAS Interaction with PI3K: More Than Just Another Effector Pathway. Genes Cancer 2011, 2, 261–274. [Google Scholar] [CrossRef] [Green Version]
- Khawaja, H.; Campbell, A.; Roberts, J.Z.; Javadi, A.; O’Reilly, P.; McArt, D.; Allen, W.L.; Majkut, J.; Rehm, M.; Bardelli, A.; et al. RALB GTPase: A critical regulator of DR5 expression and TRAIL sensitivity in KRAS mutant colorectal cancer. Cell Death Dis. 2020, 11, 1–18. [Google Scholar] [CrossRef]
- Corcoran, R.B.; Cheng, K.A.; Hata, A.N.; Faber, A.C.; Ebi, H.; Coffee, E.M.; Greninger, P.; Brown, R.D.; Godfrey, J.T.; Cohoon, T.J.; et al. Synthetic Lethal Interaction of Combined BCL-XL and MEK Inhibition Promotes Tumor Regressions in KRAS Mutant Cancer Models. Cancer Cell 2013, 23, 121–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahn, Y.; Jun, Y. Oncogenic KRAS confers chemoresistance by upregulating NRF2. Early Hum. Dev. 2007, 83, 255–262. [Google Scholar] [CrossRef]
- Oh, Y.; Jung, H.R.; Min, S.; Kang, J.; Jang, D.; Shin, S.; Kim, J.; Lee, S.E.; Sung, C.O.; Lee, W.-S.; et al. Targeting antioxidant enzymes enhances the therapeutic efficacy of the BCL-XL inhibitor ABT-263 in KRAS-mutant colorectal cancers. Cancer Lett. 2021, 497, 123–136. [Google Scholar] [CrossRef] [PubMed]
- Engelman, J.A.; Luo, J.; Cantley, L. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat. Rev. Genet. 2006, 7, 606–619. [Google Scholar] [CrossRef]
- Hao, Y.; Samuels, Y.; Li, Q.; Krokowski, D.; Guan, B.-J.; Wang, C.; Jin, Z.; Dong, B.; Cao, B.; Feng, X.; et al. Oncogenic PIK3CA mutations reprogram glutamine metabolism in colorectal cancer. Nat. Commun. 2016, 7, 11971. [Google Scholar] [CrossRef] [Green Version]
- Koundouros, N.; Karali, E.; Tripp, A.; Valle, A.; Inglese, P.; Perry, N.J.; Magee, D.J.; Virmouni, S.A.; Elder, G.A.; Tyson, A.L.; et al. Metabolic Fingerprinting Links Oncogenic PIK3CA with Enhanced Arachidonic Acid-Derived Eicosanoids. Cell 2020, 181, 1596–1611. [Google Scholar] [CrossRef]
- Franke, T.F.; Kaplan, D.R.; Cantley, L.C. PI3K: Downstream AKTion Blocks Apoptosis. Cell 1997, 88, 435–437. [Google Scholar] [CrossRef] [Green Version]
- Liu, R.; Chen, Y.; Liu, G.; Li, C.; Song, Y.; Cao, Z.; Li, W.; Hu, J.; Lu, C.; Liu, Y. PI3K/AKT pathway as a key link modulates the multidrug resistance of cancers. Cell Death Dis. 2020, 11, 1–12. [Google Scholar] [CrossRef]
- Stine, Z.E.; Walton, Z.E.; Altman, B.J.; Hsieh, A.L.; Dang, C.V. MYC, Metabolism, and Cancer MYC function. Cancer Discov. 2015, 5, 1024–1039. [Google Scholar] [CrossRef] [Green Version]
- Sarosiek, K.A.; Fraser, C.; Muthalagu, N.; Robinson, S.; Moslehi, J.; Sarosiek, K.A.; Cantlon, A.; Fisch, S.; Golomb-Mello, G.; Jeremy, A.; et al. Developmental Regulation of Mitochondrial Apoptosis by c-Myc Governs Age- and Tissue- Specific Sensitivity to Cancer Therapeutics Article Developmental Regulation of Mitochondrial Apoptosis by c-Myc Governs Age- and Tissue-Specific. Cancer Cell 2017, 31, 1–15. [Google Scholar]
- Liu, L.; Ulbrich, J.; Müller, J.; Wüstefeld, T.; Aeberhard, L.; Kress, T.R.; Muthalagu, N.; Rycak, L.; Rudalska, R.; Moll, R.; et al. Deregulated MYC expression induces dependence upon AMPK-related kinase 5. Nat. Cell Biol. 2012, 483, 608–612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dang, C.V. MYC, Metabolism, Cell Growth, and Tumorigenesis. Cold Spring Harb. Perspect. Med. 2013, 3, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Davidson, S.M.; Heiden, M.G.V. METabolic Adaptations in the Tumor MYCroenvironment. Cell Metab. 2012, 15, 131–133. [Google Scholar] [CrossRef] [Green Version]
