
Clinically Advanced Pheochromocytomas and Paragangliomas: A Comprehensive Genomic Profiling Study

 , , ,
, , ,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Methods
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lam, A.K.-Y. Update on Adrenal Tumours in 2017 World Health Organization (WHO) of Endocrine Tumours. Endocr. Pathol. 2017, 28, 213–227. [Google Scholar] [CrossRef]
- Williams, M.D.; Tischler, A.S. Update from the 4th Edition of the World Health Organization Classification of Head and Neck Tumours: Paragangliomas. Head Neck Pathol. 2017, 11, 88–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crona, J.; Lamarca, A.; Ghosal, S.; Welin, S.; Skogseid, B.; Pacak, K. Genotype–phenotype correlations in pheochromocytoma and paraganglioma: A systematic review and individual patient meta-analysis. Endocr. Relat. Cancer 2019, 26, 539–550. [Google Scholar] [CrossRef]
- Thompson, L.D.R. Pheochromocytoma of the Adrenal Gland Scaled Score (PASS) to Separate Benign from Malignant Neoplasms. Am. J. Surg. Pathol. 2002, 26, 551–566. [Google Scholar] [CrossRef] [PubMed]
- Waingankar, N.; Bratslavsky, G.; Jimenez, C.; Russo, P.; Kutikov, A. Pheochromocytoma in Urologic Practice. Eur. Urol. Focus 2016, 1, 231–240. [Google Scholar] [CrossRef] [Green Version]
- Guerrero, M.A.; Schreinemakers, J.M.; Vriens, M.R.; Suh, I.; Hwang, J.; Shen, W.T.; Gosnell, J.; Clark, O.H.; Duh, Q.-Y. Clinical Spectrum of Pheochromocytoma. J. Am. Coll. Surg. 2009, 209, 727–732. [Google Scholar] [CrossRef]
- Lenders, J.W.M.; Duh, Q.-Y.; Eisenhofer, G.; Gimenez-Roqueplo, A.-P.; Grebe, S.K.G.; Murad, M.H.; Naruse, M.; Pacak, K.; Young, W.F. Pheochromocytoma and Paraganglioma: An Endocrine Society Clinical Practice Guideline. J. Clin. Endocrinol. Metab. 2014, 99, 1915–1942. [Google Scholar] [CrossRef]
- Björklund, P.; Pacak, K.; Crona, J. Precision medicine in pheochromocytoma and paraganglioma: Current and future concepts. J. Intern. Med. 2016, 280, 559–573. [Google Scholar] [CrossRef] [Green Version]
- Frampton, G.M.; Fichtenholtz, A.; Otto, G.A.; Wang, K.; Downing, S.R.; He, J.; Schnall-Levin, M.; White, J.; Sanford, E.M.; An, P.; et al. Development and validation of a clinical cancer genomic profiling test based on massively parallel DNA sequencing. Nat Biotechnol. 2013, 31, 1023–1031. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.X.; He, Y.; Sanford, E.; Montesion, M.; Frampton, G.M.; Vignot, S.; Soria, J.-C.; Ross, J.S.; Miller, V.A.; Stephens, P.J.; et al. A computational approach to distinguish somatic vs. germline origin of genomic alterations from deep sequencing of cancer specimens without a matched normal. PLoS Comput. Biol. 2018, 14, e1005965. [Google Scholar] [CrossRef] [Green Version]
