
Protein Aggregation Patterns Inform about Breast Cancer Response to Antiestrogens and Reveal the RNA Ligase RTCB as Mediator of Acquired Tamoxifen Resistance

 ,
,  ,
,  ,
,  , and
, and 
Abstract
:Simple Summary
Abstract
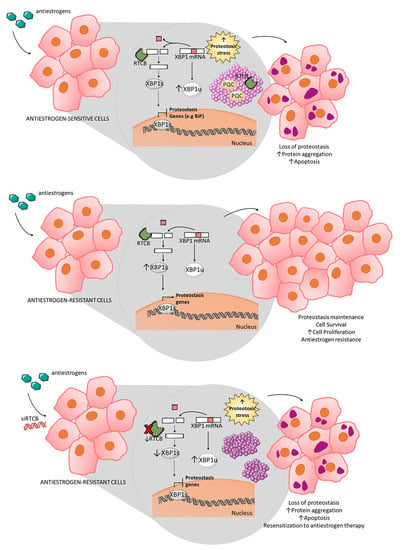
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Cell Proliferation and Transfection Assays
2.3. Insoluble Protein Isolation
2.4. SDS-PAGE Electrophoresis and Western Blot Hybridization
2.5. Aggresome Detection and Co-Localization Studies
2.6. Tryptic Digestion, Mass Spectrometry Analysis and Protein Identification
2.7. Statistical Analysis and Bioinformatics
2.8. Patient Sample Collection and Characterization
3. Results
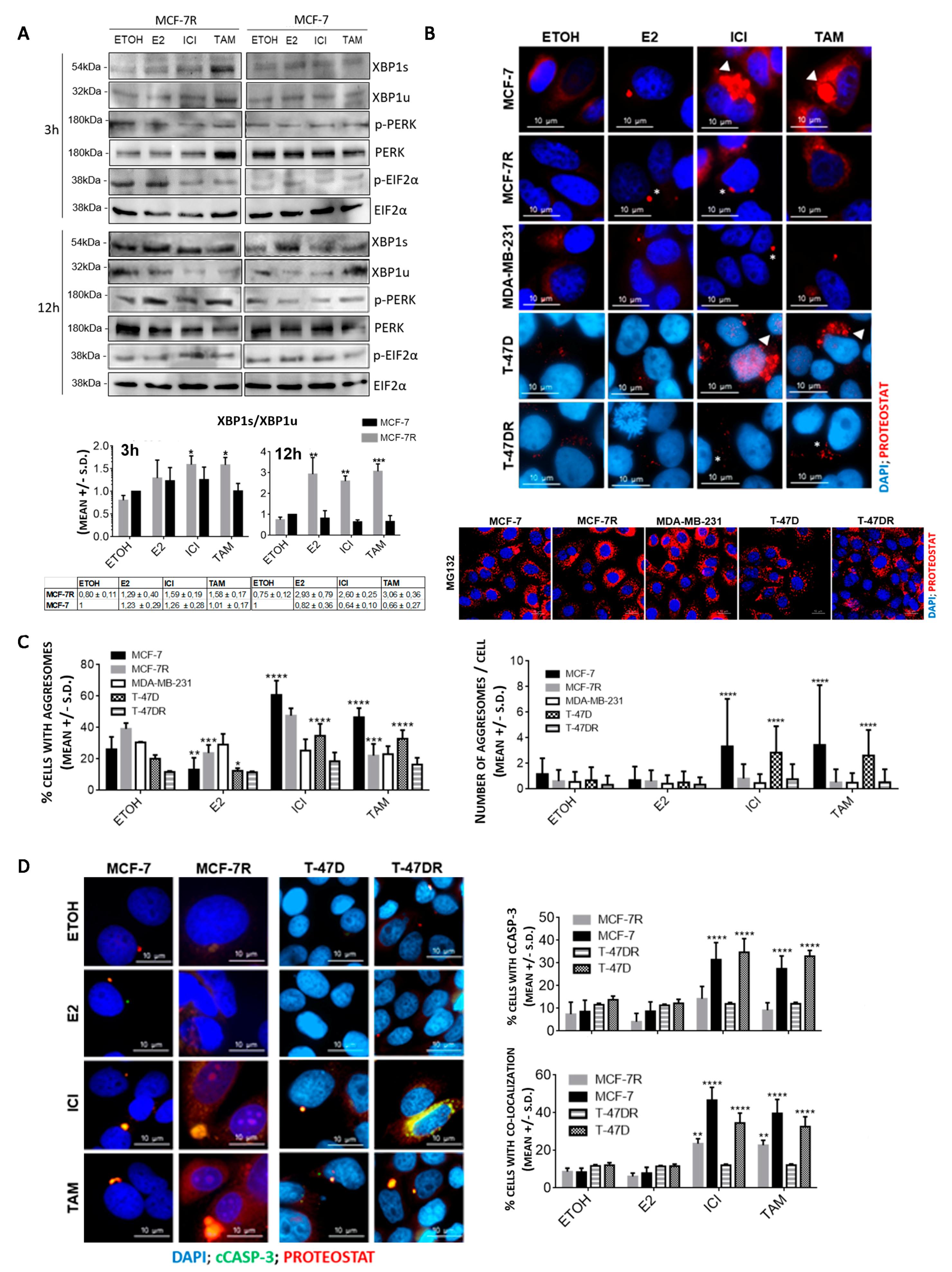
3.1. Aggresome Accumulation Correlates with Reduced UPR and Autophagy Activation in Antiestrogen-Sensitive Breast Cancer Cells
3.2. Protein Aggregation after Antiestrogen Treatment Targets Different Pathways in Sensitive and Resistant Cells
3.3. Protein Aggregation after Antiestrogen Treatment Impairs Proteasome and UPR Function
3.4. Inhibition of RTCB Expression Downregulates XBP1s and Improves Sensitivity to Tamoxifen
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Clarke, R.; Tyson, J.J.; Dixon, J.M. Endocrine resistance in breast cancer—An overview and update. Mol. Cell Endocrinol. 2015, 418, 220–234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Musgrove, E.A.; Sutherland, R.L. Biological determinants of endocrine resistance in breast cancer. Nat. Rev. Cancer 2009, 9, 631–643. [Google Scholar] [CrossRef] [PubMed]
- Direito, I.; Fardilha, M.; Helguero, L.A. Contribution of the unfolded protein response to breast and prostate tissue homeostasis and its significance to cancer endocrine response. Carcinogenesis 2019, 40, 203–215. [Google Scholar] [CrossRef]
- Gomez, B.P.; Riggins, R.B.; Shajahan, A.N.; Klimach, U.; Wang, A.; Crawford, A.C.; Zhu, Y.; Zwart, A.; Wang, M.; Clarke, R. Human X-box binding protein-1 confers both estrogen independence and antiestrogen resistance in breast cancer cell lines. FASEB J. 2007, 21, 4013–4027. [Google Scholar] [CrossRef]
- Davies, M.P.; Barraclough, D.L.; Stewart, C.; Joyce, K.A.; Eccles, R.M.; Barraclough, R.; Rudland, P.S.; Sibson, D.R. Expression and splicing of the unfolded protein response gene XBP-1 are significantly associated with clinical outcome of endocrine-treated breast cancer. Int. J. Cancer 2008, 123, 85–88. [Google Scholar] [CrossRef]
- Ming, J.; Ruan, S.; Wang, M.; Ye, D.; Fan, N.; Meng, Q.; Tian, B.; Huang, T. A novel chemical, STF-083010, reverses tamoxifen-related drug resistance in breast cancer by inhibiting IRE1/XBP1. Oncotarget 2015, 6, 40692–40703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qadir, M.A.; Kwok, B.; Dragowska, W.H.; To, K.H.; Le, D.; Bally, M.B.; Gorski, S.M. Macroautophagy inhibition sensitizes tamoxifen-resistant breast cancer cells and enhances mitochondrial depolarization. Breast Cancer Res. Treat. 2008, 112, 389–403. [Google Scholar] [CrossRef] [PubMed]
- Schoenlein, P.V.; Periyasamy-Thandavan, S.; Samaddar, J.S.; Jackson, W.H.; Barrett, J.T. Autophagy facilitates the progression of ERalpha-positive breast cancer cells to antiestrogen resistance. Autophagy 2009, 5, 400–403. [Google Scholar] [CrossRef] [Green Version]
- Cook, K.L.; Shajahan, A.N.; Wärri, A.; Jin, L.; Hilakivi-Clarke, L.A.; Clarke, R. Glucose-regulated protein 78 controls cross-talk between apoptosis and autophagy to determine antiestrogen responsiveness. Cancer Res. 2012, 72, 3337–3349. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Rucker, E.B.; Zhou, B.P. Autophagy regulation in the development and treatment of breast cancer. Acta Biochim. Biophys. Sin. 2016, 48, 60–74. [Google Scholar] [CrossRef] [Green Version]