- Elbadawy, M.; Usui, T.; Yamawaki, H.; Sasaki, K. Emerging Roles of C-Myc in Cancer Stem Cell-Related Signaling and Resistance to Cancer Chemotherapy: A Potential Therapeutic Target Against Colorectal Cancer. Int. J. Mol. Sci. 2019, 20, 2340. [Google Scholar] [CrossRef] [Green Version]
- Yue, M.; Jiang, J.; Gao, P.; Liu, H.; Qing, G. Oncogenic MYC Activates a Feedforward Regulatory Loop Promoting Essential Amino Acid Metabolism and Tumorigenesis. Cell Rep. 2017, 21, 3819–3832. [Google Scholar] [CrossRef] [Green Version]
- Zhou, W.; Feng, X.; Ren, C.; Jiang, X.; Liu, W.; Huang, W.; Liu, Z.; Li, Z.; Zeng, L.; Wang, L.; et al. Over-expression of BCAT1, a c-Myc target gene, induces cell proliferation, migration and invasion in nasopharyngeal carcinoma. Mol. Cancer 2013, 12, 53. [Google Scholar] [CrossRef] [Green Version]
- Cao, Z.; Fan-Minogue, H.; Bellovin, D.I.; Yevtodiyenko, A.; Arzeno, J.; Yang, Q.; Gambhir, S.S.; Felsher, D.W. MYC phosphorylation, activation, and tumorigenic potential in hepatocellular carcinoma are regulated by HMG-CoA reductase. Cancer Res. 2011, 71, 2286–2297. [Google Scholar] [CrossRef] [Green Version]
- Morrish, F.; Noonan, J.; Perez-Olsen, C.; Gafken, P.R.; Fitzgibbon, M.; Kelleher, J. Myc-dependent mitochondrial generation of acetyl-CoA contributes to fatty acid biosynthesis and histone acetylation during cell cycle entry. J. Biol. Chem. 2010, 285, 36267–36274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Giacomo, S.; Sollazzo, M.; Paglia, S.; Grifoni, D. MYC, cell competition, and cell death in cancer: The inseparable triad. Genes 2017, 8, 120. [Google Scholar] [CrossRef] [Green Version]
- Murphy, D.; Junttila, M.R.; Pouyet, L.; Karnezis, A.; Shchors, K.; Bui, D.A.; Brown-Swigart, L.; Johnson, L.; Evan, G.I. Distinct Thresholds Govern Myc’s Biological Output In Vivo. Cancer Cell 2008, 14, 447–457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McMahon, S.B. MYC and the Control of Apoptosis. Cold Spring Harb. Perspect. Med. 2014, 4, a014407. [Google Scholar] [CrossRef] [Green Version]
- Porter, J.R.; Fisher, B.E.; Baranello, L.; Liu, J.C.; Kambach, D.M.; Nie, Z.; Koh, W.S.; Luo, J.; Stommel, J.M.; Levens, D.; et al. Global Inhibition with Specific Activation: How p53 and MYC Redistribute the Transcriptome in the DNA Double-Strand Break Response. Mol. Cell 2017, 67, 1013–1025.e9. [Google Scholar] [CrossRef] [Green Version]
- Benassi, B.; Fanciulli, M.; Fiorentino, F.; Porrello, A.; Chiorino, G.; Loda, M.; Zupi, G.; Biroccio, A. c-Myc Phosphorylation Is Required for Cellular Response to Oxidative Stress. Mol. Cell 2006, 21, 509–519. [Google Scholar] [CrossRef]
- Sinkala, M.; Mulder, N.; Martin, D.P. Metabolic gene alterations impact the clinical aggressiveness and drug responses of 32 human cancers. Commun. Biol. 2019, 2, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Jiang, M.; Liu, S.; Lin, J.; Hao, W.; Wei, B.; Gao, Y.; Kong, C.; Yu, M.; Zhu, Y. A pan-cancer analysis of molecular characteristics and oncogenic role of hexokinase family genes in human tumors. Life Sci. 2021, 264, 118669. [Google Scholar] [CrossRef]
- Huang, J.; Yu, J.; Tu, L.; Huang, N.; Li, H.; Luo, Y. Isocitrate dehydrogenase mutations in glioma: From basic discovery to therapeutics development. Front. Oncol. 2019, 9, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Smith, A.L.M.; Whitehall, J.C.; Bradshaw, C.; Gay, D.; Robertson, F.; Blain, A.P.; Hudson, G.; Pyle, A.; Houghton, D.; Hunt, M.; et al. Age-associated mitochondrial DNA mutations cause metabolic remodeling that contributes to accelerated intestinal tumorigenesis. Nat. Cancer 2020, 1, 976–989. [Google Scholar] [CrossRef]
