
Life after Cell Death—Survival and Survivorship Following Chemotherapy

Abstract
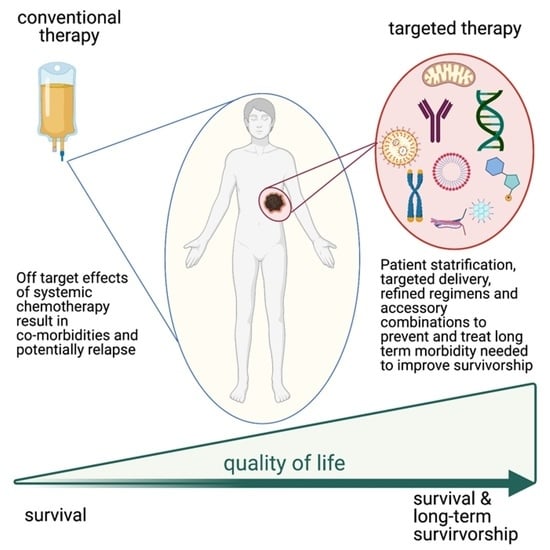
:Simple Summary
Abstract
{kind=link}
{kind=link}
{kind=link}
1. Fight to the Death
2. Relapse
3. Unintended Casualties
4. What Doesn’t Kill You…
5. Tomorrow’s Problem
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- DeVita, V.T., Jr.; Chu, E. A history of cancer chemotherapy. Cancer Res. 2008, 68, 8643–8653. [Google Scholar] [CrossRef] [Green Version]
- Von Baumgarten, L.; Illerhaus, G.; Korfel, A.; Schlegel, U.; Deckert, M.; Dreyling, M. The Diagnosis and Treatment of Primary CNS Lymphoma. Dtsch. Arztebl. Int. 2018, 115, 419–426. [Google Scholar] [CrossRef]
- Li, L.R.; Wang, L.; He, Y.Z.; Young, K.H. Current perspectives on the treatment of double hit lymphoma. Expert Rev. Hematol. 2019, 12, 507–514. [Google Scholar] [CrossRef] [PubMed]
- Schneeweiss, A.; Möbus, V.; Tesch, H.; Hanusch, C.; Denkert, C.; Lübbe, K.; Huober, J.; Klare, P.; Kümmel, S.; Untch, M.; et al. Intense dose-dense epirubicin, paclitaxel, cyclophosphamide versus weekly paclitaxel, liposomal doxorubicin (plus carboplatin in triple-negative breast cancer) for neoadjuvant treatment of high-risk early breast cancer (GeparOcto—GBG 84): A randomised phase III trial. Eur. J. Cancer 2019, 106, 181–192. [Google Scholar] [CrossRef]
- Murphy, B.L.; Day, C.N.; Hoskin, T.L.; Habermann, E.B.; Boughey, J.C. Adolescents and Young Adults with Breast Cancer have More Aggressive Disease and Treatment Than Patients in Their Forties. Ann. Surg. Oncol. 2019, 26, 3920–3930. [Google Scholar] [CrossRef]
- Fisusi, F.A.; Akala, E.O. Drug Combinations in Breast Cancer Therapy. Pharm. Nanotechnol. 2019, 7, 3–23. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, F.R.; Scagliotti, G.V.; Mulshine, J.L.; Kwon, R.; Curran, W.J., Jr.; Wu, Y.L.; Paz-Ares, L. Lung cancer: Current therapies and new targeted treatments. Lancet 2017, 389, 299–311. [Google Scholar] [CrossRef]
- Piawah, S.; Venook, A.P. Targeted therapy for colorectal cancer metastases: A review of current methods of molecularly targeted therapy and the use of tumor biomarkers in the treatment of metastatic colorectal cancer. Cancer 2019, 125, 4139–4147. [Google Scholar] [CrossRef] [PubMed]
- Letai, A. Cell Death and Cancer Therapy: Don’t Forget to Kill the Cancer Cell! Clin. Cancer Res. 2015, 21, 5015–5020. [Google Scholar] [CrossRef] [Green Version]
- Schirrmacher, V. From chemotherapy to biological therapy: A review of novel concepts to reduce the side effects of systemic cancer treatment (Review). Int. J. Oncol. 2019, 54, 407–419. [Google Scholar] [CrossRef]
- Labelle, M.; Hynes, R.O. The initial hours of metastasis: The importance of cooperative host-tumor cell interactions during hematogenous dissemination. Cancer Discov. 2012, 2, 1091–1099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dillekås, H.; Rogers, M.S.; Straume, O. Are 90% of deaths from cancer caused by metastases? Cancer Med. 2019, 8, 5574–5576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Budczies, J.; von Winterfeld, M.; Klauschen, F.; Bockmayr, M.; Lennerz, J.K.; Denkert, C.; Wolf, T.; Warth, A.; Dietel, M.; Anagnostopoulos, I.; et al. The landscape of metastatic progression patterns across major human cancers. Oncotarget 2015, 6, 570–583. [Google Scholar] [CrossRef] [Green Version]
- Middleton, J.D.; Stover, D.G.; Hai, T. Chemotherapy-Exacerbated Breast Cancer Metastasis: A Paradox Explainable by Dysregulated Adaptive-Response. Int. J. Mol. Sci. 2018, 19, 3333. [Google Scholar] [CrossRef] [Green Version]
- Karagiannis, G.S.; Pastoriza, J.M.; Wang, Y.; Harney, A.S.; Entenberg, D.; Pignatelli, J.; Sharma, V.P.; Xue, E.A.; Cheng, E.; D’Alfonso, T.M.; et al. Neoadjuvant chemotherapy induces breast cancer metastasis through a TMEM-mediated mechanism. Sci. Transl. Med. 2017, 9, eaan0026. [Google Scholar] [CrossRef] [Green Version]
- Daenen, L.G.; Roodhart, J.M.; van Amersfoort, M.; Dehnad, M.; Roessingh, W.; Ulfman, L.H.; Derksen, P.W.; Voest, E.E. Chemotherapy enhances metastasis formation via VEGFR-1-expressing endothelial cells. Cancer Res. 2011, 71, 6976–6985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, Y.S.; Jalgaonkar, S.P.; Middleton, J.D.; Hai, T. Stress-inducible gene Atf3 in the noncancer host cells contributes to chemotherapy-exacerbated breast cancer metastasis. Proc. Natl. Acad. Sci. USA 2017, 114, E7159–E7168. [Google Scholar] [CrossRef] [Green Version]
- Wang, K.; Shan, S.; Wang, S.; Gu, X.; Zhou, X.; Ren, T. HMGB1-containing nucleosome mediates chemotherapy-induced metastasis of human lung cancer. Biochem. Biophys. Res. Commun. 2018, 500, 758–764. [Google Scholar] [CrossRef] [PubMed]
- Mansouri, N.; Keklikoglou, I.; Nassiri, S.; Torchia, B.; Guichard, A.; De Palma, M. Role of extracellular vesicles in chemotherapy-induced lung metastasis. Eur. Respir. J. 2020, 56, 3944. [Google Scholar] [CrossRef]
- Wu, Q.; Yang, Z.; Nie, Y.; Shi, Y.; Fan, D. Multi-drug resistance in cancer chemotherapeutics: Mechanisms and lab approaches. Cancer Lett. 2014, 347, 159–166. [Google Scholar] [CrossRef]
- Mahdizadeh, S.; Karimi, G.; Behravan, J.; Arabzadeh, S.; Lage, H.; Kalalinia, F. Crocin suppresses multidrug resistance in MRP overexpressing ovarian cancer cell line. DARU 2016, 24, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duran, G.E.; Wang, Y.C.; Moisan, F.; Francisco, E.B.; Sikic, B.I. Decreased levels of baseline and drug-induced tubulin polymerisation are hallmarks of resistance to taxanes in ovarian cancer cells and are associated with epithelial-to-mesenchymal transition. Br. J. Cancer 2017, 116, 1318–1328. [Google Scholar] [CrossRef] [Green Version]
