
CARTmath—A Mathematical Model of CAR-T Immunotherapy in Preclinical Studies of Hematological Cancers

 , , , , and
, , , , and
Abstract
:Simple Summary
Abstract
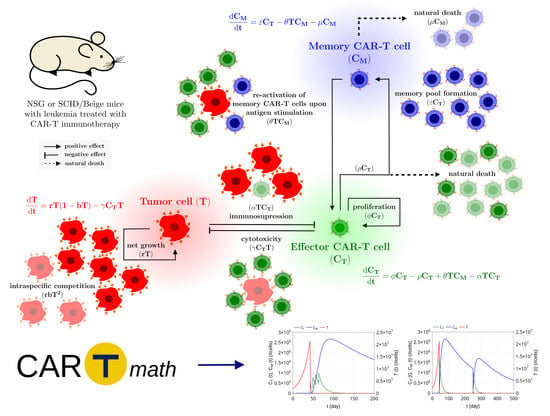
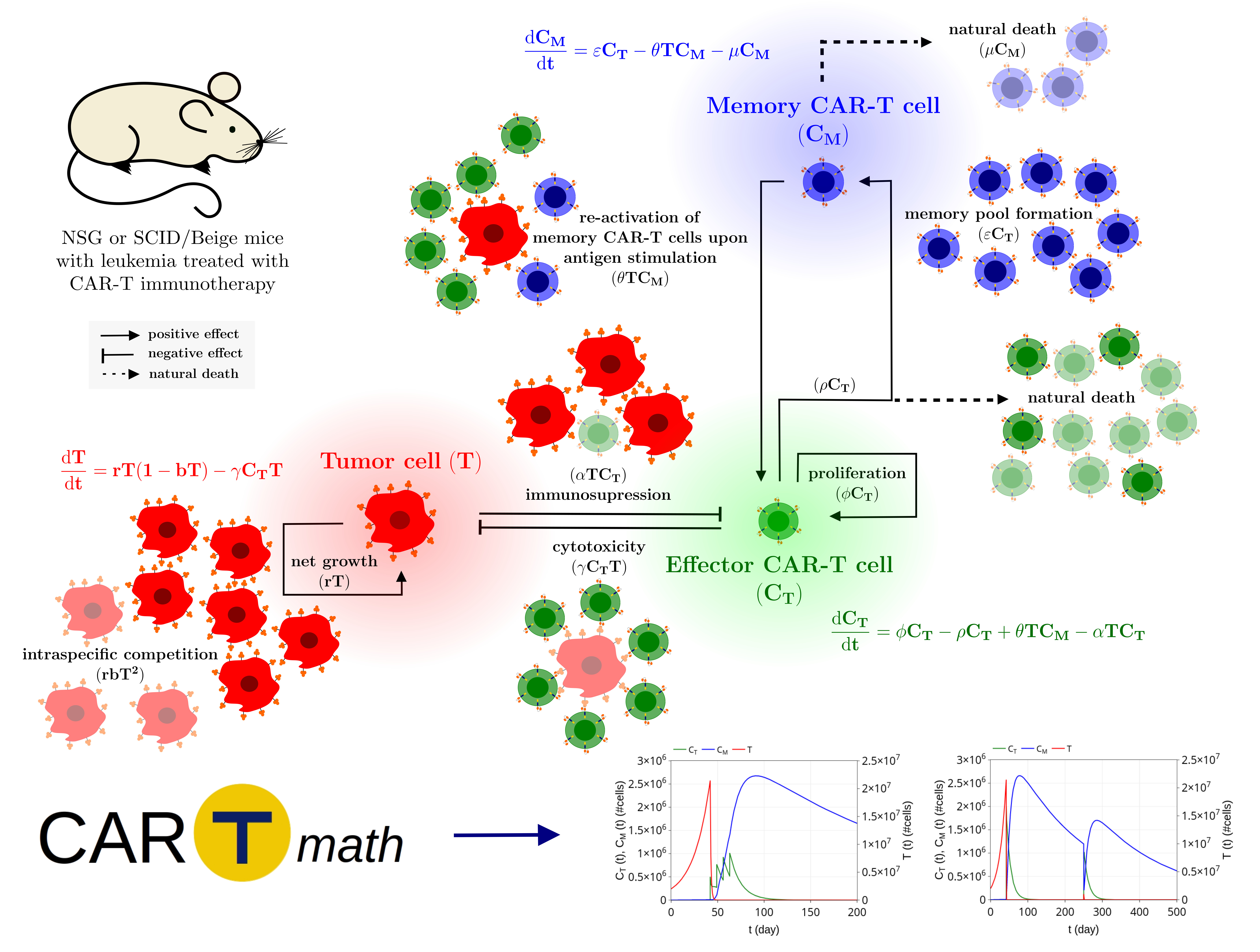
1. Introduction
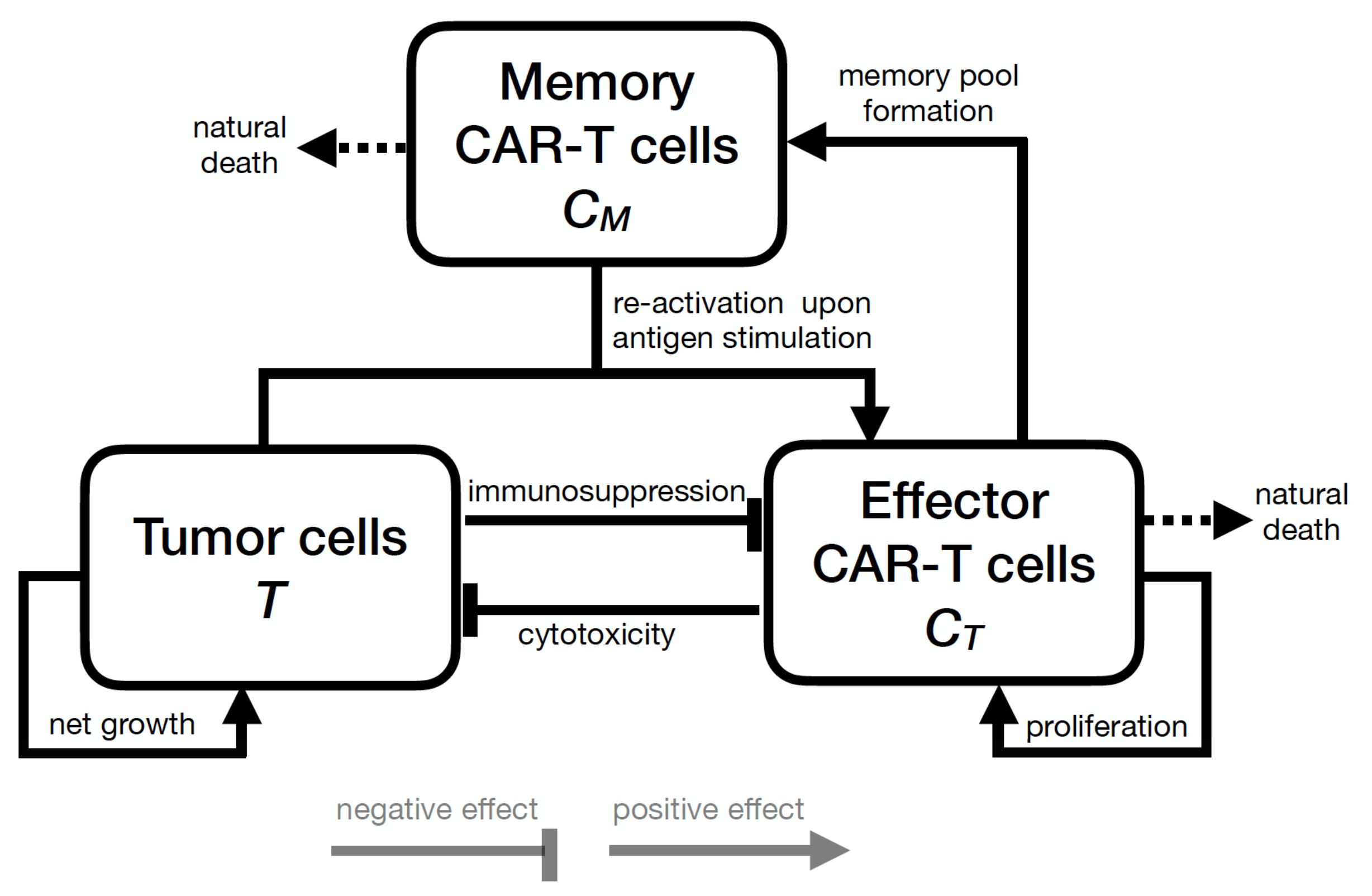
2. Mathematical Model
2.1. Model Development
2.2. In Vitro and In Vivo Data and Model Inference
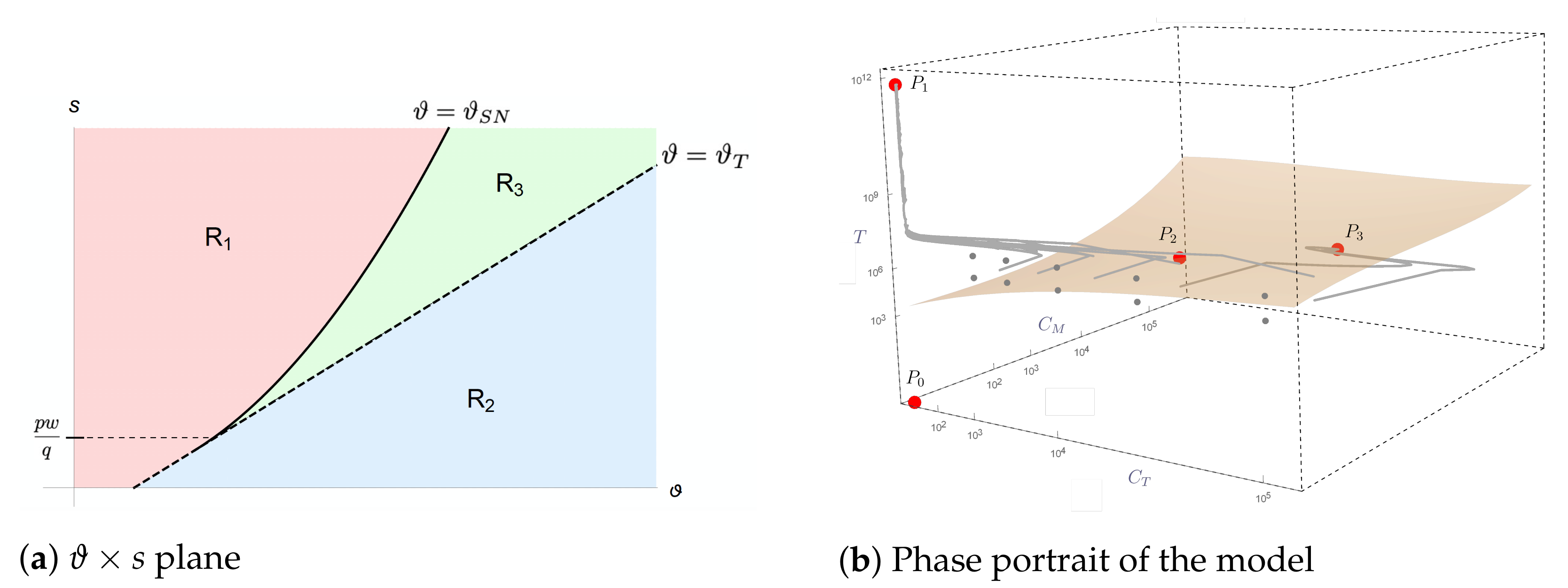
2.3. Mathematical Analysis of Model Dynamics
- (I)
- In region , the nonnegative equilibria are (which is a saddle point) and (which is locally asymptotically stable);
- (II)
- In region , there are three nonnegative equilibria, which are (saddle point), (saddle point), and (locally asymptotically stable);
- (III)
- In region , there are four nonnegative equilibria, which are (saddle point), (locally asymptotically stable), (saddle point), and (locally asymptotically stable).
