Comprehensive Cohort Analysis of Mutational Spectrum in Early Onset Breast Cancer Patients

 , , ,
, , ,
Abstract
:1. Introduction
2. Results
2.1. Structure of the EOBC Cohort
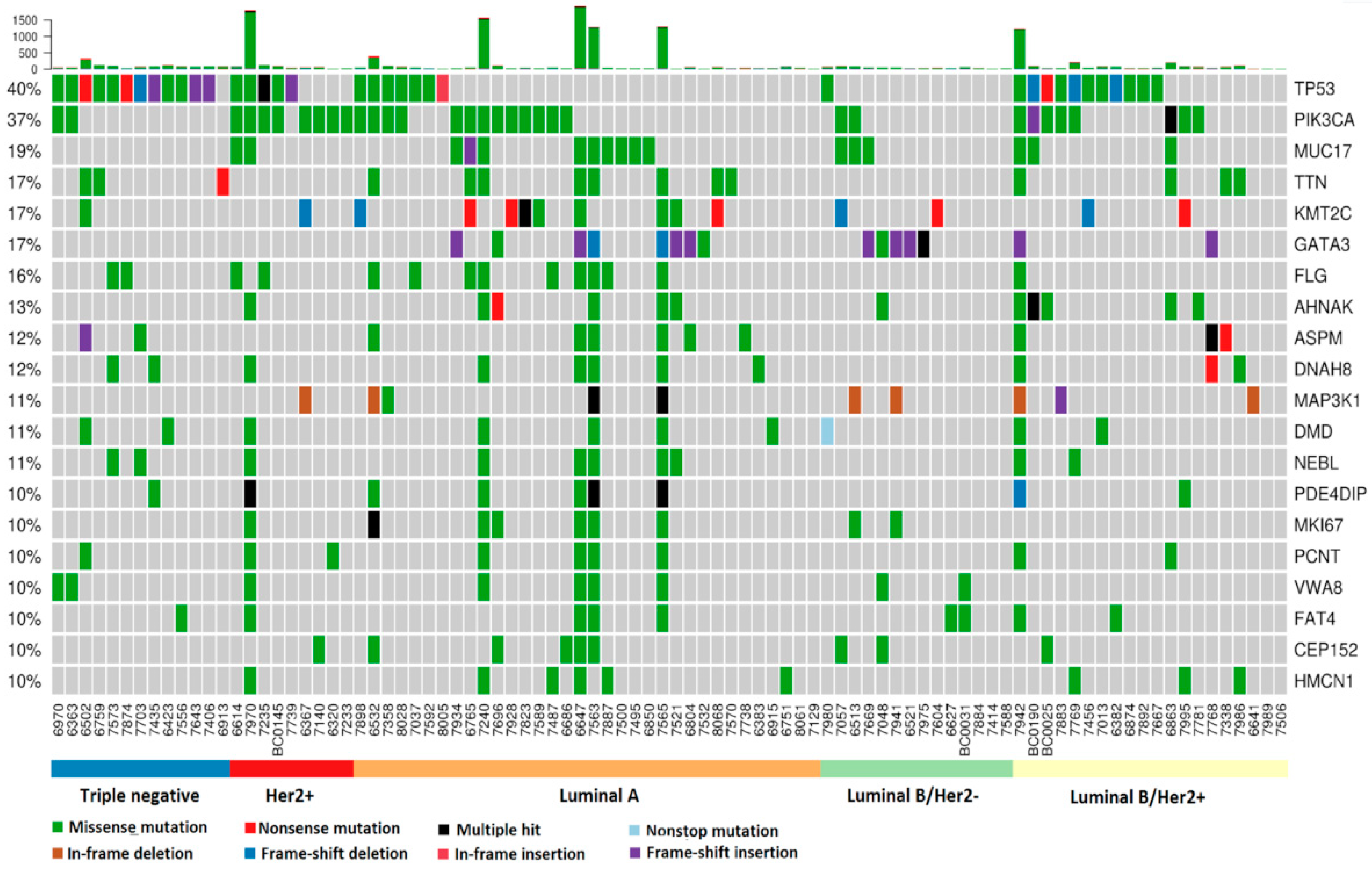
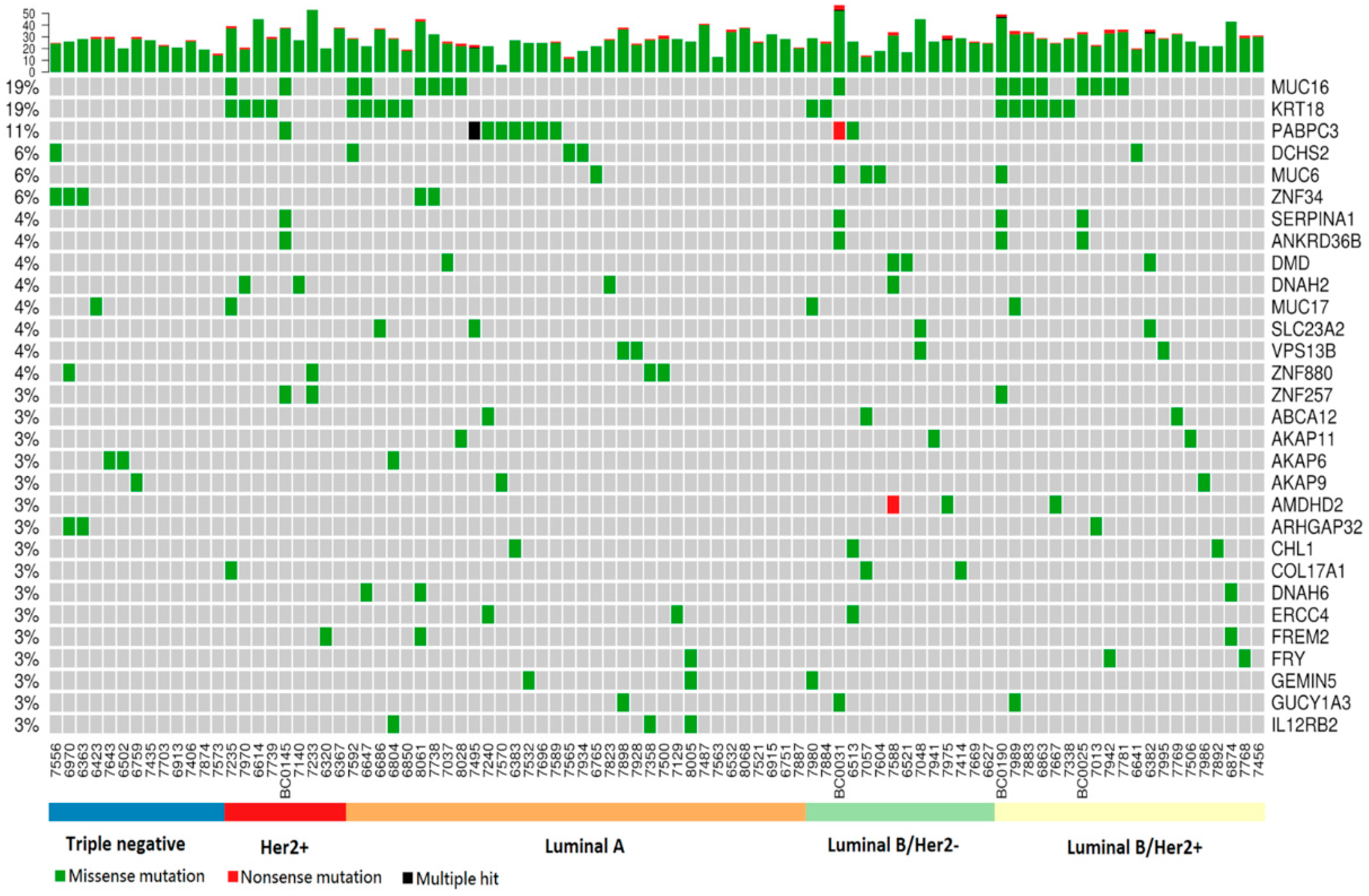
2.2. Somatic Mutation Analysis
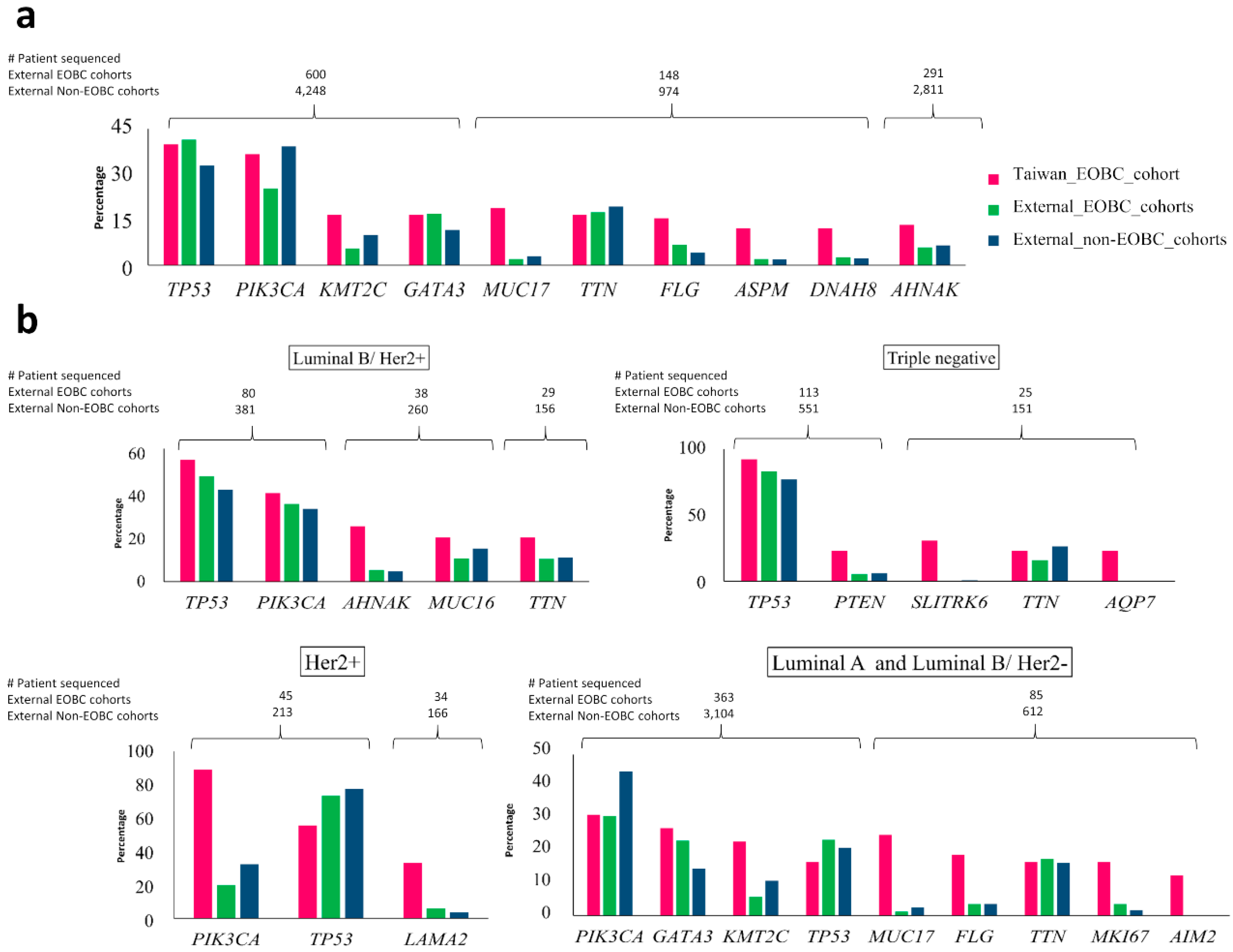
2.3. Comparison of Top Genes with Somatic Mutations in Taiwanese EOBC Cohort to Top Genes in EOBC and Non-EOBC Groups from Other Breast Cancer Cohorts
2.4. Association of Somatic Mutations with Family History of Breast Cancer
2.5. Copy Number Variation Is Associated with Subtypes
2.6. Germline Mutations
2.7. Pathway Analysis
2.8. Case Study of Sisters in the Cohort
3. Discussion
4. Materials and Methods
4.1. WGS: Tissue Sample Collection, DNA Preparation and Whole-Genome Sequencing
4.2. WES: Patient Recruitment, DNA Preparation and Whole-Exome Sequencing
4.3. Subtype Classification
4.4. Sequence Data Analysis Workflow
4.5. Somatic Mutation Analysis
4.6. Validation of Mutations
4.7. Cross-Comparison of Top Genes with Somatic Mutations between the EOBC Cohort and Other Breast Cancer Studies
4.8. Germline Mutation Analysis
4.9. Copy Number Variation Analysis
4.10. Pathway Analysis for Germline and Somatic Mutations
4.11. Ethics Approval and Consent to Participate
4.12. Availability of Data and Material