- Kujoth, G.C.; Leeuwenburgh, C.; Prolla, T.A. Mitochondrial DNA Mutations and Apoptosis in Mammalian Aging. Cancer Res. 2006, 66, 7386–7389. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Silva, J.P.; Gustafsson, C.M.; Rustin, P.; Larsson, N.-G. Increased in vivo apoptosis in cells lacking mitochondrial DNA gene expression. Proc. Natl. Acad. Sci. USA 2001, 98, 4038–4043. [Google Scholar] [CrossRef] [Green Version]
- DeBerardinis, R.J.; Lum, J.J.; Hatzivassiliou, G.; Thompson, C.B. The Biology of Cancer: Metabolic Reprogramming Fuels Cell Growth and Proliferation. Cell Metab. 2008, 7, 11–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torrence, M.E.; Manning, B.D. Nutrient Sensing in Cancer. Annu. Rev. Cancer Biol. 2018, 2, 251–269. [Google Scholar] [CrossRef]
- Denko, N.C. Hypoxia, HIF1 and glucose metabolism in the solid tumour. Nat. Rev. Cancer 2008, 8, 705–713. [Google Scholar] [CrossRef] [PubMed]
- Vaupel, P.; Mayer, A. Hypoxia in cancer: Significance and impact on clinical outcome. Cancer Metastasis Rev. 2007, 26, 225–239. [Google Scholar] [CrossRef]
- Brahimi-Horn, M.C.; Chiche, J.; Pouysségur, J. Hypoxia signalling controls metabolic demand. Curr. Opin. Cell Biol. 2007, 19, 223–229. [Google Scholar] [CrossRef]
- Semenza, G.L. Targeting HIF-1 for cancer therapy. Nat. Rev. Cancer 2003, 3, 721–732. [Google Scholar] [CrossRef] [PubMed]
- Sendoel, A.; Hengartner, M.O. Apoptotic cell death under hypoxia. Physiology 2014, 29, 168–176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okamoto, A.; Sumi, C.; Tanaka, H.; Kusunoki, M.; Iwai, T.; Nishi, K.; Matsuo, Y.; Harada, H.; Takenaga, K.; Bono, H.; et al. HIF-1-mediated suppression of mitochondria electron transport chain function confers resistance to lidocaine-induced cell death. Sci. Rep. 2017, 7, 3816. [Google Scholar] [CrossRef] [Green Version]
- Shibue, T.; Weinberg, R.A. EMT, CSCs, and drug resistance: The mechanistic link and clinical implications. Nat. Rev. Clin. Oncol. 2017, 14, 611–629. [Google Scholar] [CrossRef] [Green Version]
- Ben-Sahra, I.; Manning, B.D. mTORC1 signaling and the metabolic control of cell growth. Curr. Opin. Cell Biol. 2017, 45, 72–82. [Google Scholar] [CrossRef] [Green Version]
- Saxton, R.A.; Sabatini, D.M. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 168, 960–976. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Ng, P.K.-S.; Kucherlapati, M.; Chen, F.; Liu, Y.; Tsang, Y.H.; De Velasco, G.; Jeong, K.J.; Akbani, R.; Hadjipanayis, A.; et al. A Pan-Cancer Proteogenomic Atlas of PI3K/AKT/mTOR Pathway Alterations. Cancer Cell 2017, 31, 820–832.e3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su Yea, S.; Fruman, D.A. Achieving cancer cell death with PI3K/mTOR-targeted therapies. Ann. N. Y. Acad. Sci. 2013, 1280, 15–18. [Google Scholar] [CrossRef] [Green Version]
- González, A.; Hall, M.N.; Lin, S.-C.; Hardie, D.G. AMPK and TOR: The Yin and Yang of Cellular Nutrient Sensing and Growth Control. Cell Metab. 2020, 31, 472–492. [Google Scholar] [CrossRef]
- Wellen, K.; Thompson, C.B. Cellular Metabolic Stress: Considering How Cells Respond to Nutrient Excess. Mol. Cell 2010, 40, 323–332. [Google Scholar] [CrossRef] [Green Version]
- Castedo, M.; Ferri, K.F.; Kroemer, G. Mammalian Target of Rapamycin (mTOR): Pro- and Anti-Apoptotic. Cell Death Differ. 2002, 9, 99–100. [Google Scholar] [CrossRef]