- Forbes, S.A.; Beare, D.; Gunasekaran, P.; Leung, K.; Bindal, N.; Boutselakis, H.; Ding, M.; Bamford, S.; Cole, C.; Ward, S.; et al. COSMIC: Exploring the world’s knowledge of somatic mutations in human cancer. Nucleic Acids Res. 2015, 43, D805–D811. [Google Scholar] [CrossRef] [PubMed]
- Chalmers, Z.R.; Connelly, C.F.; Fabrizio, D.; Gay, L.; Ali, S.M.; Ennis, R.; Schrock, A.; Campbell, B.; Shlien, A.; Chmielecki, J.; et al. Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Med. 2017, 9, 34. [Google Scholar] [CrossRef] [PubMed]
- Feldman, J.M. Treatment of metastatic pheochromocytoma with streptozocin. Arch. Intern. Med. 1983, 143, 1799–1800. [Google Scholar] [CrossRef]
- Dahia, P.L.M. Pheochromocytoma and paraganglioma pathogenesis: Learning from genetic heterogeneity. Nat. Rev. Cancer 2014, 14, 108–119. [Google Scholar] [CrossRef]
- Fishbein, L.; Ben-Maimon, S.; Keefe, S.; Cengel, K.; Pryma, D.A.; Loaiza-Bonilla, A.; Fraker, D.L.; Nathanson, K.L.; Cohen, D.L. SDHB mutation carriers with malignant pheochromocytoma respond better to CVD. Endocr. Relat. Cancer 2017, 24, L51–L55. [Google Scholar] [CrossRef] [PubMed]
- Chino, J.P.; Sampson, J.H.; Tucci, D.L.; Brizel, D.M.; Kirkpatrick, J.P. Paraganglioma of the Head and Neck. Am. J. Clin. Oncol. 2009, 32, 304–307. [Google Scholar] [CrossRef]
- Venkatesan, A.M.; Locklin, J.; Lai, E.W.; Adams, K.T.; Fojo, A.T.; Pacak, K.; Wood, B.J. Radiofrequency Ablation of Metastatic Pheochromocytoma. J. Vasc. Interv. Radiol. 2009, 20, 1483–1490. [Google Scholar] [CrossRef] [Green Version]
- Oh, D.-Y.; Kim, T.-W.; Park, Y.S.; Shin, S.J.; Shin, S.H.; Song, E.-K.; Lee, H.J.; Lee, K.-W.; Bang, Y.-J. Phase 2 study of everolimus monotherapy in patients with nonfunctioning neuroendocrine tumors or pheochromocytomas/paragangliomas. Cancer 2012, 118, 6162–6170. [Google Scholar] [CrossRef]
- Watson, I.; Takahashi, K.; Futreal, P.A.; Chin, L. Emerging patterns of somatic mutations in cancer. Nat. Rev. Genet. 2013, 14, 703–718. [Google Scholar] [CrossRef] [Green Version]
- Turchini, J.; Cheung, V.K.Y.; Tischler, A.S.; De Krijger, R.R.; Gill, A.J. Pathology and genetics of phaeochromocytoma and paraganglioma. Histopathology 2018, 72, 97–105. [Google Scholar] [CrossRef] [Green Version]
