CeRNA Network Analysis Representing Characteristics of Different Tumor Environments Based on 1p/19q Codeletion in Oligodendrogliomas

 and
and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Processing Genomic and Clinical Profiles from TCGA Database
2.2. Analysis of Differentially Expressed Genes and Their Visualization
2.3. Weighted Gene Co-Expression Network Analysis
2.4. Construction of a CeRNA Network
2.5. Evaluation and Visualization of CeRNA Network
2.6. Gene Ontology and Pathway Enrichment Analysis
2.7. Statistical Analysis for Validation of the CeRNA Network
3. Results
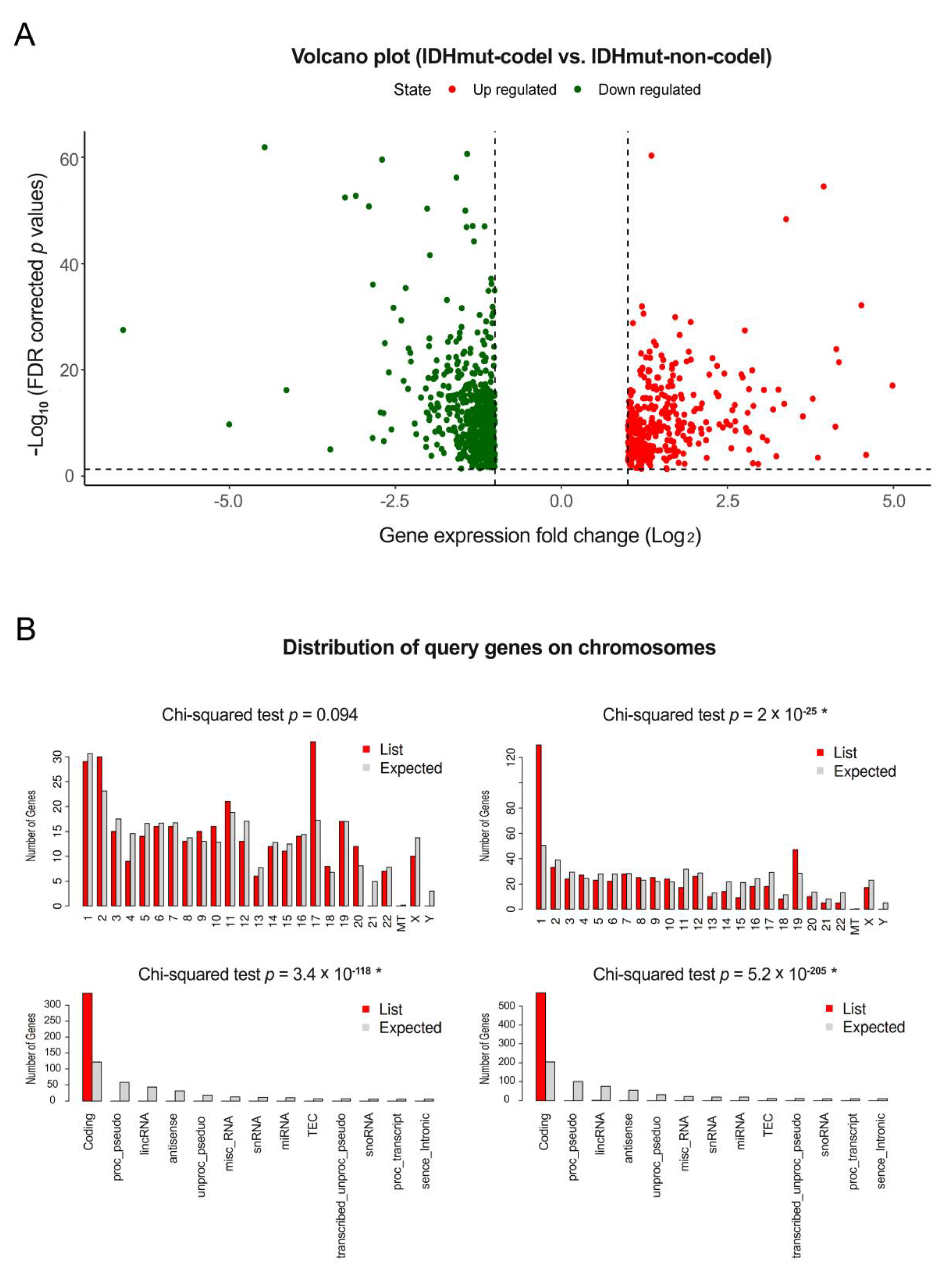
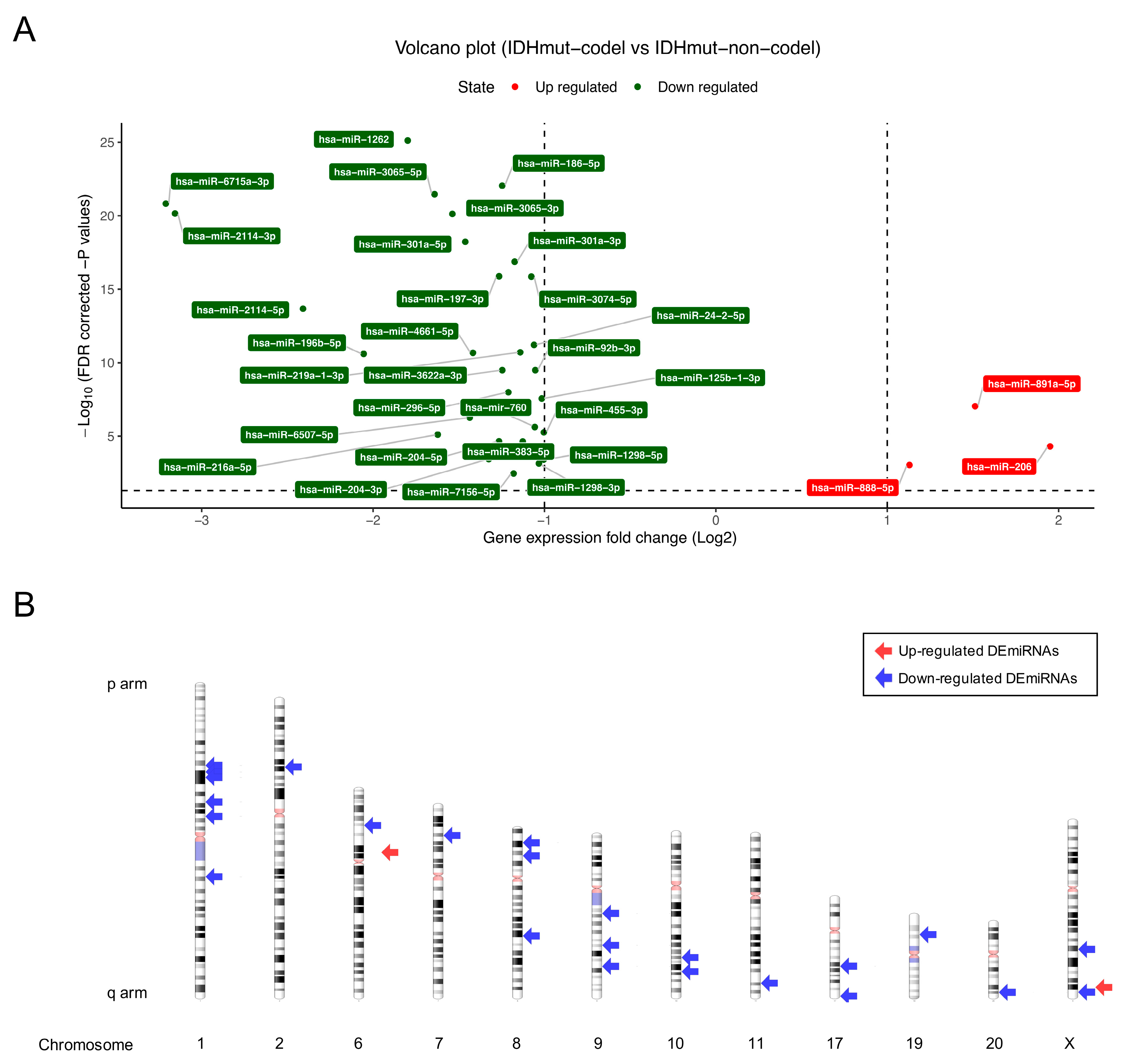
3.1. Differentially Expressed Gene Analysis of mRNAs, miRNAs, and ncRNAs
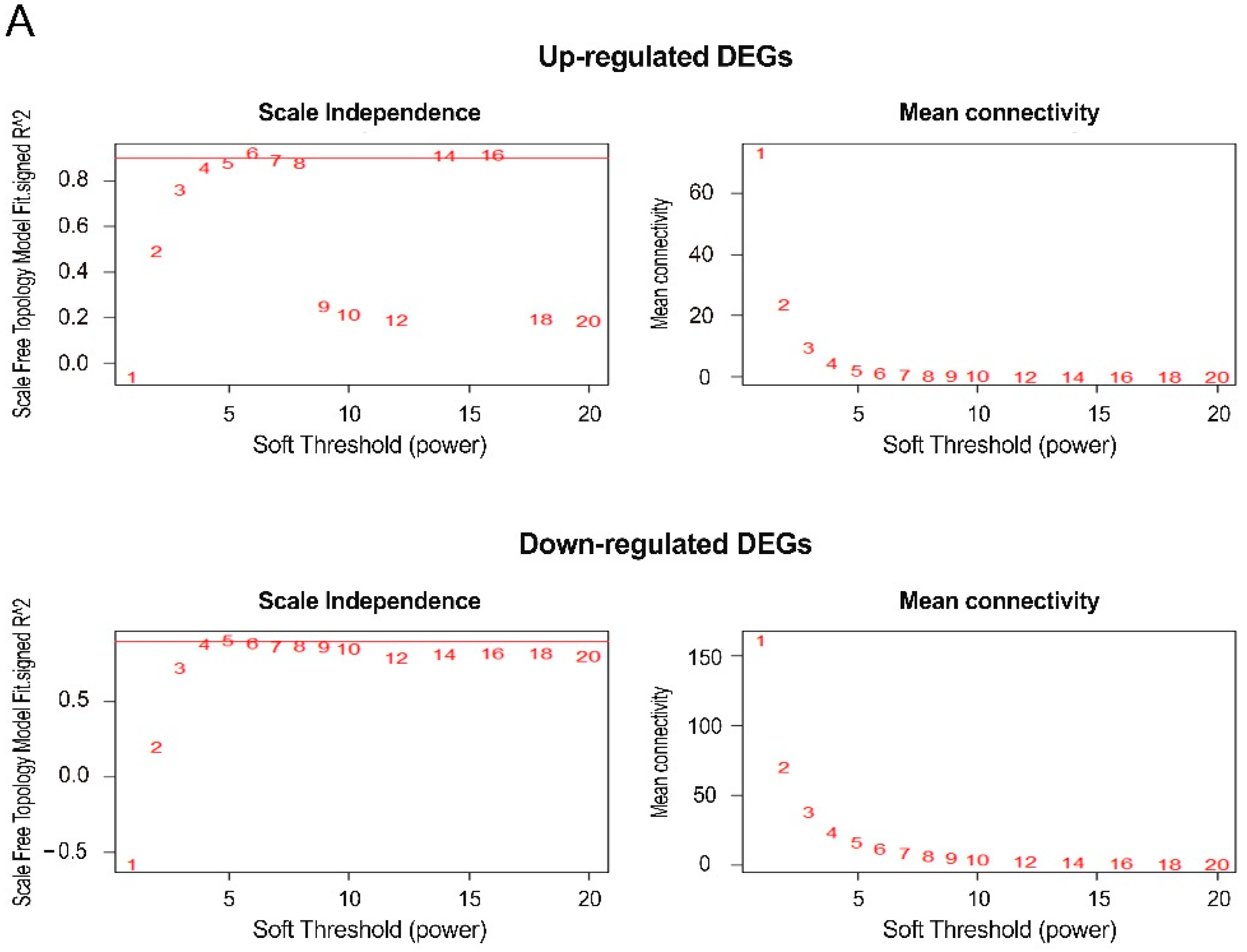
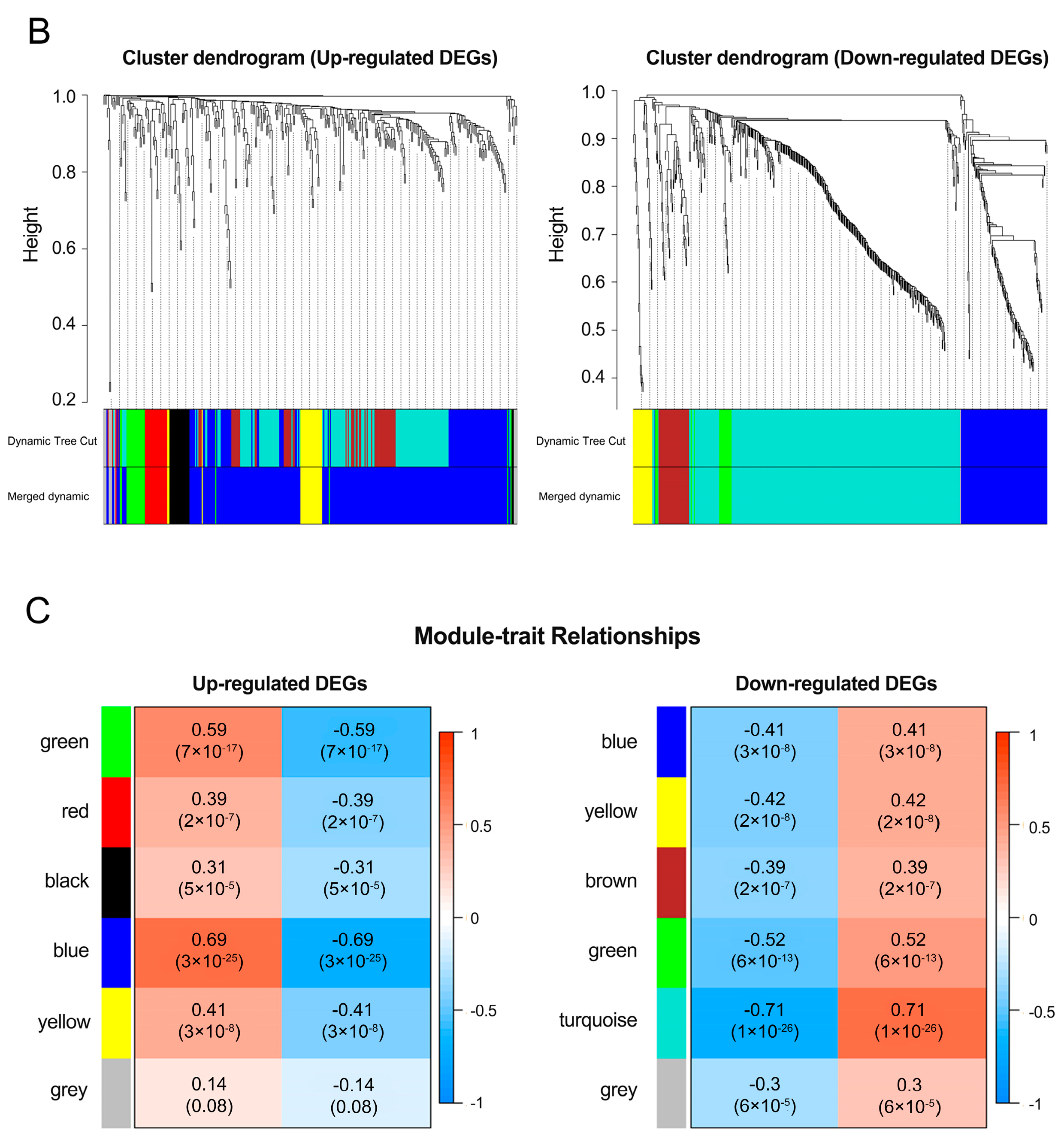
3.2. Analysis of a Weighted Co-Expression Network and Identification of Key Modules
3.3. Construction of CeRNA Network and Prediction of Function in Oligodendroglioma
3.4. Extending the CeRNA Network to Lower Grade Gliomas and Verifying the Clinical Significance through Survival Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Song, X.; Zhang, N.; Han, P.; Moon, B.S.; Lai, R.K.; Wang, K.; Lu, W. Circular RNA profile in gliomas revealed by identification tool UROBORUS. Nucleic Acids Res. 2016, 44, e87. [Google Scholar] [CrossRef]
- Xu, Y.; Geng, R.; Yuan, F.; Sun, Q.; Liu, B.; Chen, Q. Identification of differentially expressed key genes between glioblastoma and low-grade glioma by bioinformatics analysis. PeerJ 2019, 7, e6560. [Google Scholar] [CrossRef]
- Wesseling, P.; van den Bent, M.; Perry, A. Oligodendroglioma: Pathology, molecular mechanisms and markers. Acta Neuropathol. 2015, 129, 809–827. [Google Scholar] [CrossRef] [Green Version]
- Ostrom, Q.T.; Cioffi, G.; Gittleman, H.; Patil, N.; Waite, K.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS statistical report: Primary brain and other central nervous system tumors diagnosed in the United States in 2012–2016. Neuro Oncol. 2019, 21, v1–v100. [Google Scholar] [CrossRef]
- Iuchi, T.; Sugiyama, T.; Ohira, M.; Kageyama, H.; Yokoi, S.; Sakaida, T.; Hasegawa, Y.; Setoguchi, T.; Itami, M. Clinical significance of the 2016 WHO classification in Japanese patients with gliomas. Brain Tumor Pathol. 2018, 35, 71–80. [Google Scholar] [CrossRef]
- Labussiere, M.; Idbaih, A.; Wang, X.W.; Marie, Y.; Boisselier, B.; Falet, C.; Paris, S.; Laffaire, J.; Carpentier, C.; Criniere, E.; et al. All the 1p19q codeleted gliomas are mutated on IDH1 or IDH2. Neurology 2010, 74, 1886–1890. [Google Scholar] [CrossRef]
- Azar, S.; Leventoux, N.; Ripoll, C.; Rigau, V.; Goze, C.; Lorcy, F.; Bauchet, L.; Duffau, H.; Guichet, P.O.; Rothhut, B.; et al. Cellular and molecular characterization of IDH1-mutated diffuse low grade gliomas reveals tumor heterogeneity and absence of EGFR/PDGFRalpha activation. Glia 2018, 66, 239–255. [Google Scholar] [CrossRef]
- Li, Y.X.; Shi, Z.; Aibaidula, A.; Chen, H.; Tang, Q.; Li, K.K.; Chung, N.Y.; Chan, D.T.; Poon, W.S.; Mao, Y.; et al. Not all 1p/19q non-codeleted oligodendroglial tumors are astrocytic. Oncotarget 2016, 7, 64615–64630. [Google Scholar] [CrossRef]