- Zou, Z.; Tao, T.; Li, H.; Zhu, X. MTOR signaling pathway and mTOR inhibitors in cancer: Progress and challenges. Cell Biosci. 2020, 10, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Basu, A.; Sridharan, S. Regulation of anti-apoptotic Bcl-2 family protein Mcl-1 by S6 kinase 2. PLoS ONE 2017, 12, e0173854. [Google Scholar] [CrossRef] [PubMed]
- Gremke, N.; Polo, P.; Dort, A.; Schneikert, J.; Elmshäuser, S.; Brehm, C. mTOR-mediated cancer drug resistance suppresses autophagy and generates a druggable metabolic vulnerability. Nat. Commun. 2020, 11, 1–15. [Google Scholar] [CrossRef]
- Iacovelli, S.; Ricciardi, M.R.; Allegretti, M.; Mirabilii, S.; Licchetta, R.; Bergamo, P.; Rinaldo, C.; Zeuner, A.; Foà, R.; Milella, M.; et al. Co-targeting of Bcl-2 and mTOR pathway triggers synergistic apoptosis in BH3 mimetics resistant acute lymphoblastic leukemia. Oncotarget 2015, 6, 32089–32103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fulda, S. Synthetic lethality by co-targeting mitochondrial apoptosis and PI3K/Akt/mTOR signaling. Mitochondrion 2014, 19, 85–87. [Google Scholar] [CrossRef] [PubMed]
- Bergers, G.; Fendt, S.-M. The metabolism of cancer cells during metastasis. Nat. Rev. Cancer 2021, 21, 162–180. [Google Scholar] [CrossRef]
- Green, D.R.; Llambi, F. Cell Death Signaling. Cold Spring Harb. Perspect. Biol. 2015, 7, a006080. [Google Scholar] [CrossRef]
- Li, F.; Tiede, B.; Massagué, J.; Kang, Y. Beyond tumorigenesis: Cancer stem cells in metastasis. Cell Res. 2007, 17, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Carnero, A.; Lleonart, M. The hypoxic microenvironment: A determinant of cancer stem cell evolution. Insid. Cell 2015, 1, 96–105. [Google Scholar] [CrossRef]
- Kim, M.P.; Fleming, J.B.; Wang, H.; Abbruzzese, J.L.; Choi, W.; Kopetz, S.; McConkey, D.J.; Evans, D.B.; Gallick, G.E. ALDH Activity Selectively Defines an Enhanced Tumor-Initiating Cell Population Relative to CD133 Expression in Human Pancreatic Adenocarcinoma. PLoS ONE 2011, 6, e20636. [Google Scholar] [CrossRef]
- Nimmakayala, R.K.; Leon, F.; Rachagani, S.; Rauth, S.; Nallasamy, P.; Marimuthu, S.; Shailendra, G.K.; Chhonker, Y.S.; Chugh, S.; Chirravuri, R.; et al. Metabolic programming of distinct cancer stem cells promotes metastasis of pancreatic ductal adenocarcinoma. Oncogene 2021, 40, 215–231. [Google Scholar] [CrossRef]
- Kamarajugadda, S.; Stemboroski, L.; Cai, Q.; Simpson, N.E.; Nayak, S.; Tan, M.; Lu, J. Glucose Oxidation Modulates Anoikis and Tumor Metastasis. Mol. Cell. Biol. 2012, 32, 1893–1907. [Google Scholar] [CrossRef] [Green Version]
- Grassian, A.R.; Metallo, C.M.; Coloff, J.L.; Stephanopoulos, G.; Brugge, J.S. Erk regulation of pyruvate dehydrogenase flux through PDK4 modulates cell proliferation. Genes Dev. 2011, 25, 1716–1733. [Google Scholar] [CrossRef] [Green Version]
- Mason, J.A.; Davison-Versagli, C.A.; Leliaert, A.K.; Pape, D.J.; McCallister, C.; Zuo, J.; Durbin, S.M.; Buchheit, C.L.; Zhang, S.; Schafer, Z.T. Oncogenic Ras differentially regulates metabolism and anoikis in extracellular matrix-detached cells. Cell Death Differ. 2016, 23, 1271–1282. [Google Scholar] [CrossRef] [Green Version]
- Ubellacker, J.M.; Tasdogan, A.; Ramesh, V.; Shen, B.; Mitchell, E.C.; Martin-Sandoval, M.S.; Gu, Z.; McCormick, M.L.; Durham, A.B.; Spitz, D.R.; et al. Lymph protects metastasizing melanoma cells from ferroptosis. Nat. Cell Biol. 2020, 585, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Langhans, S.A. Three-dimensional In Vitro cell culture models in drug discovery and drug repositioning. Front. Pharmacol. 2018, 9, 1–14. [Google Scholar] [CrossRef]