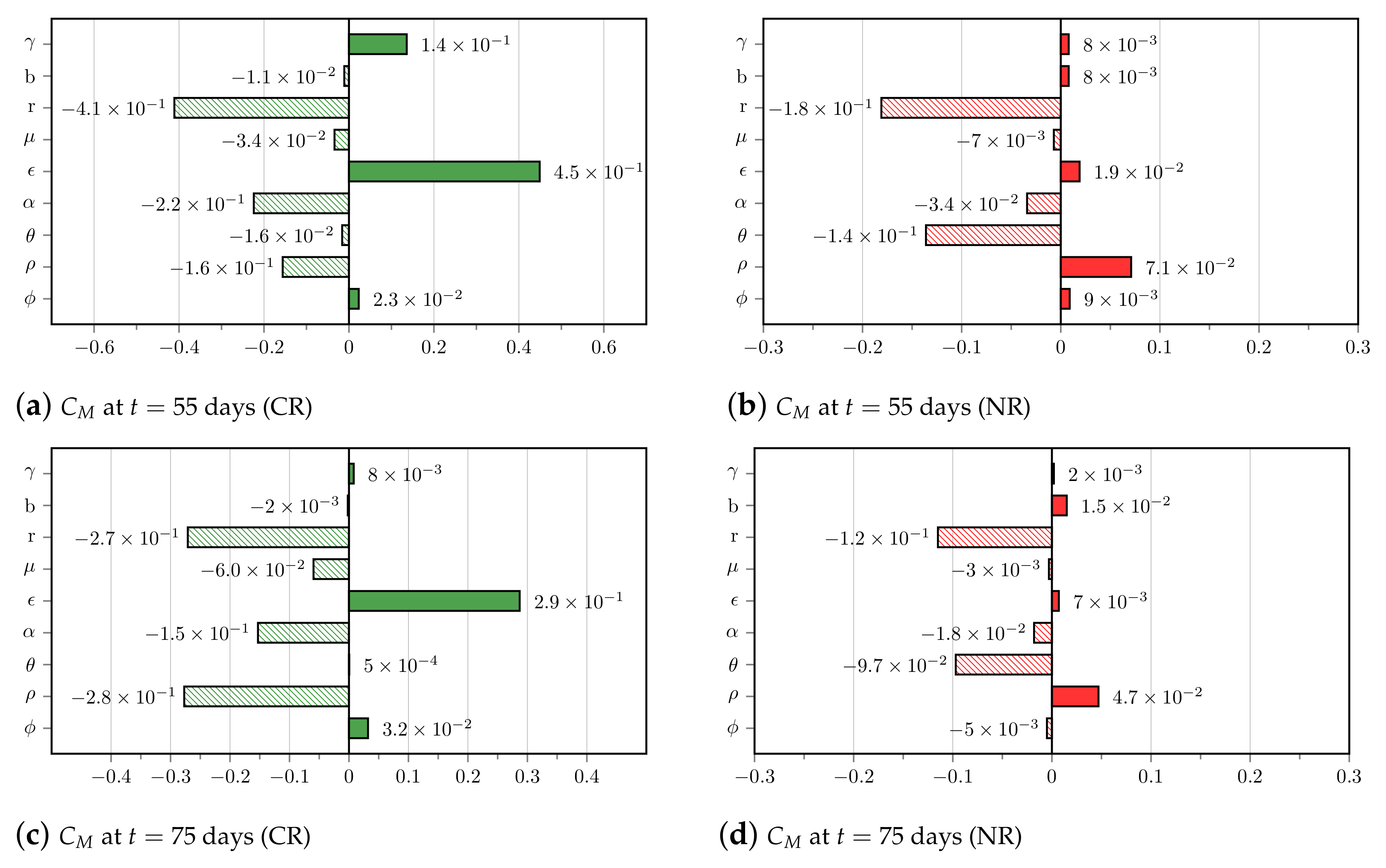
2.4. In Silico Population and Sensitivity Analysis
2.5. Model Settings and Numerical Solution
3. Results: In Silico Experiments
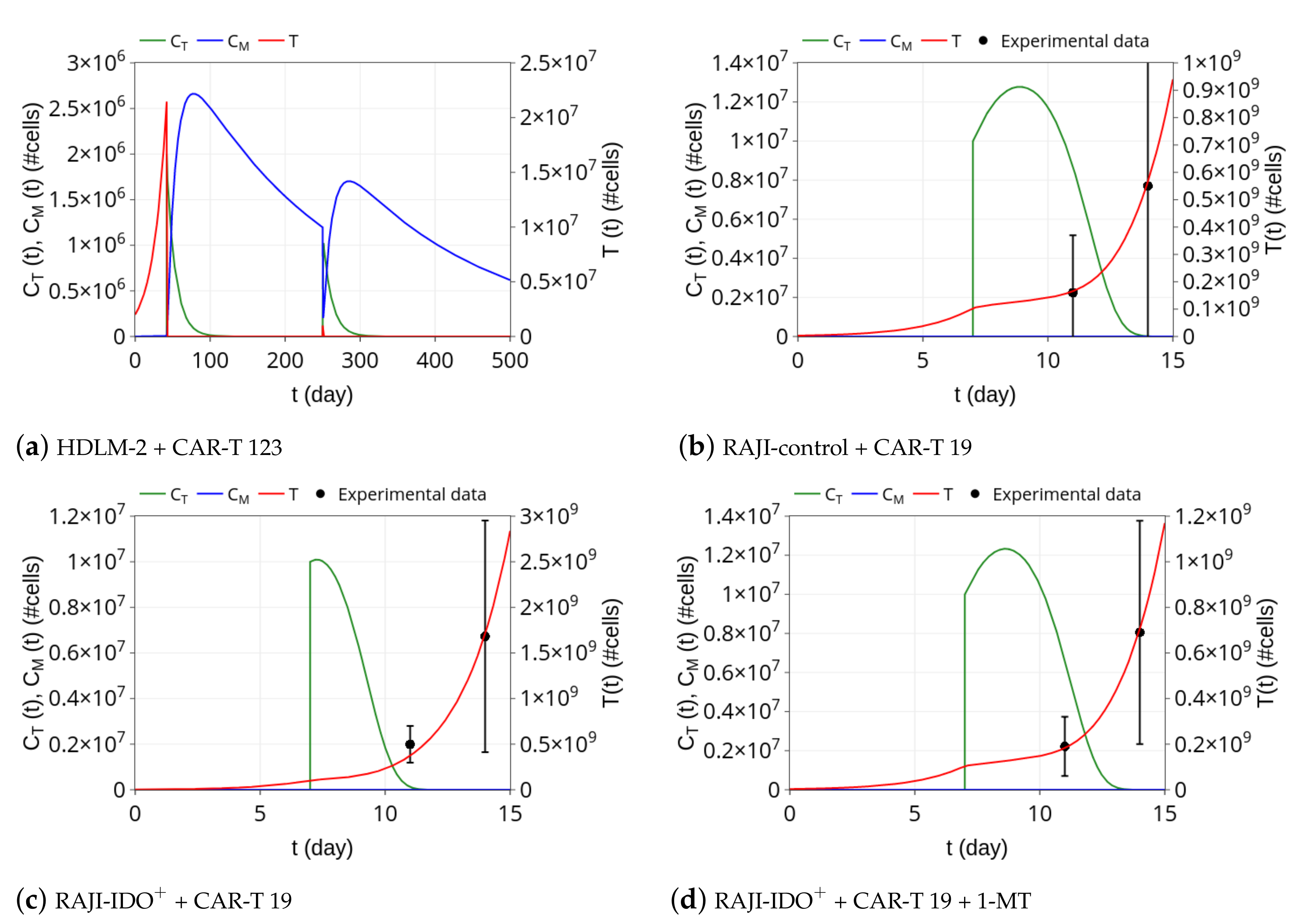
3.1. CAR-T 123 Therapy Eliminates HDLM-2 Tumors, Providing Long-Term Protection, While Immunotherapy with CAR-T 19 on RAJI Tumor Slows Down Its Growth
3.2. Insights on Immune Checkpoint Inhibitors
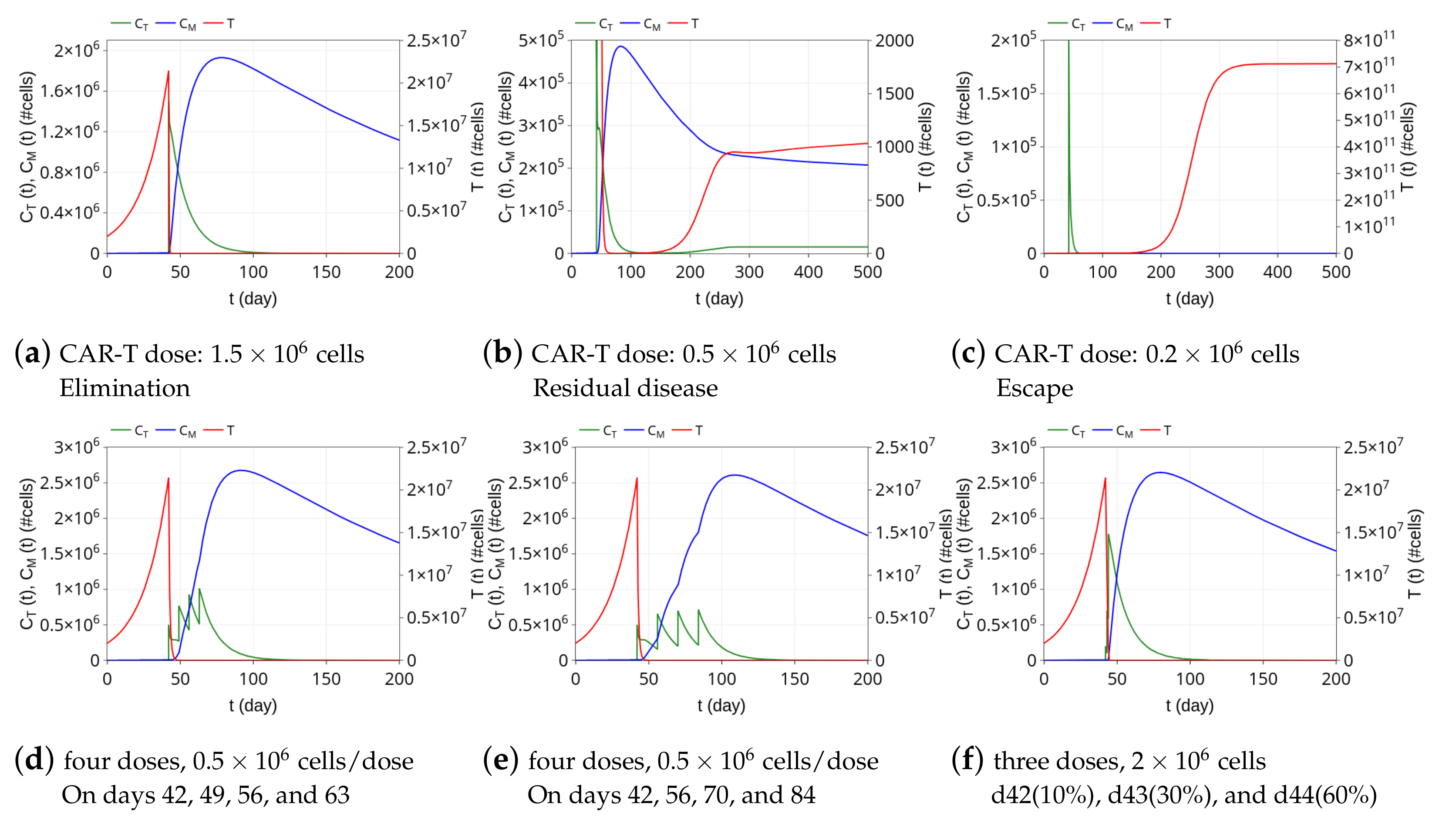
3.3. Insights on Dosing Strategies: Single and Fractionated Doses
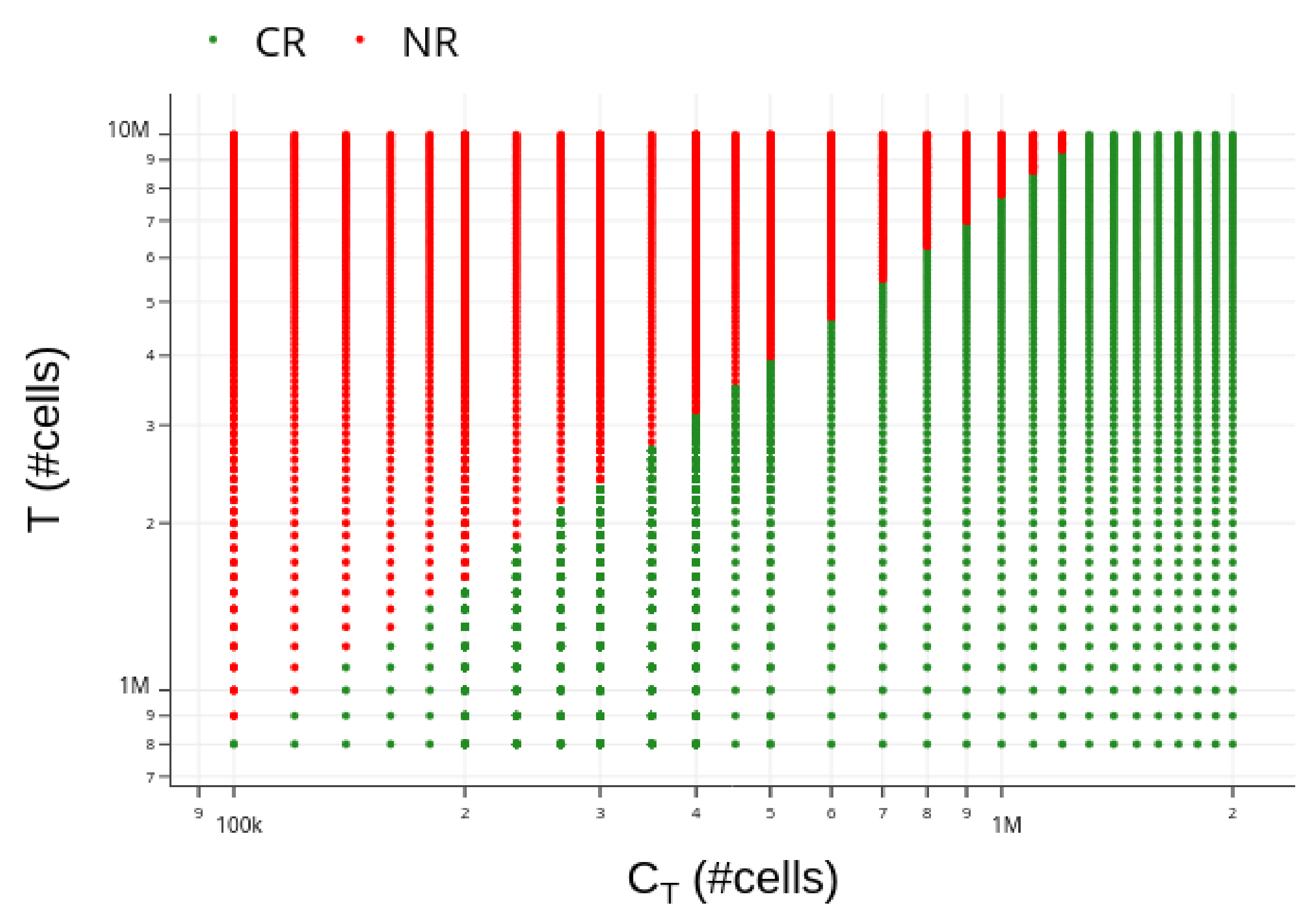
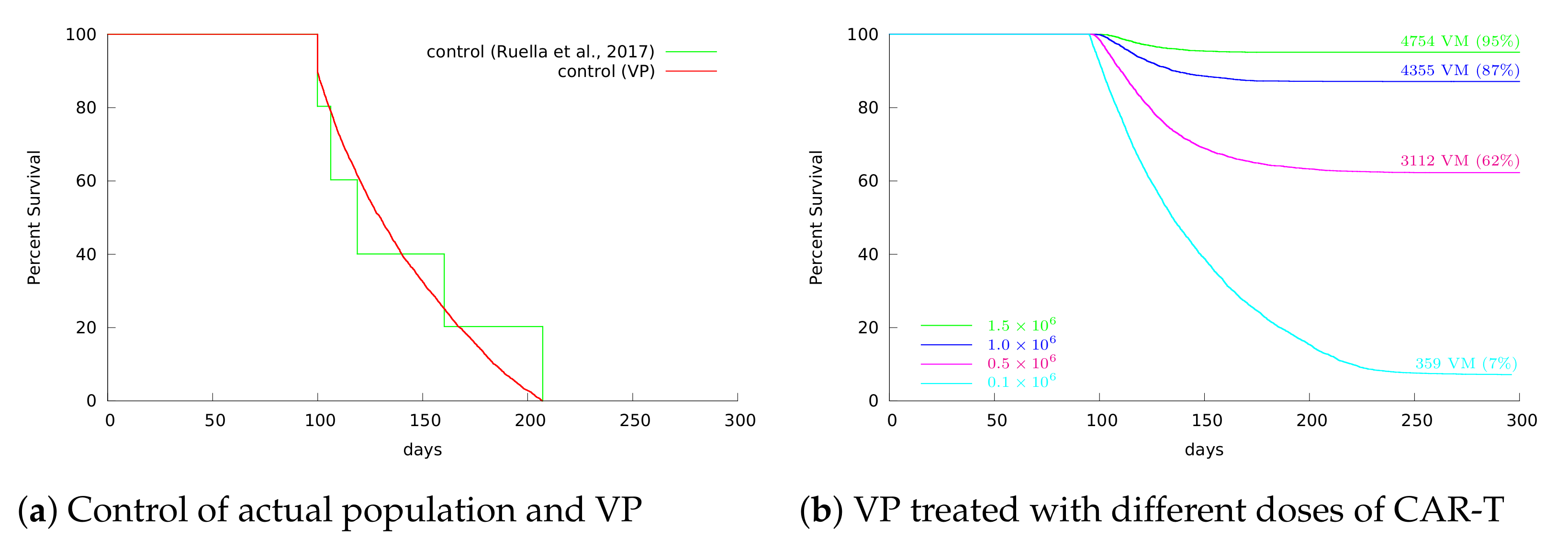
3.4. Insights on Parameter Uncertainties Impacting Treatment Outcome
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| 1-MT | 1-methyl-tryptophan |
| ALL | Acute lymphoblastic leukemia |
| AML | Acute myeloid leukemia |
| BLI | Bioluminescence imaging |
| CAR | Chimeric antigen receptor |
| CR | Complete response |
| CRS | Cytokine release syndrome |
| FDA | Food and Drug Administration |
| HL | Hodgkin lymphoma |
| ICB | Immune checkpoint blockade |
| IDO | Indoleamine 2,3-dioxygenase |
| LAG3 | Lymphocyte-activation gene 3 |
| MLE | Most a posteriori estimates |
| NR | No response |
| ODE | Ordinary differential equation |
| PD1 | Programmed cell death protein 1 |
| PD-L1 | Programmed death-ligand 1 |
| VISTA | V-domain Ig suppressor of T cell activation |
| VM | Virtual mice |
| VP | Virtual population |
References
- Maude, S.L.; Teachey, D.T.; Rheingold, S.R.; Shaw, P.A.; Aplenc, R.; Barrett, D.M.; Barker, C.S.; Callahan, C.; Frey, N.V.; Nazimuddin, F.; et al. Sustained remissions with CD19-specific chimeric antigen receptor (CAR)-modified T cells in children with relapsed/refractory ALL. J. Clin. Oncol. 2016, 34, 3011. [Google Scholar] [CrossRef]
- Sadelain, M.; Rivière, I.; Riddell, S. Therapeutic T cell engineering. Nature 2017, 545, 423–431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guedan, S.; Ruella, M.; June, C.H. Emerging Cellular Therapies for Cancer. Ann. Rev. Immunol. 2019, 37, 145–171. [Google Scholar] [CrossRef] [PubMed]
- Maude, S.L.; Teachey, D.T.; Porter, D.L.; Grupp, S.A. CD19-targeted chimeric antigen receptor T cell therapy for acute lymphoblastic leukemia. Blood 2015, 4017–4023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cummins, K.D.; Gill, S. Anti-CD123 chimeric antigen receptor T-cells (CART): An evolving treatment strategy for hematological malignancies, and a potential ace-in-the-hole against antigen-negative relapse. Leuk. Lymphoma 2018, 59, 1539–1553. [Google Scholar] [CrossRef]