- Voutsadakis, I.A. Proteasome expression and activity in cancer and cancer stem cells. Tumour Biol. 2017, 39, 1010428317692248. [Google Scholar] [CrossRef] [Green Version]
- Dubnikov, T.; Ben-Gedalya, T.; Cohen, E. Protein Quality Control in Health and Disease. Cold Spring Harb. Perspect. Biol. 2017, 9, a023523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guang, M.H.Z.; Kavanagh, E.L.; Dunne, L.P.; Dowling, P.; Zhang, L.; Lindsay, S.; Bazou, D.; Goh, C.Y.; Hanley, C.; Bianchi, G.; et al. Targeting Proteotoxic Stress in Cancer: A Review of the Role that Protein Quality Control Pathways Play in Oncogenesis. Cancers 2019, 11, 66. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Cubillos-Ruiz, J.R. Endoplasmic reticulum stress signals in the tumour and its microenvironment. Nat. Rev. Cancer 2020, 21, 71–88. [Google Scholar] [CrossRef] [PubMed]
- Hetz, C.; Chevet, E.; Harding, H.P. Targeting the unfolded protein response in disease. Nat. Rev. Drug Discov. 2013, 12, 703–719. [Google Scholar] [CrossRef] [PubMed]
- Andruska, N.; Zheng, X.; Yang, X.; Helferich, W.G.; Shapiro, D.J. Anticipatory estrogen activation of the unfolded protein response is linked to cell proliferation and poor survival in estrogen receptor α-positive breast cancer. Oncogene 2015, 34, 3760–3769. [Google Scholar] [CrossRef] [Green Version]
- Uemura, A.; Oku, M.; Mori, K.; Yoshida, H. Unconventional splicing of XBP1 mRNA occurs in the cytoplasm during the mammalian unfolded protein response. J. Cell Sci. 2009, 122, 2877–2886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Popow, J.; Englert, M.; Weitzer, S.; Schleiffer, A.; Mierzwa, B.; Mechtler, K.; Trowitzsch, S.; Will, C.L.; Lührmann, R.; Söll, D.; et al. HSPC117 is the essential subunit of a human tRNA splicing ligase complex. Science 2011, 331, 760–764. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.Y.; Fulthorpe, R.; Liss, S.N.; Edwards, E.A. Identification of estrogen-responsive genes by complementary deoxyribonucleic acid microarray and characterization of a novel early estrogen-induced gene: EEIG1. Mol. Endocrinol. 2004, 18, 402–411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Chen, X.; Gao, Y.; Wu, J.; Zeng, F.; Song, F. XBP1 induces snail expression to promote epithelial- to-mesenchymal transition and invasion of breast cancer cells. Cell Signal. 2015, 27, 82–89. [Google Scholar] [CrossRef]
- Crawford, A.C.; Riggins, R.B.; Shajahan, A.N.; Zwart, A.; Clarke, R. Co-inhibition of BCL-W and BCL2 restores antiestrogen sensitivity through BECN1 and promotes an autophagy-associated necrosis. PLoS ONE 2010, 5, e8604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samaddar, J.S.; Gaddy, V.T.; Duplantier, J.; Thandavan, S.P.; Shah, M.; Smith, M.J.; Browning, D.; Rawson, J.; Smith, S.B.; Barrett, J.T.; et al. A role for macroautophagy in protection against 4-hydroxytamoxifen-induced cell death and the development of antiestrogen resistance. Mol. Cancer Ther. 2008, 7, 2977–2987. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Groh, N.; Bühler, A.; Huang, C.; Li, K.W.; van Nierop, P.; Smit, A.B.; Fändrich, M.; Baumann, F.; David, D.C. Age-Dependent Protein Aggregation Initiates Amyloid-β Aggregation. Front. Aging Neurosci. 2017, 9, 138. [Google Scholar] [CrossRef]
- Vaquer-Alicea, J.; Diamond, M.I. Propagation of Protein Aggregation in Neurodegenerative Diseases. Annu. Rev. Biochem. 2019, 88, 785–810. [Google Scholar] [CrossRef]
- Levy, C.B.; Stumbo, A.C.; Ano Bom, A.P.; Portari, E.A.; Cordeiro, Y.; Carneiro, Y.; Silva, J.L.; De Moura-Gallo, C.V. Co-localization of mutant p53 and amyloid-like protein aggregates in breast tumors. Int. J. Biochem. Cell Biol. 2011, 43, 60–64. [Google Scholar] [CrossRef] [PubMed]