- Zoetemelk, M.; Rausch, M.; Colin, D.J.; Dormond, O.; Nowak-Sliwinska, P. Short-term 3D culture systems of various complexity for treatment optimization of colorectal carcinoma. Sci. Rep. 2019, 9, 7103. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.; Eglen, R.M. Three-Dimensional Cell Cultures in Drug Discovery and Development. SLAS Discov. Adv. Life Sci. R&D 2017, 22, 456–472. [Google Scholar] [CrossRef] [Green Version]
- Kaymak, I.; Maier, C.R.; Schmitz, W.; Campbell, A.D.; Dankworth, B.; Ade, C.P.; Walz, S.; Paauwe, M.; Kalogirou, C.; Marouf, H.; et al. Mevalonate Pathway Provides Ubiquinone to Maintain Pyrimidine Synthesis and Survival in p53-Deficient Cancer Cells Exposed to Metabolic Stress. Cancer Res. 2019, 80, 189–203. [Google Scholar] [CrossRef] [Green Version]
- Biancur, D.E.; Kapner, K.S.; Yamamoto, K.; Root, D.E.; Aguirre, A.J.; Kimmelman, A.C.; Wu, W.; Manguso, R.T.; Brown, A.; Root, D.E. Resource Functional Genomics Identifies Metabolic Vulnerabilities in Pancreatic Cancer Resource Functional Genomics Identifies Metabolic Vulnerabilities in Pancreatic Cancer. Cell Metab. 2021, 33, 1–12. [Google Scholar] [CrossRef]
- Clevers, H. Modeling Development and Disease with Organoids. Cell 2016, 165, 1586–1597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharick, J.T.; Jeffery, J.J.; Karim, M.R.; Walsh, C.M.; Esbona, K.; Cook, R.S.; Skala, M.C. Cellular Metabolic Heterogeneity In Vivo Is Recapitulated in Tumor Organoids. Neoplasia 2019, 21, 615–626. [Google Scholar] [CrossRef]
- Miranda-Gonçalves, V.; Lameirinhas, A.; Henrique, R.; Jerónimo, C. Metabolism and Epigenetic Interplay in Cancer: Regulation and Putative Therapeutic Targets. Front. Genet. 2018, 9, 427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donohoe, D.R.; Collins, L.B.; Wali, A.; Bigler, R.; Sun, W.; Bultman, S.J. The Warburg Effect Dictates the Mechanism of Butyrate-Mediated Histone Acetylation and Cell Proliferation. Mol. Cell 2012, 48, 612–626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mandal, M.; Kumar, R. Bcl-2 expression regulates sodium butyrate-induced apoptosis in human MCF-7 breast cancer cells. Cell Growth Differ. Mol. Boil. J. Am. Assoc. Cancer Res. 1996, 7, 311–318. [Google Scholar]
- Thangaraju, M.; Carswell, K.N.; Prasad, P.D.; Ganapathy, V. Colon cancer cells maintain low levels of pyruvate to avoid cell death caused by inhibition of HDAC1/HDAC3. Biochem. J. 2008, 417, 379–389. [Google Scholar] [CrossRef]
- Feng, Y.; Xiong, Y.; Qiao, T.; Li, X.; Jia, L.; Han, Y. Lactate dehydrogenase A: A key player in carcinogenesis and potential target in cancer therapy. Cancer Med. 2018, 7, 6124–6136. [Google Scholar] [CrossRef] [Green Version]
- Du, X.; Hu, H. The Roles of 2-Hydroxyglutarate. Front. Cell Dev. Biol. 2021, 9, 1–13. [Google Scholar] [CrossRef]
- McCann, C.; Crawford, N.; Majkut, J.; Holohan, C.; Armstrong, C.W.D.; Maxwell, P.J.; Ong, C.W.; LaBonte, M.J.; McDade, S.S.; Waugh, D.J.; et al. Cytoplasmic FLIP(S) and nuclear FLIP(L) mediate resistance of castrate-resistant prostate cancer to apoptosis induced by IAP antagonists. Cell Death Dis. 2018, 9, 1081. [Google Scholar] [CrossRef] [PubMed]
- Lees, A.; McIntyre, A.J.; Crawford, N.T.; Falcone, F.; McCann, C.; Holohan, C.; Quinn, G.P.; Roberts, J.Z.; Sessler, T.; Gallagher, P.F.; et al. The pseudo-caspase FLIP(L) regulates cell fate following p53 activation. Proc. Natl. Acad. Sci. USA 2020, 117, 17808–17819. [Google Scholar] [CrossRef]