- Jenkins, R.B.; Blair, H.; Ballman, K.V.; Giannini, C.; Arusell, R.M.; Law, M.; Flynn, H.; Passe, S.; Felten, S.; Brown, P.D.; et al. A t(1;19)(q10;p10) mediates the combined deletions of 1p and 19q and predicts a better prognosis of patients with oligodendroglioma. Cancer Res. 2006, 66, 9852–9861. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Yan, C.; Huang, L.; Qiu, X.; Wang, Z.; Jiang, T. Molecular prognostic factors of anaplastic oligodendroglial tumors and its relationship: A single institutional review of 77 patients from China. Neuro Oncol. 2012, 14, 109–116. [Google Scholar] [CrossRef] [Green Version]
- Gladitz, J.; Klink, B.; Seifert, M. Network-based analysis of oligodendrogliomas predicts novel cancer gene candidates within the region of the 1p/19q co-deletion. Acta Neuropathol. Commun. 2018, 6, 49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cairncross, J.G.; Wang, M.; Jenkins, R.B.; Shaw, E.G.; Giannini, C.; Brachman, D.G.; Buckner, J.C.; Fink, K.L.; Souhami, L.; Laperriere, N.J.; et al. Benefit from procarbazine, lomustine, and vincristine in oligodendroglial tumors is associated with mutation of IDH. J. Clin. Oncol. 2014, 32, 783–790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clark, V.E.; Cahill, D.P. Extent of resection versus molecular classification: What matters when? Neurosurg. Clin. N. Am. 2019, 30, 95–101. [Google Scholar] [CrossRef]
- Dubbink, H.J.; Atmodimedjo, P.N.; Kros, J.M.; French, P.J.; Sanson, M.; Idbaih, A.; Wesseling, P.; Enting, R.; Spliet, W.; Tijssen, C.; et al. Molecular classification of anaplastic oligodendroglioma using next-generation sequencing: A report of the prospective randomized EORTC Brain Tumor Group 26951 phase III trial. Neuro Oncol. 2016, 18, 388–400. [Google Scholar] [CrossRef]
- Wijnenga, M.M.J.; French, P.J.; Dubbink, H.J.; Dinjens, W.N.M.; Atmodimedjo, P.N.; Kros, J.M.; Smits, M.; Gahrmann, R.; Rutten, G.J.; Verheul, J.B.; et al. The impact of surgery in molecularly defined low-grade glioma: An integrated clinical, radiological, and molecular analysis. Neuro Oncol. 2018, 20, 103–112. [Google Scholar] [CrossRef] [Green Version]
- Van der Voort, S.R.; Incekara, F.; Wijnenga, M.M.J.; Kapas, G.; Gardeniers, M.; Schouten, J.W.; Starmans, M.P.A.; Nandoe Tewarie, R.; Lycklama, G.J.; French, P.J.; et al. Predicting the 1p/19q codeletion status of presumed low-grade glioma with an externally validated machine learning algorithm. Clin. Cancer Res. 2019, 25, 7455–7462. [Google Scholar] [CrossRef] [Green Version]
- Djebali, S.; Davis, C.A.; Merkel, A.; Dobin, A.; Lassmann, T.; Mortazavi, A.; Tanzer, A.; Lagarde, J.; Lin, W.; Schlesinger, F.; et al. Landscape of transcription in human cells. Nature 2012, 489, 101–108. [Google Scholar] [CrossRef]
- Wang, C.J.; Zhu, C.C.; Xu, J.; Wang, M.; Zhao, W.Y.; Liu, Q.; Zhao, G.; Zhang, Z.Z. The lncRNA UCA1 promotes proliferation, migration, immune escape and inhibits apoptosis in gastric cancer by sponging anti-tumor miRNAs. Mol. Cancer 2019, 18, 115. [Google Scholar] [CrossRef] [Green Version]
- Balas, M.M.; Johnson, A.M. Exploring the mechanisms behind long noncoding RNAs and cancer. Noncoding RNA Res. 2018, 3, 108–117. [Google Scholar] [CrossRef]
- Hu, X.; Yang, L.; Mo, Y.Y. Role of pseudogenes in tumorigenesis. Cancers 2018, 10, 256. [Google Scholar] [CrossRef] [Green Version]
- Wright, M.W.; Bruford, E.A. Naming ‘junk’: Human non-protein coding RNA (ncRNA) gene nomenclature. Hum. Genom. 2011, 5, 90–98. [Google Scholar] [CrossRef] [Green Version]
- Iyer, M.K.; Niknafs, Y.S.; Malik, R.; Singhal, U.; Sahu, A.; Hosono, Y.; Barrette, T.R.; Prensner, J.R.; Evans, J.R.; Zhao, S.; et al. The landscape of long noncoding RNAs in the human transcriptome. Nat. Genet. 2015, 47, 199–208. [Google Scholar] [CrossRef]