- Chiu, H.W.; Yeh, Y.L.; Ho, S.Y.; Wu, Y.H.; Wang, B.J.; Huang, W.J.; Ho, Y.S.; Wang, Y.J.; Chen, L.C.; Tu, S.H. A New Histone Deacetylase Inhibitor Enhances Radiation Sensitivity through the Induction of Misfolded Protein Aggregation and Autophagy in Triple-Negative Breast Cancer. Cancers 2019, 11, 1703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, D.; Coleman, J.; Chan, E.; Nicholson, T.P.; Dai, L.; Sheppard, P.W.; Patton, W.F. Novel cell- and tissue-based assays for detecting misfolded and aggregated protein accumulation within aggresomes and inclusion bodies. Cell Biochem. Biophys. 2011, 60, 173–185. [Google Scholar] [CrossRef] [Green Version]
- Shevchenko, A.; Tomas, H.; Havlis, J.; Olsen, J.V.; Mann, M. In-gel digestion for mass spectrometric characterization of proteins and proteomes. Nat. Protoc. 2006, 1, 2856–2860. [Google Scholar] [CrossRef]
- Oliveros, J.C. Venny. An Interactive Tool For Comparing Lists with Venn’s Diagrams. 2007–2015. Available online: https://bioinfogp.cnb.csic.es/tools/venny/index.html (accessed on 13 May 2021).
- Takalo, M.; Salminen, A.; Soininen, H.; Hiltunen, M.; Haapasalo, A. Protein aggregation and degradation mechanisms in neurodegenerative diseases. Am. J. Neurodegener Dis. 2013, 2, 1–14. [Google Scholar]
- Kaganovich, D.; Kopito, R.; Frydman, J. Misfolded proteins partition between two distinct quality control compartments. Nature 2008, 454, 1088–1095. [Google Scholar] [CrossRef]
- Ding, W.X.; Yin, X.M. Sorting, recognition and activation of the misfolded protein degradation pathways through macroautophagy and the proteasome. Autophagy 2008, 4, 141–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monaco, A.; Fraldi, A. Protein Aggregation and Dysfunction of Autophagy-Lysosomal Pathway: A Vicious Cycle in Lysosomal Storage Diseases. Front. Mol. Neurosci. 2020, 13, 37. [Google Scholar] [CrossRef]
- Liu, J.; Yue, W.; Chen, H. The correlation between autophagy and tamoxifen resistance in breast cancer. Int. J. Clin. Exp. Pathol. 2019, 12, 2066–2074. [Google Scholar] [PubMed]
- Parmar, J.H.; Cook, K.L.; Shajahan-Haq, A.N.; Clarke, P.A.; Tavassoly, I.; Clarke, R.; Tyson, J.J.; Baumann, W.T. Modelling the effect of GRP78 on anti-oestrogen sensitivity and resistance in breast cancer. Interface Focus 2013, 3, 20130012. [Google Scholar] [CrossRef] [Green Version]
- Choi, K.S. Autophagy and cancer. Exp. Mol. Med. 2012, 44, 109–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bursch, W.; Ellinger, A.; Kienzl, H.; Török, L.; Pandey, S.; Sikorska, M.; Walker, R.; Hermann, R.S. Active cell death induced by the anti-estrogens tamoxifen and ICI 164 384 in human mammary carcinoma cells (MCF-7) in culture: The role of autophagy. Carcinogenesis 1996, 17, 1595–1607. [Google Scholar] [CrossRef] [Green Version]
- Cook, K.L.; Shajahan, A.N.; Clarke, R. Autophagy and endocrine resistance in breast cancer. Expert Rev. Anticancer Ther. 2011, 11, 1283–1294. [Google Scholar] [CrossRef]
- Vera-Ramirez, L.; Vodnala, S.K.; Nini, R.; Hunter, K.W.; Green, J.E. Autophagy promotes the survival of dormant breast cancer cells and metastatic tumour recurrence. Nat. Commun. 2018, 9, 1944. [Google Scholar] [CrossRef] [Green Version]
- Lotz, G.P.; Legleiter, J. The role of amyloidogenic protein oligomerization in neurodegenerative disease. J. Mol. Med. 2013, 91, 653–664. [Google Scholar] [CrossRef]
- Shigemitsu, Y.; Hiroaki, H. Common molecular pathogenesis of disease-related intrinsically disordered proteins revealed by NMR analysis. J. Biochem. 2018, 163, 11–18. [Google Scholar] [CrossRef]
- Guo, W.; Chen, Y.; Zhou, X.; Kar, A.; Ray, P.; Chen, X.; Rao, E.J.; Yang, M.; Ye, H.; Zhu, L.; et al. An ALS-associated mutation affecting TDP-43 enhances protein aggregation, fibril formation and neurotoxicity. Nat. Struct. Mol. Biol. 2011, 18, 822–830. [Google Scholar] [CrossRef] [Green Version]