- Bolden, J.E.; Shi, W.; Jankowski, K.; Kan, C.-Y.; Cluse, L.; Martin, B.P.; MacKenzie, K.L.; Smyth, G.K.; Johnstone, R.W. HDAC inhibitors induce tumor-cell-selective pro-apoptotic transcriptional responses. Cell Death Dis. 2013, 4, e519. [Google Scholar] [CrossRef] [Green Version]
- Coloff, J.L.; Macintyre, A.; Nichols, A.G.; Liu, T.; Gallo, C.A.; Plas, D.R.; Rathmell, J.C. Akt-Dependent Glucose Metabolism Promotes Mcl-1 Synthesis to Maintain Cell Survival and Resistance to Bcl-2 Inhibition. Cancer Res. 2011, 71, 5204–5213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pradelli, L.A.; Bénéteau, M.; Chauvin, C.; Jacquin, M.A.; Marchetti, S.; Muñoz-Pinedo, C.; Auberger, P.; Pende, M.; Ricci, J.E. Glycolysis inhibition sensitizes tumor cells to death receptors-induced apoptosis by AMP kinase activation leading to Mcl-1 block in translation. Oncogene 2009, 29, 1641–1652. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Coloff, J.L.; Ferguson, E.C.; Jacobs, S.R.; Cui, K.; Rathmell, J.C. Glucose Metabolism Attenuates p53 and Puma-dependent Cell Death upon Growth Factor Deprivation. J. Biol. Chem. 2008, 283, 36344–36353. [Google Scholar] [CrossRef] [Green Version]
- Caro-Maldonado, A.; Tait, S.; Ramírez-Peinado, S.; Ricci, J.E.; Fabregat, I.; Green, D.; Muñoz-Pinedo, C. Glucose deprivation induces an atypical form of apoptosis mediated by caspase-8 in Bax-, Bak-deficient cells. Cell Death Differ. 2010, 17, 1335–1344. [Google Scholar] [CrossRef] [Green Version]
- Abu-Hamad, S.; Arbel, N.; Calo, D.; Arzoine, L.; Israelson, A.; Keinan, N.; Ben-Romano, R.; Friedman, O.; Shoshan-Barmatz, V. The VDAC1 N-terminus is essential both for apoptosis and the protective effect of anti-apoptotic proteins. J. Cell Sci. 2009, 122, 1906–1916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giménez-Cassina, A.; Danial, N.N. Regulation of mitochondrial nutrient and energy metabolism by BCL-2 family proteins. Trends Endocrinol. Metab. 2015, 26, 165–175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Danial, N.N.; Gramm, C.F.; Scorrano, L.; Zhang, C.-Y.; Krauss, S.; Ranger, A.M.; Datta, S.R.; Greenberg, M.E.; Licklider, L.J.; Lowell, B.B.; et al. BAD and glucokinase reside in a mitochondrial complex that integrates glycolysis and apoptosis. Nat. Cell Biol. 2003, 424, 952–956. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.X.; Pervaiz, S. Bcl-2 induces pro-oxidant state by engaging mitochondrial respiration in tumor cells. Cell Death Differ. 2007, 14, 1617–1627. [Google Scholar] [CrossRef] [Green Version]
- Luengo, A.; Gui, D.Y.; Heiden, M.G.V. Targeting Metabolism for Cancer Therapy. Cell Chem. Biol. 2017, 24, 1161–1180. [Google Scholar] [CrossRef] [Green Version]
- Lee, A.; Lau, P.; Kwan, Y.; Kong, S. Mitochondrial Fuel Dependence on Glutamine Drives Chemo-Resistance in the Cancer Stem Cells of Hepatocellular Carcinoma. Int. J. Mol. Sci. 2021, 22, 3315. [Google Scholar] [CrossRef]
- Masoud, R.; Reyes-Castellanos, G.; Lac, S.; Garcia, J.; Dou, S.; Shintu, L.; Hadi, N.A.; Gicquel, T.; El Kaoutari, A.; Diémé, B.; et al. Targeting Mitochondrial Complex I Overcomes Chemoresistance in High OXPHOS Pancreatic Cancer. Cell Rep. Med. 2020, 1, 100143. [Google Scholar] [CrossRef]
- Farge, T.; Saland, E.; De Toni, F.; Aroua, N.; Hosseini, M.; Perry, R.; Bosc, C.; Sugita, M.; Stuani, L.; Fraisse, M.; et al. Chemotherapy-Resistant Human Acute Myeloid Leukemia Cells Are Not Enriched for Leukemic Stem Cells but Require Oxidative Metabolism. Cancer Discov. 2017, 7, 716–735. [Google Scholar] [CrossRef] [Green Version]
- Hwang, I.T.; Chung, Y.M.; Kim, J.J.; Chung, J.S.; Kim, B.S.; Kim, H.J.; Kim, J.S.; Yoo, Y.D. Drug resistance to 5-FU linked to reactive oxygen species modulator 1. Biochem. Biophys. Res. Commun. 2007, 359, 304–310. [Google Scholar] [CrossRef]