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bonnier, P.; Romain, S.; Charpin, C.; Lejeune, C.; Tubiana, N.; Martin, P.M.; Piana, L. Age as a prognostic factor in breast cancer: Relationship to pathologic and biologic features. Int. J. Cancer 1995, 62, 138–144. [Google Scholar] [CrossRef] [PubMed]
- Anderson, W.F.; Pfeiffer, R.M.; Dores, G.M.; Sherman, M.E. Comparison of age distribution patterns for different histopathologic types of breast carcinoma. Cancer Epidemiol. Biomark. Prev. 2006, 15, 1899–1905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yankaskas, B.C. Epidemiology of breast cancer in young women. Breast Dis. 2005, 23, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Bray, F.; McCarron, P.; Parkin, D.M. The changing global patterns of female breast cancer incidence and mortality. Breast Cancer Res. 2004, 6, 229–239. [Google Scholar] [CrossRef] [Green Version]
- Banerji, S.; Cibulskis, K.; Rangel-Escareno, C.; Brown, K.K.; Carter, S.L.; Frederick, A.M.; Lawrence, M.S.; Sivachenko, A.Y.; Sougnez, C.; Zou, L.; et al. Sequence analysis of mutations and translocations across breast cancer subtypes. Nature 2012, 486, 405–409. [Google Scholar] [CrossRef] [PubMed]
- Anders, C.K.; Johnson, R.; Litton, J.; Phillips, M.; Bleyer, A. Breast cancer before age 40 years. Semin. Oncol. 2009, 36, 237–249. [Google Scholar] [CrossRef] [Green Version]