- Winton, M.J.; Igaz, L.M.; Wong, M.M.; Kwong, L.K.; Trojanowski, J.Q.; Lee, V.M. Disturbance of nuclear and cytoplasmic TAR DNA-binding protein (TDP-43) induces disease-like redistribution, sequestration, and aggregate formation. J. Biol. Chem. 2008, 283, 13302–13309. [Google Scholar] [CrossRef] [Green Version]
- Tanikawa, C.; Ueda, K.; Suzuki, A.; Iida, A.; Nakamura, R.; Atsuta, N.; Tohnai, G.; Sobue, G.; Saichi, N.; Momozawa, Y.; et al. Citrullination of RGG Motifs in FET Proteins by PAD4 Regulates Protein Aggregation and ALS Susceptibility. Cell Rep. 2018, 22, 1473–1483. [Google Scholar] [CrossRef] [Green Version]
- Monti, C.; Colugnat, I.; Lopiano, L.; Chiò, A.; Alberio, T. Network Analysis Identifies Disease-Specific Pathways for Parkinson’s Disease. Mol. Neurobiol. 2018, 55, 370–381. [Google Scholar] [CrossRef] [Green Version]
- Kipkorir, T.; Colangelo, C.M.; Manuelidis, L. Proteomic analysis of host brain components that bind to infectious particles in Creutzfeldt-Jakob disease. Proteomics 2015, 15, 2983–2998. [Google Scholar] [CrossRef] [Green Version]
- Lu, Y.; Liang, F.X.; Wang, X. A synthetic biology approach identifies the mammalian UPR RNA ligase RtcB. Mol. Cell 2014, 55, 758–770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eletto, D.; Dersh, D.; Gidalevitz, T.; Argon, Y. Protein disulfide isomerase A6 controls the decay of IRE1α signaling via disulfide-dependent association. Mol. Cell 2014, 53, 562–576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pérez-González, A.; Pazo, A.; Navajas, R.; Ciordia, S.; Rodriguez-Frandsen, A.; Nieto, A. hCLE/C14orf166 associates with DDX1-HSPC117-FAM98B in a novel transcription-dependent shuttling RNA-transporting complex. PLoS ONE 2014, 9, e90957. [Google Scholar] [CrossRef] [PubMed]
- Cross, B.C.; Bond, P.J.; Sadowski, P.G.; Jha, B.K.; Zak, J.; Goodman, J.M.; Silverman, R.H.; Neubert, T.A.; Baxendale, I.R.; Ron, D.; et al. The molecular basis for selective inhibition of unconventional mRNA splicing by an IRE1-binding small molecule. Proc. Natl. Acad. Sci. USA 2012, 109, E869–E878. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sengupta, S.; Sharma, C.G.; Jordan, V.C. Estrogen regulation of X-box binding protein-1 and its role in estrogen induced growth of breast and endometrial cancer cells. Horm. Mol. Biol. Clin. Investig. 2010, 2, 235–243. [Google Scholar] [CrossRef]
- Winklhofer, K.F.; Tatzelt, J.; Haass, C. The two faces of protein misfolding: Gain- and loss-of-function in neurodegenerative diseases. EMBO J. 2008, 27, 336–349. [Google Scholar] [CrossRef] [PubMed]
- David, D.C.; Ollikainen, N.; Trinidad, J.C.; Cary, M.P.; Burlingame, A.L.; Kenyon, C. Widespread protein aggregation as an inherent part of aging in C. elegans. PLoS Biol. 2010, 8, e1000450. [Google Scholar] [CrossRef]
- Gozal, Y.M.; Duong, D.M.; Gearing, M.; Cheng, D.; Hanfelt, J.J.; Funderburk, C.; Peng, J.; Lah, J.J.; Levey, A.I. Proteomics analysis reveals novel components in the detergent-insoluble subproteome in Alzheimer’s disease. J. Proteome Res. 2009, 8, 5069–5079. [Google Scholar] [CrossRef] [Green Version]
- Diner, I.; Nguyen, T.; Seyfried, N.T. Enrichment of Detergent-insoluble Protein Aggregates from Human Postmortem Brain. J. Vis. Exp. 2017, 128, 55835. [Google Scholar] [CrossRef]
- Savitski, M.M.; Zinn, N.; Faelth-Savitski, M.; Poeckel, D.; Gade, S.; Becher, I.; Muelbaier, M.; Wagner, A.J.; Strohmer, K.; Werner, T.; et al. Multiplexed Proteome Dynamics Profiling Reveals Mechanisms Controlling Protein Homeostasis. Cell 2018, 173, 260–274.e25. [Google Scholar] [CrossRef] [Green Version]