- Fu, Y.; Yang, G.; Zhu, F.; Peng, C.; Li, W.; Li, H.; Kim, H.-G.; Bode, A.M.; Dong, Z.; Dong, Z. Abstract 778: Antioxidants decrease the apoptotic effect of 5-Fu in colon cancer by regulating Src-dependent caspase-7 phosphorylation. Cell Death Dis. 2014, 5, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, C.-Y.; Huang, C.-Y.; Pai, Y.-C.; Lin, B.-R.; Lee, T.-C.; Liang, P.-H.; Yu, L.C.-H. Glucose Metabolites Exert Opposing Roles in Tumor Chemoresistance. Front. Oncol. 2019, 9, 1282. [Google Scholar] [CrossRef] [Green Version]
- Cruz-Bermúdez, A.; Briviesca, R.L.; Vicente-Blanco, R.J.; García-Grande, A.; Coronado, M.J.; Laine-Menéndez, S.; Palacios-Zambrano, S.; Moreno-Villa, M.R.; Ruiz-Valdepeñas, A.M.; Lendinez, C.; et al. Cisplatin resistance involves a metabolic reprogramming through ROS and PGC-1α in NSCLC which can be overcome by OXPHOS inhibition. Free Radic. Biol. Med. 2019, 135, 167–181. [Google Scholar] [CrossRef]
- Li, F.-L.; Liu, J.-P.; Bao, R.-X.; Yan, G.; Feng, X.; Xu, Y.-P.; Sun, Y.-P.; Yan, W.; Ling, Z.-Q.; Xiong, Y.; et al. Acetylation accumulates PFKFB3 in cytoplasm to promote glycolysis and protects cells from cisplatin-induced apoptosis. Nat. Commun. 2018, 9, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Circu, M.L.; Aw, T.Y. Glutathione and modulation of cell apoptosis. Biochim. Biophys. Acta Bioenerg. 2012, 1823, 1767–1777. [Google Scholar] [CrossRef] [Green Version]
- Jagust, P.; Alcalá, S.; Sainz, B., Jr.; Heeschen, C.; Sancho, P. Glutathione metabolism is essential for self-renewal and chemoresistance of pancreatic cancer stem cells. World J. Stem Cells 2020, 12, 1410–1428. [Google Scholar] [CrossRef] [PubMed]
- van Gastel, N.; Spinelli, J.B.; Sharda, A.; Schajnovitz, A.; Baryawno, N.; Rhee, C.; Oki, T.; Grace, E.; Soled, H.J.; Milosevic, J. Induction of a Timed Metabolic Collapse to Overcome Cancer Chemoresistance. SSRN Electron. J. 2020, 32, 1–13. [Google Scholar]
- Liu, M.; Wang, D.; Luo, Y.; Hu, L.; Bi, Y.; Ji, J.; Huang, H.; Wang, G.; Zhu, L.; Ma, J.; et al. Selective killing of cancer cells harboring mutant RAS by concomitant inhibition of NADPH oxidase and glutathione biosynthesis. Cell Death Dis. 2021, 12, 1–15. [Google Scholar] [CrossRef]
- Tang, L.; Wei, F.; Wu, Y.; He, Y.; Shi, L.; Xiong, F.; Gong, Z.; Guo, C.; Li, X.; Deng, H.; et al. Role of metabolism in cancer cell radioresistance and radiosensitization methods. J. Exp. Clin. Cancer Res. 2018, 37, 1–15. [Google Scholar] [CrossRef]
- Lynam-Lennon, N.; Maher, S.G.; Maguire, A.; Phelan, J.; Muldoon, C.; Reynolds, J.V.; O’Sullivan, J. Altered Mitochondrial Function and Energy Metabolism Is Associated with a Radioresistant Phenotype in Oesophageal Adenocarcinoma. PLoS ONE 2014, 9, e100738. [Google Scholar] [CrossRef]
- Kam, W.W.; Banati, R.B. Free Radical Biology and Medicine Effects of ionizing radiation on mitochondria. Free Radic. Biol. Med. 2013, 65, 607–619. [Google Scholar] [CrossRef] [PubMed]
- McCann, E.; O’Sullivan, J.; Marcone, S. Targeting cancer-cell mitochondria and metabolism to improve radiotherapy response. Transl. Oncol. 2021, 14, 100905. [Google Scholar] [CrossRef] [PubMed]
- Pitroda, S.P.; Wakim, B.T.; Sood, R.F.; Beveridge, M.G.; Beckett, M.A.; MacDermed, D.M.; Weichselbaum, R.R.; Khodarev, N.N. STAT1-dependent expression of energy metabolic pathways links tumour growth and radioresistance to the Warburg effect. BMC Med. 2009, 7, 68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, R.; Fan, M.; Candas, D.; Qin, L.; Zhang, X.; Eldridge, A.; Zou, J.X.; Zhang, T.; Juma, S.; Jin, C.; et al. CDK1-Mediated SIRT3 Activation Enhances Mitochondrial Function and Tumor Radioresistance. Mol. Cancer Ther. 2015, 14, 2090–2102. [Google Scholar] [CrossRef] [Green Version]