- Zhang, F.; Zhang, H.; Wang, Z.; Yu, M.; Tian, R.; Ji, W.; Yang, Y.; Niu, R. P-glycoprotein associates with Anxa2 and promotes invasion in multidrug resistant breast cancer cells. Biochem. Pharm. 2014, 87, 292–302. [Google Scholar] [CrossRef]
- Zheng, L.; Foley, K.; Huang, L.; Leubner, A.; Mo, G.; Olino, K.; Edil, B.H.; Mizuma, M.; Sharma, R.; Le, D.T.; et al. Tyrosine 23 phosphorylation-dependent cell-surface localization of annexin A2 is required for invasion and metastases of pancreatic cancer. PLoS ONE 2011, 6, e19390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, X.; Liu, H.; Zhang, Z.; Gu, Y.; Qiu, H.; He, Z. Annexin A2 contributes to cisplatin resistance by activation of JNK-p53 pathway in non-small cell lung cancer cells. J. Exp. Clin. Cancer Res. 2017, 36, 123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wattanawongdon, W.; Hahnvajanawong, C.; Namwat, N.; Kanchanawat, S.; Boonmars, T.; Jearanaikoon, P.; Leelayuwat, C.; Techasen, A.; Seubwai, W. Establishment and characterization of gemcitabine-resistant human cholangiocarcinoma cell lines with multidrug resistance and enhanced invasiveness. Int. J. Oncol. 2015, 47, 398–410. [Google Scholar] [CrossRef] [Green Version]
- Maria, R.M.; Altei, W.F.; Selistre-de-Araujo, H.S.; Colnago, L.A. Impact of chemotherapy on metabolic reprogramming: Characterization of the metabolic profile of breast cancer MDA-MB-231 cells using (1)H HR-MAS NMR spectroscopy. J. Pharm. Biomed. Anal. 2017, 146, 324–328. [Google Scholar] [CrossRef] [Green Version]
- Amoedo, N.D.; Obre, E.; Rossignol, R. Drug discovery strategies in the field of tumor energy metabolism: Limitations by metabolic flexibility and metabolic resistance to chemotherapy. Biochim. Biophys. Acta (BBA) Bioenerg. 2017, 1858, 674–685. [Google Scholar] [CrossRef]
- Lee, K.-m.; Giltnane, J.M.; Balko, J.M.; Schwarz, L.J.; Guerrero-Zotano, A.L.; Hutchinson, K.E.; Nixon, M.J.; Estrada, M.V.; Sánchez, V.; Sanders, M.E.; et al. MYC and MCL1 Cooperatively Promote Chemotherapy-Resistant Breast Cancer Stem Cells via Regulation of Mitochondrial Oxidative Phosphorylation. Cell Metab. 2017, 26, 633–647.e637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farge, T.; Saland, E.; de Toni, F.; Aroua, N.; Hosseini, M.; Perry, R.; Bosc, C.; Sugita, M.; Stuani, L.; Fraisse, M.; et al. Chemotherapy-Resistant Human Acute Myeloid Leukemia Cells Are Not Enriched for Leukemic Stem Cells but Require Oxidative Metabolism. Cancer Discov. 2017, 7, 716–735. [Google Scholar] [CrossRef] [Green Version]
- Oresta, B.; Pozzi, C.; Braga, D.; Hurle, R.; Lazzeri, M.; Colombo, P.; Frego, N.; Erreni, M.; Faccani, C.; Elefante, G.; et al. Mitochondrial metabolic reprogramming controls the induction of immunogenic cell death and efficacy of chemotherapy in bladder cancer. Sci. Transl. Med. 2021, 13, eaba6110. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Cao, X. Characteristics and Significance of the Pre-metastatic Niche. Cancer Cell 2016, 30, 668–681. [Google Scholar] [CrossRef] [Green Version]
- Sun, M.-Y.; Xu, B.; Wu, Q.-X.; Chen, W.-L.; Cai, S.; Zhang, H.; Tang, Q.-F. Cisplatin-Resistant Gastric Cancer Cells Promote the Chemoresistance of Cisplatin-Sensitive Cells via the Exosomal RPS3-Mediated PI3K-Akt-Cofilin-1 Signaling Axis. Front. Cell Dev. Biol. 2021, 9. [Google Scholar] [CrossRef] [PubMed]
- Bandari, S.K.; Purushothaman, A.; Ramani, V.C.; Brinkley, G.J.; Chandrashekar, D.S.; Varambally, S.; Mobley, J.A.; Zhang, Y.; Brown, E.E.; Vlodavsky, I.; et al. Chemotherapy induces secretion of exosomes loaded with heparanase that degrades extracellular matrix and impacts tumor and host cell behavior. Matrix Biol. 2018, 65, 104–118. [Google Scholar] [CrossRef]
- Wills, C.A.; Liu, X.; Chen, L.; Zhao, Y.; Dower, C.M.; Sundstrom, J.; Wang, H.-G. Chemotherapy-Induced Upregulation of Small Extracellular Vesicle-Associated PTX3 Accelerates Breast Cancer Metastasis. Cancer Res. 2021, 81, 452–463. [Google Scholar] [CrossRef] [PubMed]
- Cameron, A.C.; Touyz, R.M.; Lang, N.N. Vascular Complications of Cancer Chemotherapy. Can. J. Cardiol. 2016, 32, 852–862. [Google Scholar] [CrossRef] [Green Version]
- Sato, C.; Okuda, K.; Tamiya, H.; Yamamoto, K.; Hoshina, K.; Narumoto, O.; Urushiyama, H.; Noguchi, S.; Amano, Y.; Watanabe, K.; et al. Acute Arterial Thrombosis during Postoperative Adjuvant Cisplatin-based Chemotherapy for Completely Resected Lung Adenocarcinoma. Intern. Med. 2018, 57, 557–561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sato, Y.; Kondo, M.; Inagaki, A.; Komatsu, H.; Okada, C.; Naruse, K.; Sahashi, T.; Kuroda, J.; Ogura, H.; Uegaki, S.; et al. Highly frequent and enhanced injection site reaction induced by peripheral venous injection of fosaprepitant in anthracycline-treated patients. J. Cancer 2014, 5, 390–397. [Google Scholar] [CrossRef]
- Kong, J.; Tian, H.; Zhang, F.; Zhang, Z.; Li, J.; Liu, X.; Li, X.; Liu, J.; Li, X.; Jin, D.; et al. Extracellular vesicles of carcinoma-associated fibroblasts creates a pre-metastatic niche in the lung through activating fibroblasts. Mol. Cancer 2019, 18, 175. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.; Zhang, D.; Wu, J.Y.; Xing, K.; Yeo, E.; Li, C.; Zhang, L.; Holland, E.; Yao, L.; Qin, L.; et al. Wnt-mediated endothelial transformation into mesenchymal stem cell–like cells induces chemoresistance in glioblastoma. Sci. Transl. Med. 2020, 12, eaay7522. [Google Scholar] [CrossRef]