- Barros, L.R.C. In Search for an Ideal CAR-T Cell Antigen Target. Crit. Rev. Immunol. 2021, 41. [Google Scholar] [CrossRef] [PubMed]
- Ruella, M.; Klichinsky, M.; Kenderian, S.S.; Shestova, O.; Ziober, A.; Kraft, D.O.; Feldman, M.; Wasik, M.A.; June, C.H.; Gill, S. Overcoming the Immunosuppressive Tumor Microenvironment of Hodgkin Lymphoma Using Chimeric Antigen Receptor T Cells. Cancer Discov. 2017, 10, 1154–1167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crauste, F.; Mafille, J.; Boucinha, L.; Djebali, S.; Gandrillon, O.; Marvel, J.; Arpin, C. Identification of Nascent Memory CD8 T Cells and Modeling of Their Ontogeny. Cell Syst. 2017, 4, 306–317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, R.; Wang, M.; Liu, Q.; Wu, J.; Huang, W.; Li, X.; Du, B.; Xu, Q.; Duan, J.; Jiao, S.; et al. Sequential treatment with aT19 cells generates memory CAR-T cells and prolongs the lifespan of Raji-B-NDG mice. Cancer Lett. 2020, 469, 162–172. [Google Scholar] [CrossRef] [PubMed]
- Kimmel, G.J.; Locke, F.L.; Altrock, P.M. Response to CAR T cell therapy can be explained by ecological cell dynamics and stochastic extinction events. bioRxiv 2020. [Google Scholar] [CrossRef]
- Ninomiya, S.; Narala, N.; Huye, L.; Yagyu, S.; Savoldo, B.; Dotti, G.; Heslop, H.E.; Brenner, M.K.; Rooney, C.M.; Ramos, C.A. Tumor indoleamine 2, 3-dioxygenase (IDO) inhibits CD19-CAR T cells and is downregulated by lymphodepleting drugs. Blood 2015, 3905–3916. [Google Scholar] [CrossRef] [Green Version]
- Jacoby, E. Relapse and resistance to CAR-T cells and blinatumomab in hematologic malignancies. Clin.l Hematol. Int. 2019, 1, 79–84. [Google Scholar] [CrossRef] [Green Version]
- Shah, N.N.; Fry, T.J. Mechanisms of resistance to CAR T cell therapy. Nat. Rev. Clin. Oncol. 2019, 16, 372–385. [Google Scholar] [CrossRef]
- Antia, R.; Ganusov, V.V.; Ahmed, R. The role of models in understanding CD8+ T-cell memory. Nat. Rev. Immunol. 2005, 5, 101–111. [Google Scholar] [CrossRef]