- Roca-Portoles, A.; Rodriguez-Blanco, G.; Sumpton, D.; Cloix, C.; Mullin, M.; Mackay, G.M.; O’Neill, K.; Lemgruber, L.; Luo, X.; Tait, S.W.G. Venetoclax causes metabolic reprogramming independent of BCL-2 inhibition. Cell Death Dis. 2020, 11, 1–13. [Google Scholar] [CrossRef]
- León-Annicchiarico, C.L.; Ramírez-Peinado, S.; Domínguez-Villanueva, D.; Gonsberg, A.; Lampidis, T.J.; Munoz-Pinedo, C. ATF4 mediates necrosis induced by glucose deprivation and apoptosis induced by 2-deoxyglucose in the same cells. FEBS J. 2015, 282, 3647–3658. [Google Scholar] [CrossRef] [Green Version]
- El Mjiyad, N.; Caro-Maldonado, A.; Ramírez-Peinado, S.; Mũoz-Pinedo, C. Sugar-free approaches to cancer cell killing. Oncogene 2011, 30, 253–264. [Google Scholar] [CrossRef] [Green Version]
- Rena, G.; Hardie, D.G.; Pearson, E.R. The mechanisms of action of metformin. Diabetologia 2017, 60, 1577–1585. [Google Scholar] [CrossRef] [Green Version]
- Saraei, P.; Asadi, I.; Kakar, M.A.; Moradi-Kor, N. The beneficial effects of metformin on cancer prevention and therapy: A comprehensive review of recent advances. Cancer Manag. Res. 2019, 11, 3295–3313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fulda, S.; Galluzzi, L.; Kroemer, G. Targeting mitochondria for cancer therapy. Nat. Rev. Drug Discov. 2010, 9, 447–464. [Google Scholar] [CrossRef]
- Dong, L.; Neuzil, J. Targeting mitochondria as an anticancer strategy. Cancer Commun. 2019, 39, 63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merino, D.; Kelly, G.L.; Lessene, G.; Wei, A.H.; Roberts, A.W.; Strasser, A. BH3-Mimetic Drugs: Blazing the Trail for New Cancer Medicines. Cancer Cell 2018, 34, 879–891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Howard, A.N.; Bridges, K.A.; Meyn, R.E.; Chandra, J. ABT-737, a BH3 mimetic, induces glutathione depletion and oxidative stress. Cancer Chemother. Pharmacol. 2009, 65, 41–54. [Google Scholar] [CrossRef] [PubMed]
- Serasinghe, M.N.; Gelles, J.D.; Li, K.; Zhao, L.; Abbate, F.; Syku, M.; Mohammed, J.N.; Badal, B.; Rangel, C.A.; Hoehn, K.; et al. Dual suppression of inner and outer mitochondrial membrane functions augments apoptotic responses to oncogenic MAPK inhibition. Cell Death Dis. 2018, 9, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, F.; Kalpage, H.A.; Wang, D.; Edwards, H.; Hüttemann, M.; Ma, J.; Su, Y.; Carter, J.; Li, X.; Polin, L.; et al. Cotargeting of Mitochondrial Complex I and Bcl-2 Shows Antileukemic Activity against Acute Myeloid Leukemia Cells Reliant on Oxidative Phosphorylation. Cancers 2020, 12, 2400. [Google Scholar] [CrossRef] [PubMed]
- Roca-Portoles, A.; Tait, S.W.G. Mitochondrial quality control: From molecule to organelle. Cell. Mol. Life Sci. 2021, 78, 3853–3866. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
McCann, C.; Kerr, E.M. Metabolic Reprogramming: A Friend or Foe to Cancer Therapy? Cancers 2021, 13, 3351. https://doi.org/10.3390/cancers13133351
McCann C, Kerr EM. Metabolic Reprogramming: A Friend or Foe to Cancer Therapy? Cancers. 2021; 13(13):3351. https://doi.org/10.3390/cancers13133351
Chicago/Turabian StyleMcCann, Christopher, and Emma M. Kerr. 2021. "Metabolic Reprogramming: A Friend or Foe to Cancer Therapy?" Cancers 13, no. 13: 3351. https://doi.org/10.3390/cancers13133351