- Pignatelli, J.; Bravo-Cordero, J.J.; Roh-Johnson, M.; Gandhi, S.J.; Wang, Y.; Chen, X.; Eddy, R.J.; Xue, A.; Singer, R.H.; Hodgson, L.; et al. Macrophage-dependent tumor cell transendothelial migration is mediated by Notch1/MenaINV-initiated invadopodium formation. Sci. Rep. 2016, 6, 37874. [Google Scholar] [CrossRef] [PubMed]
- Sharma, B.; Varney, M.L.; Saxena, S.; Wu, L.; Singh, R.K. Induction of CXCR2 ligands, stem cell-like phenotype, and metastasis in chemotherapy-resistant breast cancer cells. Cancer Lett. 2016, 372, 192–200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, H.; Lin, F.; Wang, Z.; Yang, L.; Meng, J.; Ou, Z.; Shao, Z.; Di, G.; Yang, G. CXCR2 promotes breast cancer metastasis and chemoresistance via suppression of AKT1 and activation of COX2. Cancer Lett 2018, 412, 69–80. [Google Scholar] [CrossRef]
- Carlson, P.; Dasgupta, A.; Grzelak, C.A.; Kim, J.; Barrett, A.; Coleman, I.M.; Shor, R.E.; Goddard, E.T.; Dai, J.; Schweitzer, E.M.; et al. Targeting the perivascular niche sensitizes disseminated tumour cells to chemotherapy. Nat. Cell Biol. 2019, 21, 238–250. [Google Scholar] [CrossRef] [PubMed]
- Ghajar, C.M.; Peinado, H.; Mori, H.; Matei, I.R.; Evason, K.J.; Brazier, H.; Almeida, D.; Koller, A.; Hajjar, K.A.; Stainier, D.Y.; et al. The perivascular niche regulates breast tumour dormancy. Nat. Cell Biol. 2013, 15, 807–817. [Google Scholar] [CrossRef]
- Clark, A.M.; Kumar, M.P.; Wheeler, S.E.; Young, C.L.; Venkataramanan, R.; Stolz, D.B.; Griffith, L.G.; Lauffenburger, D.A.; Wells, A. A Model of Dormant-Emergent Metastatic Breast Cancer Progression Enabling Exploration of Biomarker Signatures. Mol. Cell. Proteom. 2018, 17, 619–630. [Google Scholar] [CrossRef] [Green Version]
- Bogenrieder, T.; Herlyn, M. Axis of evil: Molecular mechanisms of cancer metastasis. Oncogene 2003, 22, 6524–6536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eakin, A.J.; Mc Erlain, T.; Burke, A.; Eaton, A.; Tipping, N.; Allocca, G.; Branco, C.M. Circulating Levels of Epirubicin Cause Endothelial Senescence While Compromising Metabolic Activity and Vascular Function. Front. Cell Dev. Biol. 2020, 8, 799. [Google Scholar] [CrossRef]
- Nuver, J.; De Haas, E.C.; Van Zweeden, M.; Gietema, J.A.; Meijer, C. Vascular damage in testicular cancer patients: A study on endothelial activation by bleomycin and cisplatin in vitro. Oncol. Rep. 2010, 23, 247–253. [Google Scholar]
- Jambusaria, A.; Hong, Z.; Zhang, L.; Srivastava, S.; Jana, A.; Toth, P.T.; Dai, Y.; Malik, A.B.; Rehman, J. Endothelial heterogeneity across distinct vascular beds during homeostasis and inflammation. Elife 2020, 9. [Google Scholar] [CrossRef]
- Reiterer, M.; Branco, C.M. Endothelial cells and organ function: Applications and implications of understanding unique and reciprocal remodelling. FEBS J. 2020, 287, 1088–1100. [Google Scholar] [CrossRef]
- Mongiardi, M.P.; Radice, G.; Piras, M.; Stagni, V.; Pacioni, S.; Re, A.; Putti, S.; Ferrè, F.; Farsetti, A.; Pallini, R.; et al. Axitinib exposure triggers endothelial cells senescence through ROS accumulation and ATM activation. Oncogene 2019, 38, 5413–5424. [Google Scholar] [CrossRef]
- Brosnan, E.M.; Anders, C.K. Understanding patterns of brain metastasis in breast cancer and designing rational therapeutic strategies. Ann. Transl. Med. 2018, 6, 163. [Google Scholar] [CrossRef]
- Hader, S.N.; Zinkevich, N.; Norwood Toro, L.E.; Kriegel, A.J.; Kong, A.; Freed, J.K.; Gutterman, D.D.; Beyer, A.M. Detrimental effects of chemotherapy on human coronary microvascular function. Am. J. Physiol. Heart Circ. Physiol. 2019, 317, H705–H710. [Google Scholar] [CrossRef]
- Kalucka, J.; de Rooij, L.; Goveia, J.; Rohlenova, K.; Dumas, S.J.; Meta, E.; Conchinha, N.V.; Taverna, F.; Teuwen, L.A.; Veys, K.; et al. Single-Cell Transcriptome Atlas of Murine Endothelial Cells. Cell 2020, 180, 764–779. [Google Scholar] [CrossRef] [PubMed]
- Jin, X.; Demere, Z.; Nair, K.; Ali, A.; Ferraro, G.B.; Natoli, T.; Deik, A.; Petronio, L.; Tang, A.A.; Zhu, C.; et al. A metastasis map of human cancer cell lines. Nature 2020, 588, 331–336. [Google Scholar] [CrossRef] [PubMed]
- Doron, H.; Amer, M.; Ershaid, N.; Blazquez, R.; Shani, O.; Lahav, T.G.; Cohen, N.; Adler, O.; Hakim, Z.; Pozzi, S.; et al. Inflammatory Activation of Astrocytes Facilitates Melanoma Brain Tropism via the CXCL10-CXCR3 Signaling Axis. Cell Rep. 2019, 28, 1785–1798. [Google Scholar] [CrossRef] [Green Version]
- Imaizumi, J.; Shida, D.; Narita, Y.; Miyakita, Y.; Tanabe, T.; Takashima, A.; Boku, N.; Igaki, H.; Itami, J.; Kanemitsu, Y. Prognostic factors of brain metastases from colorectal cancer. BMC Cancer 2019, 19, 755. [Google Scholar] [CrossRef] [Green Version]
- Jin, J.; Gao, Y.; Zhang, J.; Wang, L.; Wang, B.; Cao, J.; Shao, Z.; Wang, Z. Incidence, pattern and prognosis of brain metastases in patients with metastatic triple negative breast cancer. BMC Cancer 2018, 18, 446. [Google Scholar] [CrossRef] [PubMed]
- Fitzpatrick, A.; Tutt, A. Controversial issues in the neoadjuvant treatment of triple-negative breast cancer. Adv. Med. Oncol. 2019, 11, 1758835919882581. [Google Scholar] [CrossRef]