- Konstorum, A.; Vella, A.T.; Adler, A.J.; Laubenbacher, R.C. Addressing current challenges in cancer immunotherapy with mathematical and computational modelling. J. R. Soc. Interface 2017, 14, 20170150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kronik, N.; Kogan, Y.; Vainstein, V.; Agur, Z. Improving alloreactive CTL immunotherapy for malignant gliomas using a simulation model of their interactive dynamics. Cancer Immunol. Immunother. 2008, 57, 425–439. [Google Scholar] [CrossRef] [PubMed]
- Frascoli, F.; Kim, P.S.; Hughes, B.D.; Landman, K.A. A dynamical model of tumour immunotherapy. Math. Biosci. 2014, 253, 50–62. [Google Scholar] [CrossRef]
- Qomlaqi, M.; Bahrami, F.; Ajami, M.; Hajati, J. An extended mathematical model of tumor growth and its interaction with the immune system, to be used for developing an optimized immunotherapy treatment protocol. Math. Biosci. 2017, 292, 1–9. [Google Scholar] [CrossRef]
- Arabameri, A.; Asemani, D.; Hadjati, J. A structural methodology for modeling immune-tumor interactions including pro- and anti-tumor factors for clinical applications. Math. Biosci. 2018, 304, 48–61. [Google Scholar] [CrossRef]
- Jansen, J.E.; Gaffney, E.A.; Wagg, J.; Coles, M.C. Combining Mathematical Models With Experimentation to Drive Novel Mechanistic Insights Into Macrophage Function. Front. Immunol. 2019, 10, 1283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zarnitsyna, V.I.; Handel, A.; McMaster, S.R.; Hayward, S.L.; Kohlmeier, J.E.; Antia, R. Mathematical Model Reveals the Role of Memory CD8 T Cell Populations in Recall Responses to Influenza. Front. Immunol. 2016, 7, 165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leon, K.; Garcia-Martinez, K.; Carmenate, T. Mathematical Models of the Impact of IL2 Modulation Therapies on T Cell Dynamics. Front. Immunol. 2013, 4, 439. [Google Scholar] [CrossRef] [Green Version]
- Sahoo, P.; Yang, X.; Abler, D.; Maestrini, D.; Adhikarla, V.; Frankhouser, D.; Cho, H.; Machuca, V.; Wang, D.; Barish, M.; et al. Mathematical deconvolution of CAR T-cell proliferation and exhaustion from real-time killing assay data. J. Royal Soc. Interface 2020, 17, 20190734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- León-Triana, O.; Sabir, S.; Calvo, G.F.; Belmonte-Beitia, J.; Chulián, S.; Álvaro, M.-R.; Rosa, M.; Pérez-Martínez, A.; Ramirez-Orellana, M.; Pérez-García, V.M. CAR T cell therapy in B-cell acute lymphoblastic leukaemia: Insights from mathematical models. Commun. Nonlinear Sci. Numer. Simul. 2021, 94, 105570. [Google Scholar] [CrossRef]