- Jiang, M.C.; Ni, J.J.; Cui, W.Y.; Wang, B.Y.; Zhuo, W. Emerging roles of lncRNA in cancer and therapeutic opportunities. Am. J. Cancer Res. 2019, 9, 1354–1366. [Google Scholar]
- Bartel, D.P. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef] [Green Version]
- Tay, Y.; Rinn, J.; Pandolfi, P.P. The multilayered complexity of ceRNA crosstalk and competition. Nature 2014, 505, 344–352. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Li, H.; Zheng, B.; Sun, L.; Yuan, Y.; Xing, C. Competitive endogenous RNA (ceRNA) regulation network of lncRNA-miRNA-mRNA in colorectal carcinogenesis. Dig. Dis. Sci. 2019, 64, 1868–1877. [Google Scholar] [CrossRef]
- Yao, Y.; Zhang, T.; Qi, L.; Zhou, C.; Wei, J.; Feng, F.; Liu, R.; Sun, C. Integrated analysis of co-expression and ceRNA network identifies five lncRNAs as prognostic markers for breast cancer. J. Cell Mol. Med. 2019, 23, 8410–8419. [Google Scholar] [CrossRef] [Green Version]
- Yang, R.; Xing, L.; Wang, M.; Chi, H.; Zhang, L.; Chen, J. Comprehensive analysis of differentially expressed profiles of lncRNAs/mRNAs and miRNAs with associated ceRNA networks in triple-negative breast cancer. Cell Physiol. Biochem. 2018, 50, 473–488. [Google Scholar] [CrossRef]
- Poliseno, L.; Salmena, L.; Zhang, J.; Carver, B.; Haveman, W.J.; Pandolfi, P.P. A coding-independent function of gene and pseudogene mRNAs regulates tumour biology. Nature 2010, 465, 1033–1038. [Google Scholar] [CrossRef] [Green Version]
- Guo, C.; Song, W.Q.; Sun, P.; Jin, L.; Dai, H.Y. LncRNA-GAS5 induces PTEN expression through inhibiting miR-103 in endometrial cancer cells. J. Biomed. Sci. 2015, 22, 100. [Google Scholar] [CrossRef] [Green Version]
- Yang, C.; Yuan, W.; Yang, X.; Li, P.; Wang, J.; Han, J.; Tao, J.; Li, P.; Yang, H.; Lv, Q.; et al. Circular RNA circ-ITCH inhibits bladder cancer progression by sponging miR-17/miR-224 and regulating p21, PTEN expression. Mol. Cancer 2018, 17, 19. [Google Scholar] [CrossRef] [Green Version]
- Qiao, Q.; Li, H. LncRNA FER1L4 suppresses cancer cell proliferation and cycle by regulating PTEN expression in endometrial carcinoma. Biochem. Biophys. Res. Commun. 2016, 478, 507–512. [Google Scholar] [CrossRef]
- Rinn, J.L.; Chang, H.Y. Genome regulation by long noncoding RNAs. Annu. Rev. Biochem. 2012, 81, 145–166. [Google Scholar] [CrossRef] [Green Version]
- Wang, P.; Ren, Z.; Sun, P. Overexpression of the long non-coding RNA MEG3 impairs in vitro glioma cell proliferation. J. Cell Biochem. 2012, 113, 1868–1874. [Google Scholar] [CrossRef]
- Gong, W.; Zheng, J.; Liu, X.; Ma, J.; Liu, Y.; Xue, Y. Knockdown of NEAT1 restrained the malignant progression of glioma stem cells by activating microRNA let-7e. Oncotarget 2016, 7, 62208–62223. [Google Scholar] [CrossRef] [Green Version]
- Ding, Y.; Wang, X.; Pan, J.; Ji, M.; Luo, Z.; Zhao, P.; Zhang, Y.; Wang, G. Aberrant expression of long non-coding RNAs (lncRNAs) is involved in brain glioma development. Arch. Med. Sci. 2020, 16, 177–188. [Google Scholar] [CrossRef]
- Liang, Q.; Li, X.; Guan, G.; Xu, X.; Chen, C.; Cheng, P.; Cheng, W.; Wu, A. Long non-coding RNA, HOTAIRM1, promotes glioma malignancy by forming a ceRNA network. Aging 2019, 11, 6805–6838. [Google Scholar] [CrossRef]
- Wu, Y.; Qian, Z. Long non-coding RNAs (lncRNAs) and microRNAs regulatory pathways in the tumorigenesis and pathogenesis of glioma. Discov. Med. 2019, 28, 129–138. [Google Scholar]