- Xiao, W.; Zheng, S.; Yang, A.; Zhang, X.; Zou, Y.; Tang, H.; Xie, X. Breast cancer subtypes and the risk of distant metastasis at initial diagnosis: A population-based study. Cancer Manag. Res. 2018, 10, 5329–5338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kennecke, H.; Yerushalmi, R.; Woods, R.; Cheang, M.C.; Voduc, D.; Speers, C.H.; Nielsen, T.O.; Gelmon, K. Metastatic behavior of breast cancer subtypes. J. Clin. Oncol. 2010, 28, 3271–3277. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.J.; Kim, J.S.; Park, E.S.; Lee, J.S.; Lin, Q.; Langley, R.R.; Maya, M.; He, J.; Kim, S.W.; Weihua, Z.; et al. Astrocytes upregulate survival genes in tumor cells and induce protection from chemotherapy. Neoplasia 2011, 13, 286–298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Augustin, H.G.; Koh, G.Y. Organotypic vasculature: From descriptive heterogeneity to functional pathophysiology. Science 2017, 357. [Google Scholar] [CrossRef] [Green Version]
- Yamada, T.; Egashira, N.; Bando, A.; Nishime, Y.; Tonogai, Y.; Imuta, M.; Yano, T.; Oishi, R. Activation of p38 MAPK by oxidative stress underlying epirubicin-induced vascular endothelial cell injury. Free Radic. Biol. Med. 2012, 52, 1285–1293. [Google Scholar] [CrossRef]
- Wojcik, T.; Szczesny, E.; Chlopicki, S. Detrimental effects of chemotherapeutics and other drugs on the endothelium: A call for endothelial toxicity profiling. Pharmacol. Rep. 2015, 67, 811–817. [Google Scholar] [CrossRef]
- Sonowal, H.; Pal, P.; Shukla, K.; Saxena, A.; Srivastava, S.K.; Ramana, K.V. Aldose reductase inhibitor, fidarestat prevents doxorubicin-induced endothelial cell death and dysfunction. Biochem. Pharm. 2018, 150, 181–190. [Google Scholar] [CrossRef]
- Gong, C.; Liu, B.; Yao, Y.; Qu, S.; Luo, W.; Tan, W.; Liu, Q.; Yao, H.; Zou, L.; Su, F.; et al. Potentiated DNA Damage Response in Circulating Breast Tumor Cells Confers Resistance to Chemotherapy. J. Biol. Chem. 2015, 290, 14811–14825. [Google Scholar] [CrossRef] [Green Version]
- BioRender. Available online: https://biorender.com (accessed on 5 June 2021).
- Suh, E.; Stratton, K.L.; Leisenring, W.M.; Nathan, P.C.; Ford, J.S.; Freyer, D.R.; McNeer, J.L.; Stock, W.; Stovall, M.; Krull, K.R.; et al. Late mortality and chronic health conditions in long-term survivors of early-adolescent and young adult cancers: A retrospective cohort analysis from the Childhood Cancer Survivor Study. Lancet Oncol. 2020, 21, 421–435. [Google Scholar] [CrossRef]
- Gegechkori, N.; Haines, L.; Lin, J.J. Long-Term and Latent Side Effects of Specific Cancer Types. Med. Clin. N. Am. 2017, 101, 1053–1073. [Google Scholar] [CrossRef]
- Roxburgh, C.S.; McMillan, D.C. Cancer and systemic inflammation: Treat the tumour and treat the host. Br. J. Cancer 2014, 110, 1409–1412. [Google Scholar] [CrossRef]
- Higgins, A.Y.; O’Halloran, T.D.; Chang, J.D. Chemotherapy-induced cardiomyopathy. Heart Fail. Rev. 2015, 20, 721–730. [Google Scholar] [CrossRef] [PubMed]
- Abdullah, C.S.; Alam, S.; Aishwarya, R.; Miriyala, S.; Bhuiyan, M.A.N.; Panchatcharam, M.; Pattillo, C.B.; Orr, A.W.; Sadoshima, J.; Hill, J.A.; et al. Doxorubicin-induced cardiomyopathy associated with inhibition of autophagic degradation process and defects in mitochondrial respiration. Sci. Rep. 2019, 9, 2002. [Google Scholar] [CrossRef]
- Renu, K.; Abilash, V.G.; Tirupathi, P.P.B.; Arunachalam, S. Molecular mechanism of doxorubicin-induced cardiomyopathy—An update. Eur. J. Pharm. 2018, 818, 241–253. [Google Scholar] [CrossRef] [PubMed]
- Payne, D.L.; Nohria, A. Prevention of Chemotherapy Induced Cardiomyopathy. Curr. Heart Fail. Rep. 2017, 14, 398–403. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Villani, R.M.; Wang, H.; Simpson, M.J.; Roberts, M.S.; Tang, M.; Liang, X. The role of cellular reactive oxygen species in cancer chemotherapy. J. Exp. Clin. Cancer Res. 2018, 37, 266. [Google Scholar] [CrossRef] [PubMed]
- Angsutararux, P.; Luanpitpong, S.; Issaragrisil, S. Chemotherapy-Induced Cardiotoxicity: Overview of the Roles of Oxidative Stress. Oxidative Med. Cell. Longev. 2015, 2015, 795602. [Google Scholar] [CrossRef] [Green Version]
- Jain, D.; Ahmad, T.; Cairo, M.; Aronow, W. Cardiotoxicity of cancer chemotherapy: Identification, prevention and treatment. Ann. Transl. Med. 2017, 5, 348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Todaro, M.C.; Oreto, L.; Qamar, R.; Paterick, T.E.; Carerj, S.; Khandheria, B.K. Cardioncology: State of the heart. Int. J. Cardiol. 2013, 168, 680–687. [Google Scholar] [CrossRef]
- Martins-Teixeira, M.B.; Carvalho, I. Antitumour Anthracyclines: Progress and Perspectives. ChemMedChem 2020, 15, 933–948. [Google Scholar] [CrossRef]
- Swain, S.M.; Whaley, F.S.; Ewer, M.S. Congestive heart failure in patients treated with doxorubicin: A retrospective analysis of three trials. Cancer 2003, 97, 2869–2879. [Google Scholar] [CrossRef] [PubMed]
- Khasraw, M.; Bell, R.; Dang, C. Epirubicin: Is it like doxorubicin in breast cancer? A clinical review. Breast 2012, 21, 142–149. [Google Scholar] [CrossRef]
- McCaffrey, T.A.; Tziros, C.; Lewis, J.; Katz, R.; Siegel, R.; Weglicki, W.; Kramer, J.; Mak, I.T.; Toma, I.; Chen, L.; et al. Genomic profiling reveals the potential role of TCL1A and MDR1 deficiency in chemotherapy-induced cardiotoxicity. Int. J. Biol. Sci. 2013, 9, 350–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Volkova, M.; Russell, R., 3rd. Anthracycline cardiotoxicity: Prevalence, pathogenesis and treatment. Curr. Cardiol. Rev. 2011, 7, 214–220. [Google Scholar] [CrossRef] [Green Version]