- Stein, A.M.; Grupp, S.A.; Levine, J.E.; Laetsch, T.W.; Pulsipher, M.A.; Boyer, M.W.; August, K.J.; Levine, B.L.; Tomassian, L.; Shah, S.; et al. Tisagenlecleucel Model-Based Cellular Kinetic Analysis of Chimeric Antigen Receptor-T Cells. CPT Pharmacomet. Syst. Pharmacol. 2019, 8, 285–295. [Google Scholar] [CrossRef] [Green Version]
- Toor, A.A.; Chesney, A.; Zweit, J.; Reed, J.; Hashmi, S.K. A dynamical systems perspective on chimeric antigen receptor T-cell dosing. Bone Marrow Transplant. 2019, 54, 485–489. [Google Scholar] [CrossRef]
- Ganusov, V.V. Discriminating between Different Pathways of Memory CD8+ T Cell Differentiation. J. Immunol. 2007, 179, 5006–5013. [Google Scholar] [CrossRef] [PubMed]
- Kalos, M.; Levine, B.L.; Porter, D.L.; Katz, S.; Grupp, S.A.; Bagg, A.; June, C.H. T Cells with Chimeric Antigen Receptors Have Potent Antitumor Effects and Can Establish Memory in Patients with Advanced Leukemia. Sci. Transl. Med. 2011, 3, 95ra73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sabatino, M.; Hu, J.; Sommariva, M.; Gautam, S.; Fellowes, V.; Hocker, J.D.; Dougherty, S.; Qin, H.; Klebanoff, C.A.; Fry, T.J.; et al. Generation of clinical-grade CD19-specific CAR-modified CD8+ memory stem cells for the treatment of human B-cell malignancies. Blood 2016, 128, 519–528. [Google Scholar] [CrossRef]
- Tanchot, C.; Lemonnier, F.A.; Pérarnau, B.; Freitas, A.A.; Rocha, B. Differential requirements for survival and proliferation of CD8 naive or memory T cells. Science 1997, 276, 2057–2062. [Google Scholar] [CrossRef]
- Iwai, Y.; Ishida, M.; Tanaka, Y.; Okazaki, T.; Honjo, T.; Minato, N. Involvement of PD-L1 on tumor cells in the escape from host immune system and tumor immunotherapy by PD-L1 blockade. Proc. Natl. Acad. Sci. 2002, 99, 12293–12297. [Google Scholar] [CrossRef] [Green Version]
- Alsaab, H.O.; Sau, S.; Alzhrani, R.; Tatiparti, K.; Bhise, K.; Kashaw, S.K.; Iyer, A.K. PD-1 and PD-L1 Checkpoint Signaling Inhibition for Cancer Immunotherapy: Mechanism, Combinations, and Clinical Outcome. Front. Pharmacol. 2017, 8, 561. [Google Scholar] [CrossRef]
- Burugu, S.; Dancsok, A.R.; Nielsen, T.O. Emerging targets in cancer immunotherapy. Semin. Cancer Biol. 2018, 52, 39–52. [Google Scholar] [CrossRef] [PubMed]
- Bilir, C.; Sarisozen, C. Indoleamine 2,3-dioxygenase (IDO): Only an enzyme or a checkpoint controller? J. Oncol. Sci. 2017, 3, 52–56. [Google Scholar] [CrossRef]