- Drilon, A.; Wang, L.; Arcila, M.E.; Balasubramanian, S.; Greenbowe, J.R.; Ross, J.S.; Stephens, P.J.; Lipson, D.; Miller, V.A.; Kris, M.G.; et al. Broad, Hybrid Capture–Based Next-Generation Sequencing Identifies Actionable Genomic Alterations in Lung Adenocarcinomas Otherwise Negative for Such Alterations by Other Genomic Testing Approaches. Clin. Cancer Res. 2015, 21, 3631–3639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katoh, M. Fibroblast growth factor receptors as treatment targets in clinical oncology. Nat. Rev. Clin. Oncol. 2019, 16, 105–122. [Google Scholar] [CrossRef]
- Markham, A. Erdafitinib: First Global Approval. Drugs 2019, 79, 1017–1021. [Google Scholar] [CrossRef]
- Fishbein, L.; Leshchiner, I.; Walter, V.; Danilova, L.; Robertson, A.G.; Johnson, A.R.; Lichtenberg, T.M.; Murray, B.A.; Ghayee, H.K.; Else, T.; et al. Comprehensive Molecular Characterization of Pheochromocytoma and Paraganglioma. Cancer Cell 2017, 31, 181–193. [Google Scholar] [CrossRef] [Green Version]
- Gkountakos, A.; Pilotto, S.; Mafficini, A.; Vicentini, C.; Simbolo, M.; Milella, M.; Giampaolo, T.; Aldo, S.; Emilio, B.; Vincenzo, C. Unmasking the impact of Rictor in cancer: Novel insights of mTORC2 complex. Carcinogenesis 2018, 39, 971–980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakre, N.; Wildey, G.; Behtaj, M.; Kresak, A.; Yang, M.; Fu, P.; Dowlati, A. RICTOR amplification identifies a subgroup in small cell lung cancer and predicts response to drugs targeting mTOR. Oncotarget 2016, 8, 5992–6002. [Google Scholar] [CrossRef] [Green Version]
- Tolcher, A.W.; Bendell, J.C.; Papadopoulos, K.P.; Burris, H.A.; Patnaik, A.; Jones, S.F.; Rasco, D.; Cox, D.S.; Durante, M.; Bellew, K.M.; et al. A phase IB trial of the oral MEK inhibitor trametinib (GSK1120212) in combination with everolimus in patients with advanced solid tumors. Ann. Oncol. 2015, 26, 58–64. [Google Scholar] [CrossRef]
- Chan, T.; Yarchoan, M.; Jaffee, E.; Swanton, C.; Quezada, S.; Stenzinger, A.; Peters, S. Development of tumor mutation burden as an immunotherapy biomarker: Utility for the oncology clinic. Ann. Oncol. 2019, 30, 44–56. [Google Scholar] [CrossRef] [PubMed]
- Goodman, A.M.; Piccioni, D.; Kato, S.; Boichard, A.; Wang, H.-Y.; Frampton, G.; Lippman, S.M.; Connelly, C.; Fabrizio, D.; Miller, V.; et al. Prevalence of PDL1 Amplification and Preliminary Response to Immune Checkpoint Blockade in Solid Tumors. JAMA Oncol. 2018, 4, 1237–1244. [Google Scholar] [CrossRef] [Green Version]