- Barrett-Lee, P.J.; Dixon, J.M.; Farrell, C.; Jones, A.; Leonard, R.; Murray, N.; Palmieri, C.; Plummer, C.J.; Stanley, A.; Verrill, M.W. Expert opinion on the use of anthracyclines in patients with advanced breast cancer at cardiac risk. Ann. Oncol. 2009, 20, 816–827. [Google Scholar] [CrossRef]
- Murata, T.; Yamawaki, H.; Yoshimoto, R.; Hori, M.; Sato, K.; Ozaki, H.; Karaki, H. Chronic effect of doxorubicin on vascular endothelium assessed by organ culture study. Life Sci. 2001, 69, 2685–2695. [Google Scholar] [CrossRef]
- Cao, Y.; Gong, Y.; Liu, L.; Zhou, Y.; Fang, X.; Zhang, C.; Li, Y.; Li, J. The use of human umbilical vein endothelial cells (HUVECs) as an in vitro model to assess the toxicity of nanoparticles to endothelium: A review. J. Appl. Toxicol. 2017, 37, 1359–1369. [Google Scholar] [CrossRef]
- Yamada, T.; Ueda, M.; Egashira, N.; Zukeyama, N.; Kuwahara, J.; Masuda, S. Involvement of intracellular cAMP in epirubicin-induced vascular endothelial cell injury. J. Pharm. Sci. 2016, 130, 33–37. [Google Scholar] [CrossRef] [Green Version]
- Jang, W.J.; Choi, D.Y.; Jeon, I.S. Vascular endothelial dysfunction after anthracyclines treatment in children with acute lymphoblastic leukemia. Korean J. Pediatr. 2013, 56, 130–134. [Google Scholar] [CrossRef] [Green Version]
- Dursun, B.; He, Z.; Somerset, H.; Oh, D.J.; Faubel, S.; Edelstein, C.L. Caspases and calpain are independent mediators of cisplatin-induced endothelial cell necrosis. Am. J. Physiol. Ren. Physiol. 2006, 291, F578–F587. [Google Scholar] [CrossRef] [PubMed]
- Filipowska, J.; Tomaszewski, K.A.; Niedzwiedzki, L.; Walocha, J.A.; Niedzwiedzki, T. The role of vasculature in bone development, regeneration and proper systemic functioning. Angiogenesis 2017, 20, 291–302. [Google Scholar] [CrossRef] [Green Version]
- Yao, Z.; Murali, B.; Ren, Q.; Luo, X.; Faget, D.V.; Cole, T.; Ricci, B.; Thotala, D.; Monahan, J.; van Deursen, J.M.; et al. Therapy-Induced Senescence Drives Bone Loss. Cancer Res. 2020, 80, 1171–1182. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, K.; Park, H.C.; Ratliff, B.; Addabbo, F.; Hatzopoulos, A.K.; Chander, P.; Goligorsky, M.S. Adriamycin nephropathy: A failure of endothelial progenitor cell-induced repair. Am. J. Pathol. 2010, 176, 1685–1695. [Google Scholar] [CrossRef] [PubMed]
- Kishimoto, S.; Aoki, H.; Suzuki, R.; Inoue, M.; Fukushima, S. Angiographic Evaluation of Vascular Damage in Rat Liver After Administration of Epirubicin or Miriplatin. Anticancer Res. 2018, 38, 247–251. [Google Scholar] [CrossRef] [PubMed]
- Eklund, J.W.; Trifilio, S.; Mulcahy, M.F. Chemotherapy dosing in the setting of liver dysfunction. Oncology 2005, 19, 1057–1063; discussion 1063–1064, 1069. [Google Scholar]
- Saleem, Z.; Ahmad, M.; Hashmi, F.K.; Saeed, H.; Aziz, M.T. Impairment of liver synthetic function and the production of plasma proteins in primary breast cancer patients on doxorubicincyclophosphamide (AC) protocol. Pak. J. Pharm. Sci. 2016, 29, 1555–1563. [Google Scholar]
- Ruscetti, M.; Morris, J.P.; Mezzadra, R.; Russell, J.; Leibold, J.; Romesser, P.B.; Simon, J.; Kulick, A.; Ho, Y.-j.; Fennell, M.; et al. Senescence-Induced Vascular Remodeling Creates Therapeutic Vulnerabilities in Pancreas Cancer. Cell 2020, 181, 424–441.e421. [Google Scholar] [CrossRef] [PubMed]
- Demaria, M.; O’Leary, M.N.; Chang, J.; Shao, L.; Liu, S.; Alimirah, F.; Koenig, K.; Le, C.; Mitin, N.; Deal, A.M.; et al. Cellular Senescence Promotes Adverse Effects of Chemotherapy and Cancer Relapse. Cancer Discov. 2017, 7, 165–176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cupit-Link, M.C.; Kirkland, J.L.; Ness, K.K.; Armstrong, G.T.; Tchkonia, T.; LeBrasseur, N.K.; Armenian, S.H.; Ruddy, K.J.; Hashmi, S.K. Biology of premature ageing in survivors of cancer. ESMO Open 2017, 2, e000250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deckx, L.; van den Akker, M.; Metsemakers, J.; Knottnerus, A.; Schellevis, F.; Buntinx, F. Chronic Diseases among Older Cancer Survivors. J. Cancer Epidemiol. 2012, 2012, 206414. [Google Scholar] [CrossRef] [Green Version]
- Alspach, E.; Fu, Y.; Stewart, S.A. Senescence and the pro-tumorigenic stroma. Crit. Rev. Oncog. 2013, 18, 549–558. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.; Cao, K.; Xia, Y.; Li, Y.; Hou, Y.; Wang, L.; Li, L.; Chang, L.; Li, W. Cellular senescence in ionizing radiation (Review). Oncol. Rep. 2019, 42, 883–894. [Google Scholar] [CrossRef]
- Demaria, M.; Ohtani, N.; Youssef, S.A.; Rodier, F.; Toussaint, W.; Mitchell, J.R.; Laberge, R.M.; Vijg, J.; Van Steeg, H.; Dollé, M.E.; et al. An essential role for senescent cells in optimal wound healing through secretion of PDGF-AA. Dev. Cell 2014, 31, 722–733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sabbatinelli, J.; Prattichizzo, F.; Olivieri, F.; Procopio, A.D.; Rippo, M.R.; Giuliani, A. Where Metabolism Meets Senescence: Focus on Endothelial Cells. Front. Physiol. 2019, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di, G.H.; Liu, Y.; Lu, Y.; Liu, J.; Wu, C.; Duan, H.F. IL-6 secreted from senescent mesenchymal stem cells promotes proliferation and migration of breast cancer cells. PLoS ONE 2014, 9, e113572. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Shi, C. Cellular senescence is a promising target for chronic wounds: A comprehensive review. Burns Trauma 2020, 8, tkaa021. [Google Scholar] [CrossRef] [PubMed]