- Raffo, D.; Berardi, D.E.; Pontiggia, O.; Todaro, L.; de Kier Joffé, E.B.; Simian, M. Tamoxifen selects for breast cancer cells with mammosphere forming capacity and increased growth rate. Breast Cancer Res. Treat. 2013, 142, 537–548. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, C.S.; Farnie, G.; Howell, S.J.; Clarke, R.B. Are stem-like cells responsible for resistance to therapy in breast cancer? Breast Dis. 2008, 29, 83–89. [Google Scholar] [CrossRef]
- Gong, C.; Bauvy, C.; Tonelli, G.; Yue, W.; Deloménie, C.; Nicolas, V.; Zhu, Y.; Domergue, V.; Marin-Esteban, V.; Tharinger, H.; et al. Beclin 1 and autophagy are required for the tumorigenicity of breast cancer stem-like/progenitor cells. Oncogene 2013, 32, 2261–2272. [Google Scholar] [CrossRef]
- Chaterjee, M.; van Golen, K.L. Breast cancer stem cells survive periods of farnesyl-transferase inhibitor-induced dormancy by undergoing autophagy. Bone Marrow Res. 2011, 2011, 362938. [Google Scholar] [CrossRef] [Green Version]
- Maycotte, P.; Jones, K.L.; Goodall, M.L.; Thorburn, J.; Thorburn, A. Autophagy Supports Breast Cancer Stem Cell Maintenance by Regulating IL6 Secretion. Mol. Cancer Res. 2015, 13, 651–658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolf, J.; Dewi, D.L.; Fredebohm, J.; Müller-Decker, K.; Flechtenmacher, C.; Hoheisel, J.D.; Boettcher, M. A mammosphere formation RNAi screen reveals that ATG4A promotes a breast cancer stem-like phenotype. Breast Cancer Res. 2013, 15, R109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nami, B.; Ghasemi-Dizgah, A.; Vaseghi, A. Overexpression of molecular chaperons GRP78 and GRP94 in CD44(hi)/CD24(lo) breast cancer stem cells. Bioimpacts 2016, 6, 105–110. [Google Scholar] [CrossRef]
- Chen, X.; Iliopoulos, D.; Zhang, Q.; Tang, Q.; Greenblatt, M.B.; Hatziapostolou, M.; Lim, E.; Tam, W.L.; Ni, M.; Chen, Y.; et al. XBP1 promotes triple-negative breast cancer by controlling the HIF1α pathway. Nature 2014, 508, 103–107. [Google Scholar] [CrossRef]
- Logue, S.E.; McGrath, E.P.; Cleary, P.; Greene, S.; Mnich, K.; Almanza, A.; Chevet, E.; Dwyer, R.M.; Oommen, A.; Legembre, P.; et al. Inhibition of IRE1 RNase activity modulates the tumor cell secretome and enhances response to chemotherapy. Nat. Commun. 2018, 9, 3267. [Google Scholar] [CrossRef] [PubMed]
- Govers, S.K.; Mortier, J.; Adam, A.; Aertsen, A. Protein aggregates encode epigenetic memory of stressful encounters in individual Escherichia coli cells. PLoS Biol. 2018, 16, e2003853. [Google Scholar] [CrossRef] [Green Version]
- Bufalino, M.R.; van der Kooy, D. The aggregation and inheritance of damaged proteins determines cell fate during mitosis. Cell Cycle 2014, 13, 1201–1207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harvey, Z.H.; Chen, Y.; Jarosz, D.F. Protein-Based Inheritance: Epigenetics beyond the Chromosome. Mol. Cell 2018, 69, 195–202. [Google Scholar] [CrossRef]
- Bekele, R.T.; Venkatraman, G.; Liu, R.Z.; Tang, X.; Mi, S.; Benesch, M.G.; Mackey, J.R.; Godbout, R.; Curtis, J.M.; McMullen, T.P.; et al. Oxidative stress contributes to the tamoxifen-induced killing of breast cancer cells: Implications for tamoxifen therapy and resistance. Sci. Rep. 2016, 6, 21164. [Google Scholar] [CrossRef] [Green Version]
- Radde, B.N.; Ivanova, M.M.; Mai, H.X.; Alizadeh-Rad, N.; Piell, K.; Van Hoose, P.; Cole, M.P.; Muluhngwi, P.; Kalbfleisch, T.S.; Rouchka, E.C.; et al. Nuclear respiratory factor-1 and bioenergetics in tamoxifen-resistant breast cancer cells. Exp. Cell Res. 2016, 347, 222–231. [Google Scholar] [CrossRef] [Green Version]
- Okumura, T.; Ikeda, K.; Ujihira, T.; Okamoto, K.; Horie-Inoue, K.; Takeda, S.; Inoue, S. Proteasome 26S subunit PSMD1 regulates breast cancer cell growth through p53 protein degradation. J. Biochem. 2018, 163, 19–29. [Google Scholar] [CrossRef]