- Shen, Y.C.; Chang, C.J.; Hsu, C.; Cheng, C.C.; Chiu, C.F.; Cheng, A.L. Significant difference in the trends of female breast cancer incidence between Taiwanese and Caucasian Americans: Implications from age-period-cohort analysis. Cancer Epidemiol. Biomark. Prev. 2005, 14, 1986–1990. [Google Scholar] [CrossRef] [Green Version]
- Foo, C.S.; Su, D.; Chong, C.K.; Chng, H.C.; Tay, K.H.; Low, S.C.; Tan, S.M. Breast cancer in young Asian women: Study on survival. ANZ J. Surg. 2005, 75, 566–572. [Google Scholar] [CrossRef]
- Narod, S.A. Breast cancer in young women. Nat. Rev. Clin. Oncol. 2012, 9, 460–470. [Google Scholar] [CrossRef] [Green Version]
- Chang, L.-Y.; Yang, Y.-L.; Shyu, M.-K.; Hwa, H.-L.; Hsieh, F.-J. Strategy for Breast Cancer Screening in Taiwan: Obstetrician-Gynecologists Should Actively Participate in Breast Cancer Screening. J. Med. Ultrasound 2011, 20, 1–7. [Google Scholar] [CrossRef]
- Brenner, D.R.; Brockton, N.T.; Kotsopoulos, J.; Cotterchio, M.; Boucher, B.A.; Courneya, K.S.; Knight, J.A.; Olivotto, I.A.; Quan, M.L.; Friedenreich, C.M. Breast cancer survival among young women: A review of the role of modifiable lifestyle factors. Cancer Causes Control 2016, 27, 459–472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sorlie, T.; Tibshirani, R.; Parker, J.; Hastie, T.; Marron, J.S.; Nobel, A.; Deng, S.; Johnsen, H.; Pesich, R.; Geisler, S.; et al. Repeated observation of breast tumor subtypes in independent gene expression data sets. Proc. Natl. Acad. Sci. USA 2003, 100, 8418–8423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perou, C.M.; Sorlie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; Rees, C.A.; Pollack, J.R.; Ross, D.T.; Johnsen, H.; Akslen, L.A.; et al. Molecular portraits of human breast tumours. Nature 2000, 406, 747–752. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas, N. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70. [Google Scholar] [CrossRef] [Green Version]
- Brewster, A.M.; Chavez-MacGregor, M.; Brown, P. Epidemiology, biology, and treatment of triple-negative breast cancer in women of African ancestry. Lancet Oncol. 2014, 15, 625–634. [Google Scholar] [CrossRef] [Green Version]
- Aebi, S.; Sun, Z.; Braun, D.; Price, K.N.; Castiglione-Gertsch, M.; Rabaglio, M.; Gelber, R.D.; Crivellari, D.; Lindtner, J.; Snyder, R.; et al. Differential efficacy of three cycles of CMF followed by tamoxifen in patients with ER-positive and ER-negative tumors: Long-term follow up on IBCSG Trial IX. Ann. Oncol. 2011, 22, 1981–1987. [Google Scholar] [CrossRef]
- Pharoah, P.D.; Antoniou, A.; Bobrow, M.; Zimmern, R.L.; Easton, D.F.; Ponder, B.A. Polygenic susceptibility to breast cancer and implications for prevention. Nat. Genet. 2002, 31, 33–36. [Google Scholar] [CrossRef] [PubMed]
- Nik-Zainal, S.; Davies, H.; Staaf, J.; Ramakrishna, M.; Glodzik, D.; Zou, X.; Martincorena, I.; Alexandrov, L.B.; Martin, S.; Wedge, D.C.; et al. Landscape of somatic mutations in 560 breast cancer whole-genome sequences. Nature 2016, 534, 47–54. [Google Scholar] [CrossRef]