- Xu, Q.; Long, Q.; Zhu, D.; Fu, D.; Zhang, B.; Han, L.; Qian, M.; Guo, J.; Xu, J.; Cao, L.; et al. Targeting amphiregulin (AREG) derived from senescent stromal cells diminishes cancer resistance and averts programmed cell death 1 ligand (PD-L1)-mediated immunosuppression. Aging Cell 2019, 18, e13027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duy, C.; Li, M.; Teater, M.; Meydan, C.; Garrett-Bakelman, F.E.; Lee, T.C.; Chin, C.R.; Durmaz, C.; Kawabata, K.C.; Dhimolea, E.; et al. Chemotherapy induces senescence-like resilient cells capable of initiating AML recurrence. Cancer Discov. 2021, 11, 1542–1561. [Google Scholar] [CrossRef]
- De Falco, E.; Carnevale, R.; Pagano, F.; Chimenti, I.; Fianchini, L.; Bordin, A.; Siciliano, C.; Monticolo, R.; Equitani, F.; Carrizzo, A.; et al. Role of NOX2 in mediating doxorubicin-induced senescence in human endothelial progenitor cells. Mech. Ageing Dev. 2016, 159, 37–43. [Google Scholar] [CrossRef]
- Ghosh, A.K.; Rai, R.; Park, K.E.; Eren, M.; Miyata, T.; Wilsbacher, L.D.; Vaughan, D.E. A small molecule inhibitor of PAI-1 protects against doxorubicin-induced cellular senescence. Oncotarget 2016, 7, 72443–72457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bent, E.H.; Gilbert, L.A.; Hemann, M.T. A senescence secretory switch mediated by PI3K/AKT/mTOR activation controls chemoprotective endothelial secretory responses. Genes Dev. 2016, 30, 1811–1821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evans, C.E.; Bendahl, P.O.; Belting, M.; Branco, C.; Johnson, R.S. Diverse roles of cell-specific hypoxia-inducible factor 1 in cancer-associated hypercoagulation. Blood 2016, 127, 1355–1360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Canale, M.L.; Bisceglia, I.; Lestuzzi, C.; Parrini, I. Arterial Thrombosis in Cancer: Spotlight on the Neglected Vessels. Anticancer Res. 2019, 39, 4619–4625. [Google Scholar] [CrossRef]
- Evans, C.E.; Branco-Price, C.; Johnson, R.S. HIF-mediated endothelial response during cancer progression. Int. J. Hematol. 2012, 95, 471–477. [Google Scholar] [CrossRef]
- Ten, V.S.; Pinsky, D.J. Endothelial response to hypoxia: Physiologic adaptation and pathologic dysfunction. Curr. Opin. Crit. Care 2002, 8, 245–250. [Google Scholar] [CrossRef]
- Gertler, J.P.; Weibe, D.A.; Ocasio, V.H.; Abbott, W.M. Hypoxia induces procoagulant activity in cultured human venous endothelium. J. Vasc. Surg. 1991, 13, 428–433. [Google Scholar] [CrossRef] [Green Version]
- Oppelt, P.; Betbadal, A.; Nayak, L. Approach to chemotherapy-associated thrombosis. Vasc. Med. 2015, 20, 153–161. [Google Scholar] [CrossRef] [Green Version]
- Kamath, K.; Smiyun, G.; Wilson, L.; Jordan, M.A. Mechanisms of inhibition of endothelial cell migration by taxanes. Cytoskeleton 2014, 71, 46–60. [Google Scholar] [CrossRef]
- Yamaç, D.; Elmas, Ç.; Özoğul, C.; Keskil, Z.; Dursun, A. Ultrastructural Damage in Vascular Endothelium in Rats Treated with Paclitaxel and Doxorubicin. Ultrastruct. Pathol. 2006, 30, 103–110. [Google Scholar] [CrossRef] [PubMed]
- Palmer, A.C.; Sorger, P.K. Combination Cancer Therapy Can Confer Benefit via Patient-to-Patient Variability without Drug Additivity or Synergy. Cell 2017, 171, 1678–1691.e13. [Google Scholar] [CrossRef] [Green Version]
- Doll, D.C.; List, A.F.; Greco, F.A.; Hainsworth, J.D.; Hande, K.R.; Johnson, D.H. Acute vascular ischemic events after cisplatin-based combination chemotherapy for germ-cell tumors of the testis. Ann. Intern. Med. 1986, 105, 48–51. [Google Scholar] [CrossRef]
- Moore, R.A.; Adel, N.; Riedel, E.; Bhutani, M.; Feldman, D.R.; Tabbara, N.E.; Soff, G.; Parameswaran, R.; Hassoun, H. High incidence of thromboembolic events in patients treated with cisplatin-based chemotherapy: A large retrospective analysis. J. Clin. Oncol. 2011, 29, 3466–3473. [Google Scholar] [CrossRef]
- Licciardello, J.T.; Moake, J.L.; Rudy, C.K.; Karp, D.D.; Hong, W.K. Elevated plasma von Willebrand factor levels and arterial occlusive complications associated with cisplatin-based chemotherapy. Oncology 1985, 42, 296–300. [Google Scholar] [CrossRef] [PubMed]
- Lechner, D.; Kollars, M.; Gleiss, A.; Kyrle, P.A.; Weltermann, A. Chemotherapy-induced thrombin generation via procoagulant endothelial microparticles is independent of tissue factor activity. J. Thromb. Haemost. 2007, 5, 2445–2452. [Google Scholar] [CrossRef] [PubMed]
- Shim, K.; MacKenzie, M.J.; Winquist, E. Chemotherapy-associated osteonecrosis in cancer patients with solid tumours: A systematic review. Drug Saf. 2008, 31, 359–371. [Google Scholar] [CrossRef]
- Levine, M.N.; Gent, M.; Hirsh, J.; Arnold, A.; Goodyear, M.D.; Hryniuk, W.; De Pauw, S. The thrombogenic effect of anticancer drug therapy in women with stage II breast cancer. N. Engl. J. Med. 1988, 318, 404–407. [Google Scholar] [CrossRef] [PubMed]
- Swystun, L.L.; Shin, L.Y.; Beaudin, S.; Liaw, P.C. Chemotherapeutic agents doxorubicin and epirubicin induce a procoagulant phenotype on endothelial cells and blood monocytes. J. Thromb. Haemost. 2009, 7, 619–626. [Google Scholar] [CrossRef]
- Brown, T.; Sykes, D.; Allen, A.R. Implications of Breast Cancer Chemotherapy-Induced Inflammation on the Gut, Liver, and Central Nervous System. Biomedicines 2021, 9, 189. [Google Scholar] [CrossRef] [PubMed]