- Whitesell, L.; Santagata, S.; Mendillo, M.L.; Lin, N.U.; Proia, D.A.; Lindquist, S. HSP90 empowers evolution of resistance to hormonal therapy in human breast cancer models. Proc. Natl. Acad. Sci. USA 2014, 111, 18297–18302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, L.; Yan, J.; Zhu, J.; Zhong, H.; Lu, Q.; Wang, Z.; Huang, C.; Ye, Q. Ligand-independent activation of estrogen receptor alpha by XBP-1. Nucleic Acids Res. 2003, 31, 5266–5274. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Uniprot IDs | Entry Name | Protein Name | Gene Names |
|---|---|---|---|
| TAM | |||
| P42765 | THIM_HUMAN | 3-ketoacyl-CoA thiolase, mitochondrial | ACAA2 |
| P22695 | QCR2_HUMAN | Cytochrome b-c1 complex subunit 2, mitochondrial | UQCRC2 |
| Q9GZR7 | DDX24_HUMAN | ATP-dependent RNA helicase DDX24 | DDX24 |
| P21926 | CD9_HUMAN | CD9 antigen | CD9 MIC3 TSPAN29 GIG2 |
| O75607 | NPM3_HUMAN | Nucleoplasmin-3 | NPM3 |
| Q96I24-1 | FUBP3_HUMAN | Far upstream element-binding protein 3 | FUBP3 FBP3 |
| Q92552-2 | RT27_HUMAN | 28S ribosomal protein S27, mitochondrial | MRPS27 KIAA0264 |
| Q14974 | IMB1_HUMAN | Importin subunit beta-1 | KPNB1 NTF97 |
| P46087-4 | NOP2_HUMAN | Probable 28S rRNA | NOP2 NOL1 NSUN1 |
| P07910-2 | HNRPC_HUMAN | Heterogeneous nuclear ribonucleoproteins C1/C2 | HNRNPC HNRPC |
| Q9P0M6 | H2AW_HUMAN | Core histone macro-H2A.2 | MACROH2A2 H2AFY2 |
| O00571 | DDX3X_HUMAN | ATP-dependent RNA helicase DDX3X | DDX3X DBX DDX3 |
| P07741-1 | APT_HUMAN | Adenine phosphoribosyltransferase | APRT |
| P31040 | SDHA_HUMAN | Succinate dehydrogenase [ubiquinone] flavoprotein subunit, mitochondrial | SDHA SDH2 SDHF |
| O95793 | STAU1_HUMAN | Double-stranded RNA-binding protein Staufen homolog 1 | STAU1 STAU |
| Q8WXF1 | PSPC1_HUMAN | Paraspeckle component 1 | PSPC1 PSP1 |
| P19338 | NUCL_HUMAN | Nucleolin | NCL |
| P30041 | PRDX6_HUMAN | Peroxiredoxin-6 | PRDX6 AOP2 KIAA0106 |
| Q5SSJ5-1 | HP1B3_HUMAN | Heterochromatin protein 1-binding protein 3 | HP1BP3 |
| P52943-2 | CRIP2_HUMAN | Cysteine-rich protein 2 | CRIP2 CRP2 |
| Q96DI7-2 | SNR40_HUMAN | U5 small nuclear ribonucleoprotein 40 kDa protein | SNRNP40 PRP8BP SFP38 |
| P06748 | NPM_HUMAN | Nucleophosmin | NPM1 NPM |
| Q15233 | NONO_HUMAN | Non-POU domain-containing octamer-binding protein | NONO NRB54 |
| P35659-1 | DEK_HUMAN | Protein DEK | DEK |
| Q9HCD5 | NCOA5_HUMAN | Nuclear receptor coactivator 5 | NCOA5 KIAA1637 |
| P62081 | RS7_HUMAN | 40S ribosomal protein S7 | RPS7 |
| Q12905 | ILF2_HUMAN | Interleukin enhancer-binding factor 2 | ILF2 NF45 PRO3063 |
| Q9BTV4 | TMM43_HUMAN | Transmembrane protein 43 | TMEM43 UNQ2564/PRO6244 |
| P09467 | F16P1_HUMAN | Fructose-1,6-bisphosphatase 1 | FBP1 FBP |
| Q15084-2 | PDIA6_HUMAN | Protein disulfide-isomerase A6 | PDIA6 ERP5 P5 TXNDC7 |
| O75367-1 | H2AY_HUMAN | Core histone macro-H2A.1 | MACROH2A1 H2AFY |
| Q02543 | RL18A_HUMAN | 60S ribosomal protein L18a | RPL18A |
| Q15427 | SF3B4_HUMAN | Splicing factor 3B subunit 4 | SF3B4 SAP49 |
| P52597 | HNRPF_HUMAN | Heterogeneous nuclear ribonucleoprotein F | HNRNPF HNRPF |
| Q96GQ7 | DDX27_HUMAN | Probable ATP-dependent RNA helicase DDX27 | DDX27 cPERP-F RHLP |
| Q14651 | PLSI_HUMAN | Plastin-1 | PLS1 |
| Q07065 | CKAP4_HUMAN | Cytoskeleton-associated protein 4 | CKAP4 |
| Q9Y3I0 | RTCB_HUMAN | RNA-splicing ligase RtcB homolog | RTCB C22orf28 HSPC117 |
| P46777 | RL5_HUMAN | 60S ribosomal protein L5 | RPL5 MSTP030 |
| Q9BQG0-2 | MBB1A_HUMAN | Myb-binding protein 1A | MYBBP1A P160 |
| P12956 | XRCC6_HUMAN | X-ray repair cross-complementing protein 6 | XRCC6 G22P1 |
| O60506 | HNRPQ_HUMAN | Heterogeneous nuclear ribonucleoprotein Q | SYNCRIP HNRPQ NSAP1 |
| Q9UBU9-1 | NXF1_HUMAN | Nuclear RNA export factor 1 | NXF1 TAP |
| Q9UHX1-1 | PUF60_HUMAN | Poly(U)-binding-splicing factor PUF60 | PUF60 FIR ROBPI SIAHBP1 |
| P13010 | XRCC5_HUMAN | X-ray repair cross-complementing protein 5 | XRCC5 G22P2 |