- Colaprico, A.; Silva, T.C.; Olsen, C.; Garofano, L.; Cava, C.; Garolini, D.; Sabedot, T.S.; Malta, T.M.; Pagnotta, S.M.; Castiglioni, I.; et al. TCGAbiolinks: An R/Bioconductor package for integrative analysis of TCGA data. Nucleic Acids Res. 2016, 44, e71. [Google Scholar] [CrossRef]
- Silva, T.C.; Colaprico, A.; Olsen, C.; D’Angelo, F.; Bontempi, G.; Ceccarelli, M.; Noushmehr, H. TCGA Workflow: Analyze cancer genomics and epigenomics data using Bioconductor packages. F1000Res 2016, 5, 1542. [Google Scholar] [CrossRef]
- Xu, T.; Su, N.; Liu, L.; Zhang, J.; Wang, H.; Zhang, W.; Gui, J.; Yu, K.; Li, J.; Le, T.D. miRBaseConverter: An R/Bioconductor package for converting and retrieving miRNA name, accession, sequence and family information in different versions of miRBase. BMC Bioinf. 2018, 19, 514. [Google Scholar] [CrossRef] [Green Version]
- Ge, S.X.; Jung, D.; Yao, R. ShinyGO: A graphical gene-set enrichment tool for animals and plants. Bioinformatics 2020, 36, 2628–2629. [Google Scholar] [CrossRef] [PubMed]
- Coordinators, N.R. Database resources of the national center for biotechnology information. Nucleic Acids Res. 2016, 44, D7–D19. [Google Scholar] [CrossRef] [Green Version]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinformatics 2008, 9, 559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, Y.; Liu, T.; Cui, T.; Wang, Z.; Zhang, Y.; Tan, P.; Huang, Y.; Yu, J.; Wang, D. RNAInter in 2020: RNA interactome repository with increased coverage and annotation. Nucleic Acids Res. 2020, 48, D189–D197. [Google Scholar] [CrossRef] [PubMed]
- Li, J.H.; Liu, S.; Zhou, H.; Qu, L.H.; Yang, J.H. starBase v2.0: Decoding miRNA-ceRNA, miRNA-ncRNA and protein-RNA interaction networks from large-scale CLIP-Seq data. Nucleic Acids Res. 2014, 42, D92–D97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tokar, T.; Pastrello, C.; Rossos, A.E.M.; Abovsky, M.; Hauschild, A.C.; Tsay, M.; Lu, R.; Jurisica, I. mirDIP 4.1-integrative database of human microRNA target predictions. Nucleic Acids Res. 2018, 46, D360–D370. [Google Scholar] [CrossRef]
- Karagkouni, D.; Paraskevopoulou, M.D.; Chatzopoulos, S.; Vlachos, I.S.; Tastsoglou, S.; Kanellos, I.; Papadimitriou, D.; Kavakiotis, I.; Maniou, S.; Skoufos, G.; et al. DIANA-TarBase v8: A decade-long collection of experimentally supported miRNA-gene interactions. Nucleic Acids Res. 2018, 46, D239–D245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sticht, C.; De La Torre, C.; Parveen, A.; Gretz, N. miRWalk: An online resource for prediction of microRNA binding sites. PLoS ONE 2018, 13, e0206239. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Bindea, G.; Mlecnik, B.; Hackl, H.; Charoentong, P.; Tosolini, M.; Kirilovsky, A.; Fridman, W.H.; Pages, F.; Trajanoski, Z.; Galon, J. ClueGO: A Cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics 2009, 25, 1091–1093. [Google Scholar] [CrossRef] [Green Version]
- Tang, Z.; Li, C.; Kang, B.; Gao, G.; Li, C.; Zhang, Z. GEPIA: A web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 2017, 45, W98–W102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization classification of tumors of the central nervous system: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pardo, L.A.; Stuhmer, W. The roles of K(+) channels in cancer. Nat. Rev. Cancer 2014, 14, 39–48. [Google Scholar] [CrossRef] [PubMed]
- Prevarskaya, N.; Skryma, R.; Shuba, Y. Ion channels in cancer: Are cancer hallmarks oncochannelopathies? Physiol. Rev. 2018, 98, 559–621. [Google Scholar] [CrossRef] [Green Version]