- Théate, I.; van Baren, N.; Pilotte, L.; Moulin, P.; Larrieu, P.; Renauld, J.C.; Hervé, C.; Gutierrez-Roelens, I.; Marbaix, E.; Sempoux, C.; et al. Extensive Profiling of the Expression of the Indoleamine 2,3-Dioxygenase 1 Protein in Normal and Tumoral Human Tissues. Cancer Immunol. Res. 2015, 3, 161–172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanel, A.; Fonseca, S.G.; Rebeka Bordi, B.Y.D.; Zeidan, J.; Shi, Y.; Benne, C.; Sékaly, R.P. Cellular and molecular mechanisms of memory T-cell survival. Expert Rev. Vaccines 2009, 8, 299–312. [Google Scholar] [CrossRef]
- Enderling, H.; Chaplain, M.A. Mathematical Modeling of Tumor Growth and Treatment. Curr. Pharm. Des. 2014, 20, 4934–4940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murphy, H.; Jaafari, H.; Dobrovolny, H.M. Differences in predictions of ODE models of tumor growth: A cautionary example. BMC Cancer 2016, 16, 163. [Google Scholar] [CrossRef] [Green Version]
- Geyer, M.B.; Brentjens, R.J. Review: Current clinical applications of chimeric antigen receptor (CAR) modified T cells. Cytotherapy 2016, 18, 1393–1409. [Google Scholar] [CrossRef] [Green Version]
- Hammerl, D.; Rieder, D.; Martens, J.W.; Trajanoski, Z.; Debets, R. Adoptive T Cell Therapy: New Avenues Leading to Safe Targets and Powerful Allies. Trends Immunol. 2018, 39, 921–936. [Google Scholar] [CrossRef]
- Benmebarek, M.R.; Karches, C.H.; Cadilha, B.L.; Lesch, S.; Endres, S.; Kobold, S. Killing Mechanisms of Chimeric Antigen Receptor (CAR) T Cells. Int. J. Mol. Sci. 2019, 20, 1283. [Google Scholar] [CrossRef] [Green Version]
- Scholler, J.; Brady, T.L.; Binder-Scholl, G.; Hwang, W.T.; Plesa, G.; Hege, K.M.; Vogel, A.N.; Kalos, M.; Riley, J.L.; Deeks, S.G.; et al. Decade-long safety and function of retroviral-modified chimeric antigen receptor T cells. Sci. Translacional Med. 2012, 132, 132ra53. [Google Scholar] [CrossRef] [Green Version]
- Allen, A.B.; Gazit, Z.; Su, S.; Stevens, H.Y.; Guldberg, R.E. In vivo bioluminescent tracking of mesenchymal stem cells within large hydrogel constructs. Tissue Eng. Part C 2012, 20, 806–816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gertner-Dardenne, J. Standard 4-hours Chromium-51 (51Cr) Release Assay. Bio-protocol 2012, 2, e201. [Google Scholar] [CrossRef]
- G3Data. G3Data Graph Analyzer. Version 1.5.4.. 2011. Available online: https://github.com/pn2200/g3data (accessed on 13 July 2018).