- Mounier, N.M.; Abdel-Maged, A.E.; Wahdan, S.A.; Gad, A.M.; Azab, S.S. Chemotherapy-induced cognitive impairment (CICI): An overview of etiology and pathogenesis. Life Sci. 2020, 258, 118071. [Google Scholar] [CrossRef]
- Manchon, J.F.; Dabaghian, Y.; Uzor, N.E.; Kesler, S.R.; Wefel, J.S.; Tsvetkov, A.S. Levetiracetam mitigates doxorubicin-induced DNA and synaptic damage in neurons. Sci. Rep. 2016, 6, 25705. [Google Scholar] [CrossRef]
- Cheruku, S.P.; Ramalingayya, G.V.; Chamallamudi, M.R.; Biswas, S.; Nandakumar, K.; Nampoothiri, M.; Gourishetti, K.; Kumar, N. Catechin ameliorates doxorubicin-induced neuronal cytotoxicity in in vitro and episodic memory deficit in in vivo in Wistar rats. Cytotechnology 2018, 70, 245–259. [Google Scholar] [CrossRef]
- Boykoff, N.; Moieni, M.; Subramanian, S.K. Confronting chemobrain: An in-depth look at survivors’ reports of impact on work, social networks, and health care response. J. Cancer Surviv. 2009, 3, 223–232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loman, B.R.; Jordan, K.R.; Haynes, B.; Bailey, M.T.; Pyter, L.M. Chemotherapy-induced neuroinflammation is associated with disrupted colonic and bacterial homeostasis in female mice. Sci. Rep. 2019, 9, 16490. [Google Scholar] [CrossRef] [Green Version]
- McLeary, F.; Davis, A.; Rudrawar, S.; Perkins, A.; Anoopkumar-Dukie, S. Mechanisms underlying select chemotherapeutic-agent-induced neuroinflammation and subsequent neurodegeneration. Eur. J. Pharm. 2019, 842, 49–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, P.; Talukder, M.A.H.; Gao, F. Oxidative Stress and Microvessel Barrier Dysfunction. Front. Physiol. 2020, 11, 472. [Google Scholar] [CrossRef]
- Nome, M.E.; Euceda, L.R.; Jabeen, S.; Debik, J.; Bathen, T.F.; Giskeødegård, G.F.; Taskén, K.A.; Mælandsmo, G.M.; Halvorsen, B.; Yndestad, A.; et al. Serum levels of inflammation-related markers and metabolites predict response to neoadjuvant chemotherapy with and without bevacizumab in breast cancers. Int. J. Cancer 2020, 146, 223–235. [Google Scholar] [CrossRef]
- Christie, L.A.; Acharya, M.M.; Parihar, V.K.; Nguyen, A.; Martirosian, V.; Limoli, C.L. Impaired cognitive function and hippocampal neurogenesis following cancer chemotherapy. Clin. Cancer Res. 2012, 18, 1954–1965. [Google Scholar] [CrossRef] [Green Version]
- Han, R.; Yang, Y.M.; Dietrich, J.; Luebke, A.; Mayer-Proschel, M.; Noble, M. Systemic 5-fluorouracil treatment causes a syndrome of delayed myelin destruction in the central nervous system. J. Biol. 2008, 7, 12. [Google Scholar] [CrossRef] [Green Version]
- Van der Willik, K.D.; Koppelmans, V.; Hauptmann, M.; Compter, A.; Ikram, M.A.; Schagen, S.B. Inflammation markers and cognitive performance in breast cancer survivors 20 years after completion of chemotherapy: A cohort study. Breast Cancer Res. 2018, 20, 135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nudelman, K.N.; McDonald, B.C.; Wang, Y.; Smith, D.J.; West, J.D.; O’Neill, D.P.; Zanville, N.R.; Champion, V.L.; Schneider, B.P.; Saykin, A.J. Cerebral Perfusion and Gray Matter Changes Associated With Chemotherapy-Induced Peripheral Neuropathy. J. Clin. Oncol. 2016, 34, 677–683. [Google Scholar] [CrossRef] [Green Version]
- Mueller, K.; Thiel, F.; Beutner, F.; Teren, A.; Frisch, S.; Ballarini, T.; Möller, H.E.; Ihle, K.; Thiery, J.; Schuler, G.; et al. Brain Damage With Heart Failure: Cardiac Biomarker Alterations and Gray Matter Decline. Circ. Res. 2020, 126, 750–764. [Google Scholar] [CrossRef]
- Wang, F.; Cao, Y.; Ma, L.; Pei, H.; Rausch, W.D.; Li, H. Dysfunction of Cerebrovascular Endothelial Cells: Prelude to Vascular Dementia. Front. Aging Neurosci. 2018, 10, 376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Makker, P.G.; Duffy, S.S.; Lees, J.G.; Perera, C.J.; Tonkin, R.S.; Butovsky, O.; Park, S.B.; Goldstein, D.; Moalem-Taylor, G. Characterisation of Immune and Neuroinflammatory Changes Associated with Chemotherapy-Induced Peripheral Neuropathy. PLoS ONE 2017, 12, e0170814. [Google Scholar] [CrossRef]
- Kandula, T.; Farrar, M.A.; Cohn, R.J.; Mizrahi, D.; Carey, K.; Johnston, K.; Kiernan, M.C.; Krishnan, A.V.; Park, S.B. Chemotherapy-Induced Peripheral Neuropathy in Long-term Survivors of Childhood Cancer: Clinical, Neurophysiological, Functional, and Patient-Reported Outcomes. JAMA Neurol. 2018, 75, 980–988. [Google Scholar] [CrossRef]
- Shim, H.S.; Bae, C.; Wang, J.; Lee, K.H.; Hankerd, K.M.; Kim, H.K.; Chung, J.M.; La, J.H. Peripheral and central oxidative stress in chemotherapy-induced neuropathic pain. Mol. Pain 2019, 15, 1744806919840098. [Google Scholar] [CrossRef] [Green Version]
- Fukuda, Y.; Li, Y.; Segal, R.A. A Mechanistic Understanding of Axon Degeneration in Chemotherapy-Induced Peripheral Neuropathy. Front. Neurosci. 2017, 11, 481. [Google Scholar] [CrossRef]
- Scott, A.R.; Stoltzfus, K.C.; Tchelebi, L.T.; Trifiletti, D.M.; Lehrer, E.J.; Rao, P.; Bleyer, A.; Zaorsky, N.G. Trends in Cancer Incidence in US Adolescents and Young Adults, 1973-2015. JAMA Netw. Open 2020, 3, e2027738. [Google Scholar] [CrossRef] [PubMed]
- Mauri, G.; Sartore-Bianchi, A.; Russo, A.G.; Marsoni, S.; Bardelli, A.; Siena, S. Early-onset colorectal cancer in young individuals. Mol. Oncol. 2019, 13, 109–131. [Google Scholar] [CrossRef] [Green Version]
- Houghton, P.J.; Kurmasheva, R.T. Challenges and Opportunities for Childhood Cancer Drug Development. Pharm. Rev. 2019, 71, 671–697. [Google Scholar] [CrossRef] [PubMed]