- Prevarskaya, N.; Skryma, R.; Shuba, Y. Ion channels and the hallmarks of cancer. Trends Mol. Med. 2010, 16, 107–121. [Google Scholar] [CrossRef]
- Talley, E.M.; Solorzano, G.; Lei, Q.; Kim, D.; Bayliss, D.A. Cns distribution of members of the two-pore-domain (KCNK) potassium channel family. J. Neurosci. 2001, 21, 7491–7505. [Google Scholar] [CrossRef]
- Kanjhan, R.; Pow, D.V.; Noakes, P.G.; Bellingham, M.C. The two-pore domain K+ channel TASK-1 is closely associated with brain barriers and meninges. J. Mol. Histol. 2010, 41, 315–323. [Google Scholar] [CrossRef]
- Williams, S.; Bateman, A.; O’Kelly, I. Altered expression of two-pore domain potassium (K2P) channels in cancer. PLoS ONE 2013, 8, e74589. [Google Scholar] [CrossRef]
- Hansen, J.B. Towards selective Kir6.2/SUR1 potassium channel openers, medicinal chemistry and therapeutic perspectives. Curr. Med. Chem. 2006, 13, 361–376. [Google Scholar] [CrossRef]
- De Franco, E.; Saint-Martin, C.; Brusgaard, K.; Knight Johnson, A.E.; Aguilar-Bryan, L.; Bowman, P.; Arnoux, J.B.; Larsen, A.R.; May, S.; Greeley, S.A.W.; et al. Update of variants identified in the pancreatic beta-cell KATP channel genes KCNJ11 and ABCC8 in individuals with congenital hyperinsulinism and diabetes. Hum. Mutat. 2020, 41, 884–905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, D.; Liu, H.; Ruzi, A.; Gao, G.; Nasir, A.; Liu, Y.; Yang, F.; Wu, F.; Xu, G.; Li, Y.X. Modeling congenital hyperinsulinism with ABCC8-deficient human embryonic stem cells generated by CRISPR/Cas9. Sci. Rep. 2017, 7, 3156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, G.M.; Sung, M.W.; Shyng, S.L. Pharmacological chaperones of ATP-sensitive potassium channels: Mechanistic insight from cryoEM structures. Mol. Cell Endocrinol. 2020, 502, 110667. [Google Scholar] [CrossRef] [PubMed]
- Unal, S.; Gonulal, D.; Ucakturk, A.; Siyah Bilgin, B.; Flanagan, S.E.; Gurbuz, F.; Tayfun, M.; Elmaogullari, S.; Arasli, A.; Demirel, F.; et al. A novel homozygous mutation in the KCNJ11 gene of a neonate with congenital hyperinsulinism and successful management with sirolimus. J. Clin. Res. Pediatr. Endocrinol. 2016, 8, 478–481. [Google Scholar] [CrossRef]
- Song, J.; Yang, Y.; Mauvais-Jarvis, F.; Wang, Y.P.; Niu, T. KCNJ11, ABCC8 and TCF7L2 polymorphisms and the response to sulfonylurea treatment in patients with type 2 diabetes: A bioinformatics assessment. BMC Med. Genet. 2017, 18, 64. [Google Scholar] [CrossRef]
- Poloz, Y.; Stambolic, V. Obesity and cancer, a case for insulin signaling. Cell Death Dis. 2015, 6, e2037. [Google Scholar] [CrossRef] [Green Version]
- Yu, H.; Rohan, T. Role of the insulin-like growth factor family in cancer development and progression. J. Natl. Cancer Inst. 2000, 92, 1472–1489. [Google Scholar] [CrossRef]
- Lu, P.; Takai, K.; Weaver, V.M.; Werb, Z. Extracellular matrix degradation and remodeling in development and disease. Cold Spring Harb. Perspect Biol. 2011, 3. [Google Scholar] [CrossRef]
- Leins, A.; Riva, P.; Lindstedt, R.; Davidoff, M.S.; Mehraein, P.; Weis, S. Expression of tenascin-C in various human brain tumors and its relevance for survival in patients with astrocytoma. Cancer 2003, 98, 2430–2439. [Google Scholar] [CrossRef]
- Xia, S.; Lal, B.; Tung, B.; Wang, S.; Goodwin, C.R.; Laterra, J. Tumor microenvironment tenascin-C promotes glioblastoma invasion and negatively regulates tumor proliferation. Neuro Oncol. 2016, 18, 507–517. [Google Scholar] [CrossRef] [Green Version]