- Kato, S.; Goodman, A.; Walavalkar, V.; Barkauskas, D.A.; Sharabi, A.; Kurzrock, R. Hyperprogressors after Immunotherapy: Analysis of Genomic Alterations Associated with Accelerated Growth Rate. Clin. Cancer Res. 2017, 23, 4242–4250. [Google Scholar] [CrossRef] [Green Version]
- Buffet, A.; Ben Aim, L.; Leboulleux, S.; Drui, D.; Vezzosi, D.; Libé, R.; Ajzenberg, C.; Bernardeschi, D.; Cariou, B.; Chabolle, F.; et al. Positive Impact of Genetic Test on the Management and Outcome of Patients with Paraganglioma and/or Pheochromocytoma. J. Clin. Endocrinol. Metab. 2019, 104, 1109–1118. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 1.3 to 2.4 Mutations/Mb | CA-Pheo | CA-Para |
|---|---|---|
| Number of Cases | 45 | 83 |
| Age in years (range) | 53 (7–78) | 48 (10–80) |
| Males/Females | 21/24 | 50/33 |
| Sample Type Used for Sequencing | Primary Tumor 27% Metastatic Site 73% | Primary tumor 17% Metastatic Site 83% |
| GA per tumor | 2.3 | 1.9 |
| Predicted Frequency of Germline GA | 31% | 43% |
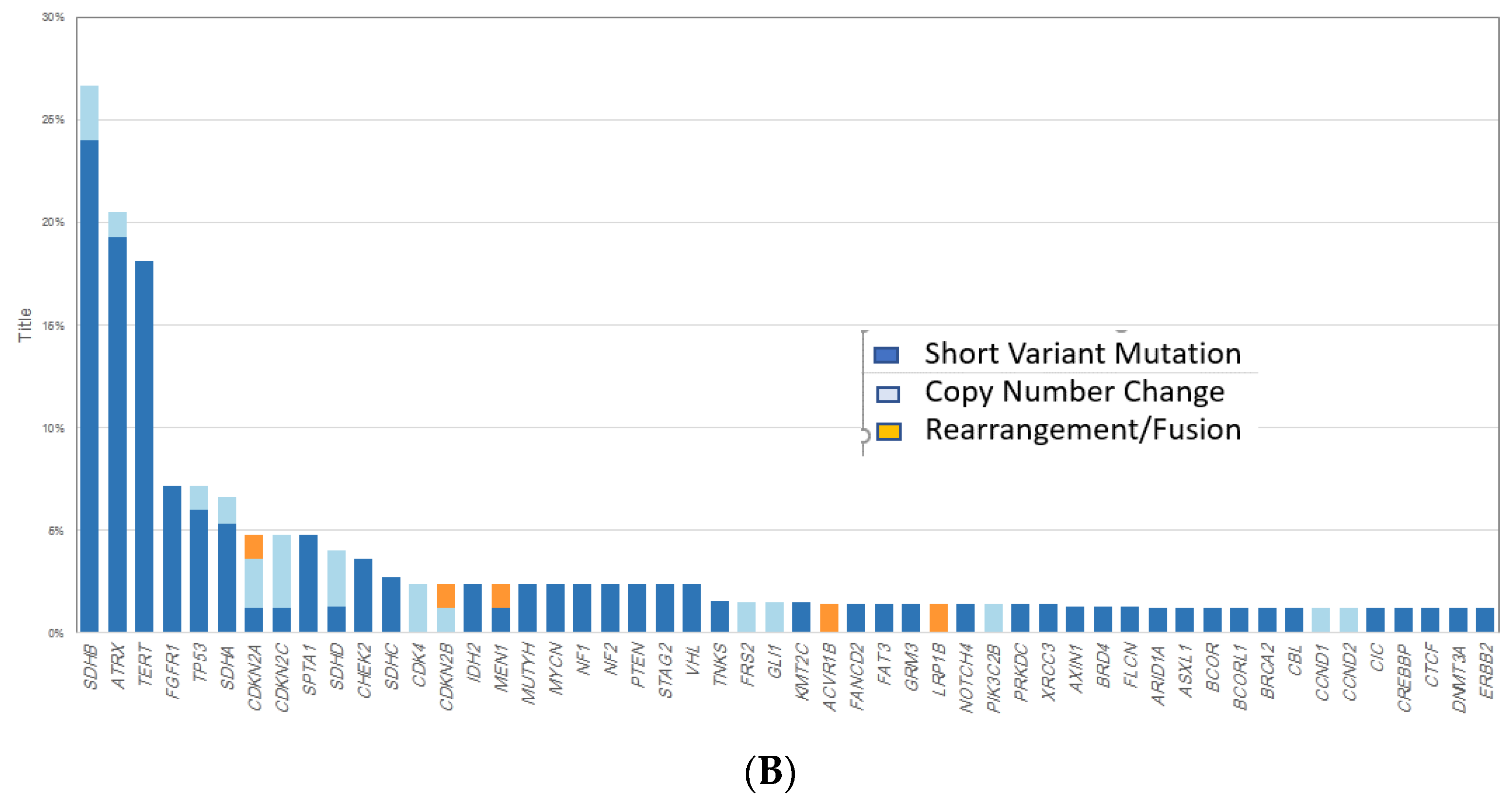
| Most common Untargetable GA | ATRX (24%) TP53 (20%) SDHB (16%) CTNNB1 (7%) VHL (7%) CDKN2A/2B, PIK3R2, NOTCH2 and MEN1 (all 4%) | SDHB (27%) ATRX (21%) TERT (18%) TP53 (7%) SDHA (7%) |
| Most common Targetable GA | RET (9%) NF1 (11%) FGFR1 (7%) | FGFR1 (7%) NF1 (2%) PTEN (2%) NF2 (2%) CDK4 (2%) |
| CD274 amplification | 0% | 0% |
| PBRM1 GA | 2% | 1% |
| MSI | 0% | 0% |
| Median TMB mut/Mb | 1.7 | 1.2 |
| TMB ≥ 10/20 mut/Mb | 2%/0% | 6%/2% |
| PD-L1 Expression low/high | 0%/0% | 14%/0% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bratslavsky, G.; Sokol, E.S.; Daneshvar, M.; Necchi, A.; Shapiro, O.; Jacob, J.; Liu, N.; Sanford, T.S.; Pinkhasov, R.; Goldberg, H.; et al. Clinically Advanced Pheochromocytomas and Paragangliomas: A Comprehensive Genomic Profiling Study. Cancers 2021, 13, 3312. https://doi.org/10.3390/cancers13133312
Bratslavsky G, Sokol ES, Daneshvar M, Necchi A, Shapiro O, Jacob J, Liu N, Sanford TS, Pinkhasov R, Goldberg H, et al. Clinically Advanced Pheochromocytomas and Paragangliomas: A Comprehensive Genomic Profiling Study. Cancers. 2021; 13(13):3312. https://doi.org/10.3390/cancers13133312
Chicago/Turabian StyleBratslavsky, Gennady, Ethan S. Sokol, Michael Daneshvar, Andrea Necchi, Oleg Shapiro, Joseph Jacob, Nick Liu, Tom S. Sanford, Ruben Pinkhasov, Hanan Goldberg, and et al. 2021. "Clinically Advanced Pheochromocytomas and Paragangliomas: A Comprehensive Genomic Profiling Study" Cancers 13, no. 13: 3312. https://doi.org/10.3390/cancers13133312