- Allen, R.; Rieger, T.; Musante, C. Efficient Generation and Selection of Virtual Populations in Quantitative Systems Pharmacology Models. CPT Pharmacomet. Syst. Pharmacol. 2016, 5, 140–146. [Google Scholar] [CrossRef] [Green Version]
- Cassidy, T.; Craig, M. Determinant of combination GM-CSF immunotherapy and oncolytic virotherapy success identified through in silico treatment personalization. PLoS Comput. Biol. 2019, 15, e1007495. [Google Scholar] [CrossRef]
- Wang, H.; Sové, R.J.; Jafarnejad, M.; Rahmeh, S.; Jaffee, E.M.; Stearns, V.; Torres, E.T.R.; Connolly, R.M.; Popel, A.S. Conducting a Virtual Clinical Trial in HER2-Negative Breast Cancer Using a Quantitative Systems Pharmacology Model With an Epigenetic Modulator and Immune Checkpoint Inhibitors. Front. Bioeng. Biotechnol. 2020, 8, 141. [Google Scholar] [CrossRef] [PubMed]
- Faires, J.D.; Burden, R.L. Numerical Methods; Cengage Learning: Belmont, CA, USA, 2003. [Google Scholar]
- Paixão, E.A.; Naozuka, G.T.; Valli, A.M.P.; Barros, L.R.C.; Almeida, R.C. CARTmath. 2020. Available online: https://github.com/tmglncc/CARTmath (accessed on 4 May 2021). [CrossRef]
- Poorebrahim, M.; Melief, J.; de Coaña, Y.P.; Wickström, S.L.; Cid-Arregui, A.; Kiessling, R. Counteracting CAR T cell dysfunction. Oncogene 2021, 40, 421–435. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Kaur, G.; Sankin, A.I.; Chen, F.; Guan, F.; Zang, X. Immune checkpoint blockade and CAR-T cell therapy in hematologic malignancies. J. Hematol. Oncol. 2019, 12, 59. [Google Scholar] [CrossRef] [PubMed]
- Dunn, G.P.; Old, L.J.; Schreiber, R.D. Cancer Immunoediting: From immunosurveillance to tumor escape. Ann. Rev. Immunol. 2004, 22, 329–360. [Google Scholar] [CrossRef] [PubMed]
- Frey, N.V.; Shaw, P.A.; Hexner, E.O.; Gill, S.; Marcucci, K.; Luger, S.M.; Mangan, J.K.; Grupp, S.A.; Maude, S.L.; Ericson, S.; et al. Optimizing chimeric antigen receptor (CAR) T cell therapy for adult patients with relapsed or refractory (r/r) acute lymphoblastic leukemia (ALL). J. Clin. Oncol. 2016, 34, 7002. [Google Scholar] [CrossRef]
- Maude, S.L.; Laetsch, T.W.; Buechner, J.; Rives, S.; Boyer, M.; Bittencourt, H.; Bader, P.; Verneris, M.R.; Stefanski, H.E.; Myers, G.D.; et al. Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. N. Engl. J. Med. 2018, 378, 439–448. [Google Scholar] [CrossRef]
- Ghorashian, S.; Kramer, A.M.; Onuoha, S.; Wright, G.; Bartram, J.; Richardson, R.; Amrolia, P.J. Enhanced CAR T cell expansion and prolonged persistence in pediatric patients with ALL treated with a low-affinity CD19 CAR. Nat. Med. 2019, 25, 1408–1414. [Google Scholar] [CrossRef] [PubMed]
- de Macedo Abdo, L.; Barros, L.R.C.; Viegas, M.S.; Marques, L.V.C.; de Sousa Ferreira, P.; Chicaybam, L.; Bonamino, M.H. Development of CAR-T Cell Therapy for B-ALL Using a Point-of-Care Approach. Oncoimmunology 2020, 9. [Google Scholar] [CrossRef] [Green Version]
- Chicaybam, L.; Abdo, L.; Carneiro, M.; Peixoto, B.; Viegas, M.; de Sousa, P.; Fornazin, M.C.; Spago, M.C.; Albertoni Laranjeira, A.B.; de Campos-Lima, P.O.; et al. CAR T Cells Generated Using Sleeping Beauty Transposon Vectors and Expanded with an EBV-Transformed Lymphoblastoid Cell Line Display Antitumor Activity In Vitro and In Vivo. Human Gene Ther. 2019, 30, 511–522. [Google Scholar] [CrossRef]
- Levin, A.G.; Kronik, N.; Shiloach, T.; Waks, T.; Eshhar, Z.; Vainstein, V. Less is more: Reducing the number of administered chimeric antigen receptor T cells in a mouse model using a mathematically guided approach. Cancer Immunol. Immunother. 2020, 69. [Google Scholar] [CrossRef]
- Hanson, S.; Grimes, D.R.; Taylor-King, J.P.; Bauer, B.; Warman, P.I.; Frankenstein, Z.; Kaznatcheev, A.; Bonassar, M.J.; Cannataro, V.L.; Motawe, Z.Y.; et al. Toxicity Management in CAR T cell therapy for B-ALL: Mathematical modelling as a new avenue for improvement. bioRXiv 2016. [Google Scholar] [CrossRef] [Green Version]
- León-Triana, O.; Pérez-Martínez, A.; Ramírez-Orellana, M.; Pérez-García, V.M. Dual-Target CAR-Ts with On- and Off-Tumour Activity May Override Immune Suppression in Solid Cancers: A Mathematical Proof of Concept. Cancers 2021, 13, 703. [Google Scholar] [CrossRef]
- Majzner, R.G.; Mackall, C.L. Tumor Antigen Escape from CAR T-cell Therapy. Cancer Discov. 2018, 8, 1219–1226. [Google Scholar] [CrossRef] [Green Version]
- Xu, X.; Sun, Q.; Liang, X.; Chen, Z.; Zhang, X.; Zhou, X.; Li, M.; Tu, H.; Liu, Y.; Tu, S.; et al. Mechanisms of Relapse After CD19 CAR T-Cell Therapy for Acute Lymphoblastic Leukemia and Its Prevention and Treatment Strategies. Front. Immunol. 2019, 10, 2664. [Google Scholar] [CrossRef] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Unit | Meaning |
|---|---|---|
| day | Proliferation rate of effector CAR-T cells | |
| day | Reduction rate of effector CAR-T cells, encompassing the natural death of these cells and their differentiation into memory CAR-T cells | |
| (cell · day) | Conversion coefficient of memory CAR-T cells into effector CAR-T cells due to interaction with tumor cells | |
| (cell · day) | Inhibition/expansion coefficient of effector CAR-T cells due to interaction with tumor cells | |
| day | Effective conversion rate of effector CAR-T cells into memory CAR-T cells | |
| day | Death rate of memory CAR-T cells | |
| r | day | Maximum growth rate of tumor cells |
| b | cell | Inverse of the tumor carrying capacity |
| (cell · day) | Cytotoxic coefficient induced by effector CAR-T cells | |
| Restriction | Meaning | |
| Effector CAR-T cells decay to zero in the absence of tumor cells | ||
| Healthy donor CAR-T cells proliferate in vivo and differentiate into memory CAR-T cells | ||
| Parameter | HDLM-2 + CAR-T 123 | RAJI-Control + CAR-T 19 |
|---|---|---|
| day | day | |
| day | day | |
| day | day | |
| (cell · day) | (cell · day) | |
| (cell · day) | (cell · day) | |
| day | day | |
| r | day | day |
| b | cell | 0 cell |
| (cell · day) | (cell · day) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barros, L.R.C.; Paixão, E.A.; Valli, A.M.P.; Naozuka, G.T.; Fassoni, A.C.; Almeida, R.C. CARTmath—A Mathematical Model of CAR-T Immunotherapy in Preclinical Studies of Hematological Cancers. Cancers 2021, 13, 2941. https://doi.org/10.3390/cancers13122941
Barros LRC, Paixão EA, Valli AMP, Naozuka GT, Fassoni AC, Almeida RC. CARTmath—A Mathematical Model of CAR-T Immunotherapy in Preclinical Studies of Hematological Cancers. Cancers. 2021; 13(12):2941. https://doi.org/10.3390/cancers13122941
Chicago/Turabian StyleBarros, Luciana R. C., Emanuelle A. Paixão, Andrea M. P. Valli, Gustavo T. Naozuka, Artur C. Fassoni, and Regina C. Almeida. 2021. "CARTmath—A Mathematical Model of CAR-T Immunotherapy in Preclinical Studies of Hematological Cancers" Cancers 13, no. 12: 2941. https://doi.org/10.3390/cancers13122941