- Sarkar, S.; Zemp, F.J.; Senger, D.; Robbins, S.M.; Yong, V.W. ADAM-9 is a novel mediator of tenascin-C-stimulated invasiveness of brain tumor-initiating cells. Neuro Oncol. 2015, 17, 1095–1105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El Ayachi, I.; Baeza, N.; Fernandez, C.; Colin, C.; Scavarda, D.; Pesheva, P.; Figarella-Branger, D. KIAA0510, the 3′-untranslated region of the tenascin-R gene, and tenascin-R are overexpressed in pilocytic astrocytomas. Neuropathol. Appl. Neurobiol. 2010, 36, 399–410. [Google Scholar] [CrossRef] [PubMed]
- Wong, K.K.; Rostomily, R.; Wong, S.T.C. Prognostic gene discovery in glioblastoma patients using deep learning. Cancers 2019, 11, 53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarkar, S.; Nuttall, R.K.; Liu, S.; Edwards, D.R.; Yong, V.W. Tenascin-C stimulates glioma cell invasion through matrix metalloproteinase-12. Cancer Res. 2006, 66, 11771–11780. [Google Scholar] [CrossRef] [Green Version]
- Sugimoto, W.; Itoh, K.; Hirata, H.; Abe, Y.; Torii, T.; Mitsui, Y.; Budirahardja, Y.; Tanaka, N.; Kawauchi, K. MMP24 as a target of YAP is a potential prognostic factor in cancer patients. Bioengineering 2020, 7, 18. [Google Scholar] [CrossRef] [Green Version]
- Llano, E.; Pendas, A.M.; Freije, J.P.; Nakano, A.; Knauper, V.; Murphy, G.; Lopez-Otin, C. Identification and characterization of human MT5-MMP, a new membrane-bound activator of progelatinase a overexpressed in brain tumors. Cancer Res. 1999, 59, 2570–2576. [Google Scholar]
- Hagemann, C.; Anacker, J.; Ernestus, R.I.; Vince, G.H. A complete compilation of matrix metalloproteinase expression in human malignant gliomas. World J. Clin. Oncol. 2012, 3, 67–79. [Google Scholar] [CrossRef]
- Qu, M.; Yu, J.; Liu, H.; Ren, Y.; Ma, C.; Bu, X.; Lan, Q. The candidate tumor suppressor gene SLC8A2 inhibits invasion, angiogenesis and growth of glioblastoma. Mol. Cells 2017, 40, 761–772. [Google Scholar] [CrossRef] [Green Version]
- Gao, J.; Yin, X.; Yu, X.; Dai, C.; Zhou, F. Long noncoding LINC01551 promotes hepatocellular carcinoma cell proliferation, migration, and invasion by acting as a competing endogenous RNA of microRNA-122-5p to regulate ADAM10 expression. J. Cell Biochem. 2019, 120, 16393–16407. [Google Scholar] [CrossRef]
- Yue, X.; Cao, D.; Lan, F.; Pan, Q.; Xia, T.; Yu, H. MiR-301a is activated by the Wnt/beta-catenin pathway and promotes glioma cell invasion by suppressing SEPT7. Neuro Oncol. 2016, 18, 1288–1296. [Google Scholar] [CrossRef] [Green Version]
- Zheng, J.Z.; Huang, Y.N.; Yao, L.; Liu, Y.R.; Liu, S.; Hu, X.; Liu, Z.B.; Shao, Z.M. Elevated miR-301a expression indicates a poor prognosis for breast cancer patients. Sci. Rep. 2018, 8, 2225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, W.Q.; Chen, J.; Fang, W.; Yang, X.Q.; Zhu, A.; Zhang, D.; Zhong, H.L.; Yang, B.; Luo, Z. LncRNA INHBA-AS1 promotes cell growth, migration, and invasion of oral squamous cell carcinoma by sponging miR-143-3p. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 1821–1828. [Google Scholar] [CrossRef] [PubMed]
- Xie, K.; Chen, M.; Zhu, M.; Wang, C.; Qin, N.; Liang, C.; Song, C.; Dai, J.; Jin, G.; Shen, H.; et al. A polymorphism in miR-1262 regulatory region confers the risk of lung cancer in Chinese population. Int. J. Cancer 2017, 141, 958–966. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Mu, S.; Zhao, Q.; Xue, L.; Wang, S. Identification of differentially expressed microRNAs and the potential of microRNA-455-3p as a novel prognostic biomarker in glioma. Oncol. Lett. 2019, 18, 6150–6156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhan, T.; Huang, X.; Tian, X.; Chen, X.; Ding, Y.; Luo, H.; Zhang, Y. Downregulation of microRNA-455-3p links to proliferation and drug resistance of pancreatic cancer cells via targeting TAZ. Mol. Ther. Nucleic Acids 2018, 10, 215–226. [Google Scholar] [CrossRef] [Green Version]