| P22087 | FBRL_HUMAN | rRNA 2’-O-methyltransferase fibrillarin | FBL FIB1 FLRN |
| Q9NP79 | VTA1_HUMAN | Vacuolar protein sorting-associated protein VTA1 homolog | VTA1 C6orf55 HSPC228 My012 |
| O76021 | RL1D1_HUMAN | Ribosomal L1 domain-containing protein 1 | RSL1D1 CATX11 CSIG PBK1 |
| P36578 | RL4_HUMAN | 60S ribosomal protein L4 | RPL4 RPL1 |
| P62269 | RS18_HUMAN | 40S ribosomal protein S18 | RPS18 D6S218E |
| P13804-1 | ETFA_HUMAN | Electron transfer flavoprotein subunit alpha, mitochondrial | ETFA |
| O43390-1 | HNRPR_HUMAN | Heterogeneous nuclear ribonucleoprotein R | HNRNPR HNRPR |
| P37837 | TALDO_HUMAN | Transaldolase | TALDO1 TAL TALDO TALDOR |
| P11387 | TOP1_HUMAN | DNA topoisomerase 1 | TOP1 |
| ICI | |||
| P27797 | CALR_HUMAN | Calreticulin | CALR CRTC |
| P05386 | RLA1_HUMAN | 60S acidic ribosomal protein P1 | RPLP1 RRP1 |
| Q92499 | DDX1_HUMAN | ATP-dependent RNA helicase DDX1 | DDX1 |
| Q13263 | TIF1B_HUMAN | Transcription intermediary factor 1-beta | TRIM28 KAP1 RNF96 TIF1B |
| P50395-1 | GDIB_HUMAN | Rab GDP dissociation inhibitor beta | GDI2 RABGDIB |
| P21266 | GSTM3_HUMAN | Glutathione S-transferase Mu 3 | GSTM3 GST5 |
| P62847-4 | RS24_HUMAN | 40S ribosomal protein S24 | RPS24 |
| P80404 | GABT_HUMAN | 4-aminobutyrate aminotransferase, mitochondrial | ABAT GABAT |
| P21964-1 | COMT_HUMAN | Catechol O-methyltransferase | COMT |
| Q16643-3 | DREB_HUMAN | Drebrin | DBN1 D0S117E |
| Q14258 | TRI25_HUMAN | E3 ubiquitin/ISG15 ligase TRIM25 | TRIM25 EFP RNF147 ZNF147 |
| P55060-1 | XPO2_HUMAN | Exportin-2 | CSE1L CAS XPO2 |
| Q9UKD2 | MRT4_HUMAN | mRNA turnover protein 4 homolog | MRTO4 C1orf33 MRT4 |
| P07237 | PDIA1_HUMAN | Protein disulfide-isomerase | P4HB ERBA2L PDI PDIA1 |
| P07384 | CAN1_HUMAN | Calpain-1 catalytic subunit | CAPN1 CANPL1 PIG30 |
| O94826 | TOM70_HUMAN | Mitochondrial import receptor subunit TOM70 | TOMM70 KIAA0719 TOM70 |
| O75955 | FLOT1_HUMAN | Flotillin-1 | FLOT1 |
| P62851 | RS25_HUMAN | 40S ribosomal protein S25 | RPS25 |
| P06396 | GELS_HUMAN | Gelsolin | GSN |
| Q9UL46 | PSME2_HUMAN | Proteasome activator complex subunit 2 | PSME2 |
| P13489 | RINI_HUMAN | Ribonuclease inhibitor | RNH1 PRI RNH |
| P22314 | UBA1_HUMAN | Ubiquitin-like modifier-activating enzyme 1 | UBA1 A1S9T UBE1 |
| TAM or ICI | |||
| Q14257-2 | RCN2_HUMAN | Reticulocalbin-2 | RCN2 ERC55 |
| P82979 | SARNP_HUMAN | SAP domain-containing ribonucleoprotein | SARNP HCC1 HSPC316 |
| Q99873 | ANM1_HUMAN | Protein arginine N-methyltransferase 1 | PRMT1 HMT2 HRMT1L2 IR1B4 |
| Q16181 | SEPT7_HUMAN | Septin-7 | SEPTIN7 CDC10 SEPT7 |
| P41250 | GARS_HUMAN | Glycine-tRNA ligase | GARS1 GARS |
| Q99832 | TCPH_HUMAN | T-complex protein 1 subunit beta | CCT7 CCTH NIP7-1 |
| P17480-1 | UBF1_HUMAN | Nucleolar transcription factor 1 | UBTF UBF UBF1 |
| P55735-1 | SEC13_HUMAN | Protein SEC13 homolog | SEC13 D3S1231E SEC13A |
| Q96QR8 | PURB_HUMAN | Transcriptional activator protein Pur-beta | PURB |
| P25787 | PSA2_HUMAN | Proteasome subunit alpha type-2 | PSMA2 HC3 PSC3 |
| Q99497 | PARK7_HUMAN | Parkinson disease protein 7 | PARK7 |
| Q08945 | SSRP1_HUMAN | FACT complex subunit SSRP1 | SSRP1 FACT80 |
| P27824-2 | CALX_HUMAN | Calnexin | CANX |
| P82650 | RT22_HUMAN | 28S ribosomal protein S22, mitochondrial | MRPS22 C3orf5 RPMS22 |
| P12532 | KCRU_HUMAN | Creatine kinase U-type, mitochondrial | CKMT1A CKMT; CKMT1B |
| P31939 | PUR9_HUMAN | Bifunctional purine biosynthesis protein ATIC | ATIC PURH OK/SW-cl.86 |
| Q6PJT7-1 | ZC3HE_HUMAN | Zinc finger CCCH domain-containing protein 14 | ZC3H14 |
| Q9BVI4 | NOC4L_HUMAN | Nucleolar complex protein 4 homolog | NOC4L |
| Q8WXX5 | DNJC9_HUMAN | DnaJ homolog subfamily C member 9 | DNAJC9 |
| Q9NXF1 | TEX10_HUMAN | Testis-expressed protein 10 | TEX10 L18 Nbla10363 |
| O60762 | DPM1_HUMAN | Dolichol-phosphate mannosyltransferase subunit 1 | DPM1 |