- Kocián, P.; Svobodová, I.; Krejčí, D.; Blaha, M.; Gürlich, R.; Dušek, L.; Hoch, J.; Whitley, A. Is colorectal cancer a more aggressive disease in young patients? A population-based study from the Czech Republic. Cancer Epidemiol. 2019, 63, 101621. [Google Scholar] [CrossRef]
- Haupt, R.; Essiaf, S.; Dellacasa, C.; Ronckers, C.M.; Caruso, S.; Sugden, E.; Zadravec Zaletel, L.; Muraca, M.; Morsellino, V.; Kienesberger, A.; et al. The ‘Survivorship Passport’ for childhood cancer survivors. Eur. J. Cancer 2018, 102, 69–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bleyer, A.; Barr, R.; Hayes-Lattin, B.; Thomas, D.; Ellis, C.; Anderson, B. The distinctive biology of cancer in adolescents and young adults. Nat. Rev. Cancer 2008, 8, 288–298. [Google Scholar] [CrossRef] [PubMed]
- Anders, C.K.; Hsu, D.S.; Broadwater, G.; Acharya, C.R.; Foekens, J.A.; Zhang, Y.; Wang, Y.; Marcom, P.K.; Marks, J.R.; Febbo, P.G.; et al. Young age at diagnosis correlates with worse prognosis and defines a subset of breast cancers with shared patterns of gene expression. J. Clin. Oncol. 2008, 26, 3324–3330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, H.M.; Irish, W.; Vohra, N.A.; Wong, J.H. Refining breast cancer prognosis by incorporating age at diagnosis into clinical prognostic staging: Introduction of a novel online calculator. Breast Cancer Res. Treat. 2021. [Google Scholar] [CrossRef]
- Nassar, A.H.; Mouw, K.W.; Jegede, O.; Shinagare, A.B.; Kim, J.; Liu, C.-J.; Pomerantz, M.; Harshman, L.C.; Van Allen, E.M.; Wei, X.X.; et al. A model combining clinical and genomic factors to predict response to PD-1/PD-L1 blockade in advanced urothelial carcinoma. Br. J. Cancer 2020, 122, 555–563. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, A.J.; Li, X.; Tucker, M.; Li, S.; Mu, X.J.; Eng, K.W.; Sboner, A.; Rubin, M.; Gerstein, M. Molecular medicine tumor board: Whole-genome sequencing to inform on personalized medicine for a man with advanced prostate cancer. Prostate Cancer Prostatic Dis. 2021. [Google Scholar] [CrossRef] [PubMed]
- Gambardella, V.; Tarazona, N.; Cejalvo, J.M.; Lombardi, P.; Huerta, M.; Roselló, S.; Fleitas, T.; Roda, D.; Cervantes, A. Personalized Medicine: Recent Progress in Cancer Therapy. Cancers 2020, 12, 1009. [Google Scholar] [CrossRef] [Green Version]
- Costa, R.L.B.; Czerniecki, B.J. Clinical development of immunotherapies for HER2+ breast cancer: A review of HER2-directed monoclonal antibodies and beyond. npj Breast Cancer 2020, 6, 10. [Google Scholar] [CrossRef] [Green Version]
- Gravalos, C.; Jimeno, A. HER2 in gastric cancer: A new prognostic factor and a novel therapeutic target. Ann. Oncol. 2008, 19, 1523–1529. [Google Scholar] [CrossRef] [PubMed]
- Ravasco, P. Nutrition in Cancer Patients. J. Clin. Med. 2019, 8, 1211. [Google Scholar] [CrossRef] [Green Version]
- Redondo-Blanco, S.; Fernández, J.; Gutiérrez-del-Río, I.; Villar, C.J.; Lombó, F. New Insights toward Colorectal Cancer Chemotherapy Using Natural Bioactive Compounds. Front. Pharmacol. 2017, 8. [Google Scholar] [CrossRef] [Green Version]
- Khiati, S.; Dalla Rosa, I.; Sourbier, C.; Ma, X.; Rao, V.A.; Neckers, L.M.; Zhang, H.; Pommier, Y. Mitochondrial Topoisomerase I (Top1mt) Is a Novel Limiting Factor of Doxorubicin Cardiotoxicity. Clin. Cancer Res. 2014, 20, 4873–4881. [Google Scholar] [CrossRef] [Green Version]
- Fendt, S.M.; Frezza, C.; Erez, A. Targeting Metabolic Plasticity and Flexibility Dynamics for Cancer Therapy. Cancer Discov. 2020, 10, 1797–1807. [Google Scholar] [CrossRef] [PubMed]
- Olusanya, T.O.B.; Haj Ahmad, R.R.; Ibegbu, D.M.; Smith, J.R.; Elkordy, A.A. Liposomal Drug Delivery Systems and Anticancer Drugs. Molecules 2018, 23, 907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- La-Beck, N.M.; Liu, X.; Wood, L.M. Harnessing Liposome Interactions With the Immune System for the Next Breakthrough in Cancer Drug Delivery. Front. Pharmacol. 2019, 10. [Google Scholar] [CrossRef] [Green Version]
- Merino, M.; Lozano, T.; Casares, N.; Lana, H.; Troconiz, I.F.; Ten Hagen, T.L.M.; Kochan, G.; Berraondo, P.; Zalba, S.; Garrido, M.J. Dual activity of PD-L1 targeted Doxorubicin immunoliposomes promoted an enhanced efficacy of the antitumor immune response in melanoma murine model. J. Nanobiotechnol. 2021, 19, 102. [Google Scholar] [CrossRef] [PubMed]
- Leach, A.; Smyth, P.; Ferguson, L.; Steven, J.; Greene, M.K.; Branco, C.M.; McCann, A.P.; Porter, A.; Barelle, C.J.; Scott, C.J. Anti-DLL4 VNAR targeted nanoparticles for targeting of both tumour and tumour associated vasculature. Nanoscale 2020, 12, 14751–14763. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Lim, H.K.; Tan, S.J.; Gautam, A.; Hou, H.W.; Ng, K.W.; Tan, N.S.; Tay, C.Y. Potent-By-Design: Amino Acids Mimicking Porous Nanotherapeutics with Intrinsic Anticancer Targeting Properties. Small 2020, 16, e2003757. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mc Erlain, T.; Burke, A.; Branco, C.M. Life after Cell Death—Survival and Survivorship Following Chemotherapy. Cancers 2021, 13, 2942. https://doi.org/10.3390/cancers13122942
Mc Erlain T, Burke A, Branco CM. Life after Cell Death—Survival and Survivorship Following Chemotherapy. Cancers. 2021; 13(12):2942. https://doi.org/10.3390/cancers13122942
Chicago/Turabian StyleMc Erlain, Tamara, Aileen Burke, and Cristina M. Branco. 2021. "Life after Cell Death—Survival and Survivorship Following Chemotherapy" Cancers 13, no. 12: 2942. https://doi.org/10.3390/cancers13122942