- Han, B.W.; Ye, H.; Wei, P.P.; He, B.; Han, C.; Chen, Z.H.; Chen, Y.Q.; Wang, W.T. Global identification and characterization of lncRNAs that control inflammation in malignant cholangiocytes. BMC Genom. 2018, 19, 735. [Google Scholar] [CrossRef]
- Li, T.; Pan, H.; Li, R. The dual regulatory role of miR-204 in cancer. Tumour Biol. 2016, 37, 11667–11677. [Google Scholar] [CrossRef] [Green Version]
- Ni, J.S.; Zheng, H.; Huang, Z.P.; Hong, Y.G.; Ou, Y.L.; Tao, Y.P.; Wang, M.C.; Wang, Z.G.; Yang, Y.; Zhou, W.P. MicroRNA-197-3p acts as a prognostic marker and inhibits cell invasion in hepatocellular carcinoma. Oncol. Lett. 2019, 17, 2317–2327. [Google Scholar] [CrossRef] [Green Version]
- Wang, F.; Jiang, H.; Wang, S.; Chen, B. Dual functional microRNA-186-5p targets both FGF2 and RelA to suppress tumorigenesis of glioblastoma multiforme. Cell Mol. Neurobiol. 2017, 37, 1433–1442. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
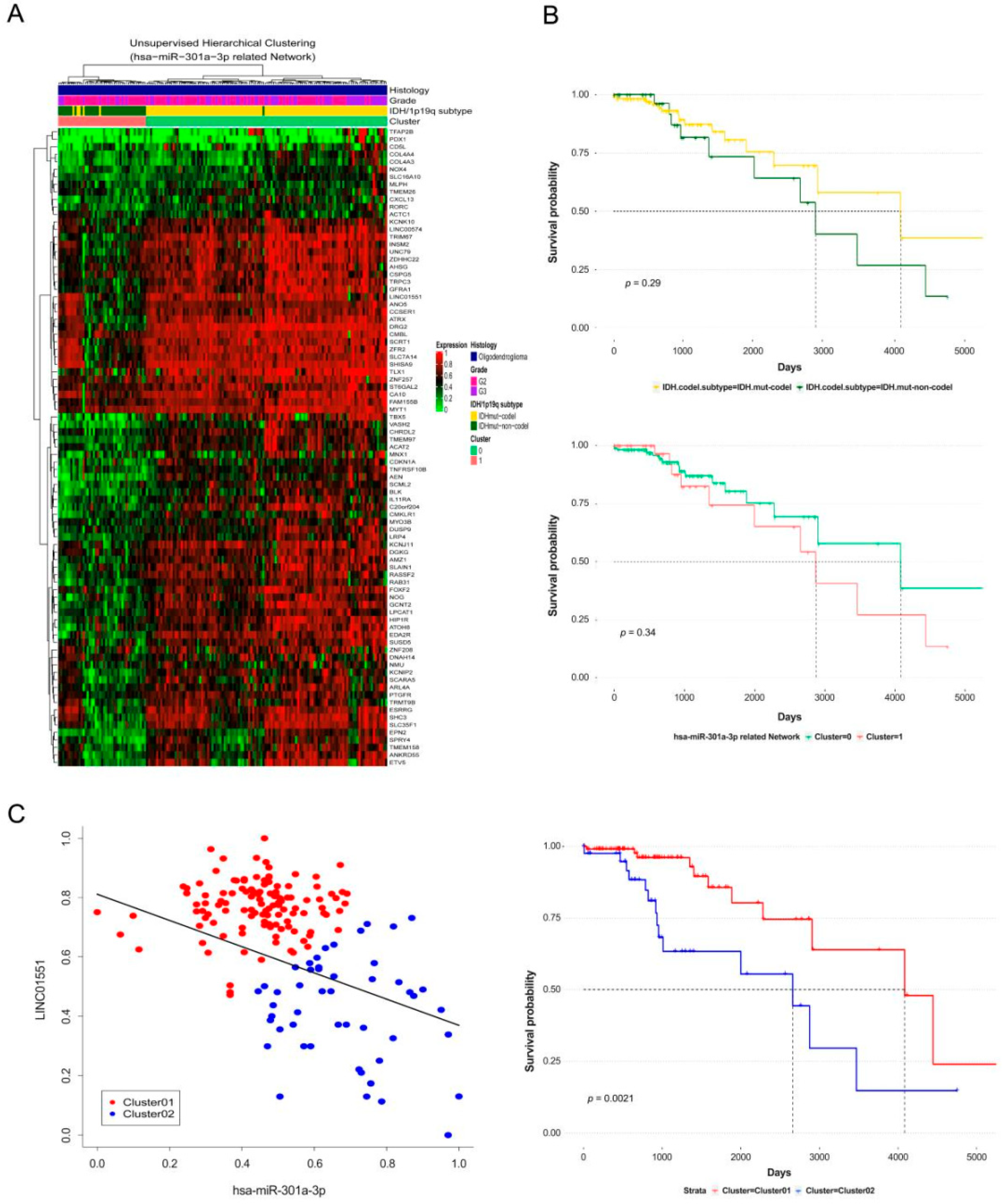{kind=link}
| Network | Source miRNAs | No. of Target Coding Genes | No. of Target Non-Coding Genes | ARI | |
|---|---|---|---|---|---|
| Oligodenroglioma (k * = 2) | Lower Grade Glioma (k * = 3) | ||||
| Network01 | hsa-miR-296-5p | 78 | 6 | 0.921 | 0.806 |
| Network02 | hsa-miR-455-3p | 68 | 1 | 0.896 | 0.888 |
| Network03 | hsa-miR-760 | 92 | 1 | 0.896 | 0.789 |
| Network04 | hsa-miR-1298-5p | 43 | 4 | 0.896 | 0.761 |
| Network05 | hsa-miR-197-3p | 78 | 5 | 0.872 | 0.770 |
| Network06 | hsa-miR-301a-5p | 25 | 2 | 0.872 | 0.728 |
| Network07 | hsa-miR-1262 | 62 | 2 | 0.872 | 0.724 |
| Network08 | hsa-miR-186-5p | 99 | 9 | 0.871 | 0.747 |
| Network09 | hsa-miR-301a-3p | 83 | 2 | 0.847 | 0.834 |
| Network10 | hsa-miR-383-5p | 97 | 1 | 0.824 | 0.714 |
| Network11 | hsa-miR-2114-3p | 30 | 2 | 0.801 | 0.497 |
| Network12 | hsa-miR-204-5p | 138 | 7 | 0.801 | 0.756 |
| Network13 | hsa-miR-7156-5p | 31 | 6 | 0.800 | 0.594 |
| Network14 | hsa-miR-92b-3p | 76 | 4 | 0.778 | 0.704 |
| Network15 | hsa-miR-3074-5p | 31 | 9 | 0.778 | 0.637 |
| Network16 | hsa-miR-1298-3p | 30 | 2 | 0.692 | 0.671 |
| Subtype | miRNA Symbol | CeRNA Symbol | First Cluster | Second Cluster | p | FDR | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| Median | 95% CI * | No. | Median | 95% CI * | No. | |||||
| OD | hsa-miR-186-5p | INHBA-AS1 | 4084 | 2907 to NA | 113 | 2660 | 1011 to NA | 48 | 0.002 | 0.023 |
| hsa-miR-301a-3p | LINC01551 | 4084 | 2907 to NA | 115 | 2660 | 1011 to NA | 46 | 0.002 | 0.023 | |
| hsa-miR-186-5p | ZNF876P | 4084 | 2907 to NA | 102 | 2660 | 1886 to NA | 59 | 0.023 | 0.168 | |
| hsa-miR-186-5p | TM4SF19-AS1 | 4084 | 2907 to NA | 107 | 2660 | 1886 to NA | 54 | 0.033 | 0.183 | |
| hsa-miR-186-5p | FZD10-AS1 | 4084 | 2907 to NA | 80 | 2660 | 1886 to NA | 81 | 0.055 | 0.211 | |
| hsa-miR-204-5p | NXF4 | 4084 | 2907 to NA | 98 | 2660 | 1886 to NA | 63 | 0.058 | 0.211 | |
| hsa-miR-301a-5p | SMCR5 | 4084 | 2907 to NA | 99 | 2875 | 1886 to NA | 62 | 0.113 | 0.356 | |
| hsa-miR-186-5p | LPAL2 | 4084 | 2907 to NA | 104 | 2660 | 1886 to NA | 57 | 0.150 | 0.366 | |
| hsa-miR-197-3p | SMCR5 | 4084 | 2907 to NA | 92 | 2875 | 2000 to NA | 69 | 0.148 | 0.366 | |