| Q16630-2 | CPSF6_HUMAN | Cleavage and polyadenylation specificity factor subunit 6 | CPSF6 CFIM68 |
| Q86UP2-1 | KTN1_HUMAN | Kinectin | KTN1 CG1 KIAA0004 |
| P49591 | SYSC_HUMAN | Serine-tRNA ligase, cytoplasmic | SARS1 SARS SERS |
| Q9UN86 | G3BP2_HUMAN | Ras GTPase-activating protein-binding protein 2 | G3BP2 KIAA0660 |
| Q96T37-1 | RBM15_HUMAN | RNA-binding protein 15 | RBM15 OTT OTT1 |
| Q92530 | PSMF1_HUMAN | Proteasome inhibitor PI31 subunit | PSMF1 |
| P23381 | SYWC_HUMAN | Tryptophan--tRNA ligase, cytoplasmic | WARS1 IFI53 WARS WRS |
| Q8NE71-1 | ABCF1_HUMAN | ATP-binding cassette sub-family F member 1 | ABCF1 ABC50 |
| P61353 | RL27_HUMAN | 60S ribosomal protein L27 | RPL27 |
| P55084 | ECHB_HUMAN | Trifunctional enzyme subunit beta, mitochondrial | HADHB MSTP029 |
| P09622 | DLDH_HUMAN | Dihydrolipoyl dehydrogenase, mitochondrial | DLD GCSL LAD PHE3 |
| Q53GS9 | SNUT2_HUMAN | U4/U6.U5 tri-snRNP-associated protein 2 | USP39 CGI-21 HSPC332 |
| P30101 | PDIA3_HUMAN | Protein disulfide-isomerase A3 | PDIA3 ERP57 ERP60 GRP58 |
| O14579-1 | COPE_HUMAN | Coatomer subunit epsilon | COPE |
| Q9UHB9 | SRP68_HUMAN | Signal recognition particle subunit SRP68 | SRP68 |
| Q96EP5 | DAZP1_HUMAN | DAZ-associated protein 1 | DAZAP1 |
| P17987 | TCPA_HUMAN | T-complex protein 1 subunit alpha | TCP1 CCT1 CCTA |
| MCF-7 | TAM | ICI | ||
|---|---|---|---|---|
| Term | # Proteins | p Value | # Proteins | p Value |
| hsa03010:Ribosome | 25 | 3.89 × 10−19 | 6 | 0.011 |
| hsa03040:Spliceosome | 23 | 5.88 × 10−17 | 11 | 6.71 × 10−7 |
| hsa03015:mRNA surveillance pathway | 6 | 0.015 | ||
| hsa03050:Proteasome | 4 | 0.033 | 6 | 7.03 × 10−5 |
| hsa03008:Ribosome biogenesis in eukaryotes | 5 | 0.050 | ||
| hsa01130:Biosynthesis of antibiotics | 8 | 0.053 | ||
| hsa03013:RNA transport | 7 | 0.057 | ||
| hsa00020:Citrate cycle (TCA cycle) | 3 | 0.083 | ||
| hsa00280:Valine, leucine and isoleucine degradation | 4 | 0.011 | ||
| hsa01130:Biosynthesis of antibiotics | 7 | 0.019 | ||
| hsa04141:Protein processing in endoplasmic reticulum | 6 | 0.027 | ||
| hsa04612:Antigen processing and presentation | 4 | 0.040 | ||
| MCF-7R | TAM | ICI | ||
| Term | # Proteins | p Value | # Proteins | p Value |
| hsa03010:Ribosome | 39 | 2.12 × 10−39 | 17 | 1.17 × 10−10 |
| hsa03040:Spliceosome | 22 | 3.91 × 10−16 | 29 | 2.10 × 10−25 |
| hsa03008:Ribosome biogenesis in eukaryotes | 5 | 0.045 | ||
| hsa05168:Herpes simplex infection | 7 | 0.063 | 8 | 0.020 |
| hsa05412:Arrhythmogenic right ventricular cardiomyopathy (ARVC) | 4 | 0.084 | ||
| hsa05016:Huntington’s disease | 10 | 0.002 | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Direito, I.; Monteiro, L.; Melo, T.; Figueira, D.; Lobo, J.; Enes, V.; Moura, G.; Henrique, R.; Santos, M.A.S.; Jerónimo, C.; et al. Protein Aggregation Patterns Inform about Breast Cancer Response to Antiestrogens and Reveal the RNA Ligase RTCB as Mediator of Acquired Tamoxifen Resistance. Cancers 2021, 13, 3195. https://doi.org/10.3390/cancers13133195
Direito I, Monteiro L, Melo T, Figueira D, Lobo J, Enes V, Moura G, Henrique R, Santos MAS, Jerónimo C, et al. Protein Aggregation Patterns Inform about Breast Cancer Response to Antiestrogens and Reveal the RNA Ligase RTCB as Mediator of Acquired Tamoxifen Resistance. Cancers. 2021; 13(13):3195. https://doi.org/10.3390/cancers13133195
Chicago/Turabian StyleDireito, Inês, Liliana Monteiro, Tânia Melo, Daniela Figueira, João Lobo, Vera Enes, Gabriela Moura, Rui Henrique, Manuel A. S. Santos, Carmen Jerónimo, and et al. 2021. "Protein Aggregation Patterns Inform about Breast Cancer Response to Antiestrogens and Reveal the RNA Ligase RTCB as Mediator of Acquired Tamoxifen Resistance" Cancers 13, no. 13: 3195. https://doi.org/10.3390/cancers13133195