| hsa-miR-197-3p | CD81-AS1 | 4084 | 2907 to NA | 107 | 2660 | 1886 to NA | 54 | 0.235 | 0.518 | |
| hsa-miR-301a-5p | FZD10-AS1 | 2907 | 2875 to NA | 85 | 2660 | 1886 to NA | 76 | 0.286 | 0.571 | |
| hsa-miR-186-5p | LINC01460 | 4084 | 2907 to NA | 97 | 2875 | 2000 to NA | 64 | 0.364 | 0.575 | |
| hsa-miR-197-3p | LINC02076 | 4084 | 2907 to NA | 98 | 2875 | 2000 to NA | 63 | 0.366 | 0.575 | |
| hsa-miR-760 | LPAL2 | 3470 | 2907 to NA | 76 | 2875 | 2000 to NA | 85 | 0.349 | 0.575 | |
| hsa-miR-204-5p | TM4SF19-AS1 | 4084 | 4084 to NA | 81 | 2875 | 2000 to NA | 80 | 0.410 | 0.602 | |
| hsa-miR-204-5p | EPN2-AS1 | 3470 | 2660 to NA | 86 | 2875 | 2000 to NA | 75 | 0.472 | 0.615 | |
| hsa-miR-204-5p | SMCR5 | 4084 | 2660 to NA | 86 | 2875 | 2000 to NA | 75 | 0.479 | 0.615 | |
| hsa-miR-204-5p | GUCY1B2 | 4084 | 2907 to NA | 101 | 2875 | 1401 to NA | 60 | 0.503 | 0.615 | |
| hsa-miR-92b-3p | TBX5-AS1 | 4084 | 2660 to NA | 71 | 2907 | 2282 to NA | 90 | 0.620 | 0.717 | |
| hsa-miR-197-3p | FZD10-AS1 | 3470 | 2907 to NA | 95 | 2875 | 2000 to NA | 66 | 0.694 | 0.763 | |
| hsa-miR-7156-5p | MROCKI | 4084 | 1886 to NA | 68 | 3470 | 2875 to NA | 93 | 0.831 | 0.871 | |
| hsa-miR-204-5p | MROCKI | 3470 | 2875 to NA | 89 | 2907 | 2000 to NA | 72 | 0.933 | 0.933 | |
| LGG | hsa-miR-1262 | CISTR | 4445 | 4084 to NA | 211 | 2000 | 1762 to 2875 | 256 | 0.000 | 0.000 |
| hsa-miR-186-5p | INHBA-AS1 | 4084 | 2907 to NA | 213 | 2286 | 1933 to 4412 | 254 | 0.000 | 0.002 | |
| hsa-miR-3074-5p | INHBA-AS1 | 4084 | 2875 to NA | 254 | 2286 | 1915 to NA | 213 | 0.000 | 0.002 | |
| hsa-miR-186-5p | LPAL2 | 4084 | 2907 to NA | 174 | 2433 | 1933 to 4412 | 293 | 0.002 | 0.005 | |
| hsa-miR-186-5p | ZNF876P | 4084 | 2907 to NA | 178 | 2433 | 1933 to 4412 | 289 | 0.006 | 0.012 | |
| hsa-miR-455-3p | LINC01586 | 4068 | 2907 to NA | 251 | 2286 | 1915 to NA | 216 | 0.005 | 0.012 | |
| hsa-miR-186-5p | LINC01460 | 4084 | 2907 to NA | 181 | 2433 | 1933 to 4412 | 286 | 0.014 | 0.026 | |
| hsa-miR-186-5p | TM4SF19-AS1 | 4084 | 2907 to NA | 186 | 2433 | 1933 to 4412 | 281 | 0.020 | 0.032 | |
| hsa-miR-204-5p | GUCY1B2 | 3470 | 1891 to NA | 230 | 2907 | 2286 to NA | 237 | 0.023 | 0.033 | |
| hsa-miR-301a-3p | LINC01551 | 4084 | 3470 to NA | 249 | 2235 | 1915 to 3978 | 218 | 0.029 | 0.037 | |
| hsa-miR-204-5p | NXF4 | 4084 | 2907 to NA | 214 | 2433 | 1933 to 4068 | 253 | 0.072 | 0.085 | |
| hsa-miR-301a-5p | FZD10-AS1 | 3470 | 2875 to NA | 216 | 2433 | 1933 to NA | 251 | 0.203 | 0.220 | |
| hsa-miR-197-3p | CD81-AS1 | 4068 | 2907 to NA | 202 | 2660 | 1933 to 4445 | 265 | 0.399 | 0.399 | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ahn, J.W.; Park, Y.; Kang, S.J.; Hwang, S.J.; Cho, K.G.; Lim, J.; Kwack, K. CeRNA Network Analysis Representing Characteristics of Different Tumor Environments Based on 1p/19q Codeletion in Oligodendrogliomas. Cancers 2020, 12, 2543. https://doi.org/10.3390/cancers12092543
Ahn JW, Park Y, Kang SJ, Hwang SJ, Cho KG, Lim J, Kwack K. CeRNA Network Analysis Representing Characteristics of Different Tumor Environments Based on 1p/19q Codeletion in Oligodendrogliomas. Cancers. 2020; 12(9):2543. https://doi.org/10.3390/cancers12092543
Chicago/Turabian StyleAhn, Ju Won, YoungJoon Park, Su Jung Kang, So Jung Hwang, Kyung Gi Cho, JaeJoon Lim, and KyuBum Kwack. 2020. "CeRNA Network Analysis Representing Characteristics of Different Tumor Environments Based on 1p/19q Codeletion in Oligodendrogliomas" Cancers 12, no. 9: 2543. https://doi.org/10.3390/cancers12092543