- Ahn, S.G.; Yoon, C.I.; Lee, J.H.; Lee, H.S.; Park, S.E.; Cha, Y.J.; Cha, C.; Bae, S.J.; Lee, K.A.; Jeong, J. Low PR in ER(+)/HER2(-) breast cancer: High rates of TP53 mutation and high SUV. Endocr. Relat. Cancer 2018. [Google Scholar] [CrossRef]
- Niyomnaitham, S.; Parinyanitikul, N.; Roothumnong, E.; Jinda, W.; Samarnthai, N.; Atikankul, T.; Suktitipat, B.; Thongnoppakhun, W.; Limwongse, C.; Pithukpakorn, M. Tumor mutational profile of triple negative breast cancer patients in Thailand revealed distinctive genetic alteration in chromatin remodeling gene. PeerJ 2019, 7. [Google Scholar] [CrossRef]
- Dite, G.S.; Jenkins, M.A.; Southey, M.C.; Hocking, J.S.; Giles, G.G.; McCredie, M.R.; Venter, D.J.; Hopper, J.L. Familial risks, early-onset breast cancer, and BRCA1 and BRCA2 germline mutations. J. Natl. Cancer Inst. 2003, 95, 448–457. [Google Scholar] [CrossRef] [Green Version]
- Cui, J.; Hopper, J.L. Why are the majority of hereditary cases of early-onset breast cancer sporadic? A simulation study. Cancer Epidemiol. Biomark. Prev. 2000, 9, 805–812. [Google Scholar] [PubMed]
- Loizidou, M.; Marcou, Y.; Anastasiadou, V.; Newbold, R.; Hadjisavvas, A.; Kyriacou, K. Contribution of BRCA1 and BRCA2 germline mutations to the incidence of early-onset breast cancer in Cyprus. Clin. Genet. 2007, 71, 165–170. [Google Scholar] [CrossRef]
- Walsh, P.C. Re: Increased cancer risks for relatives of very early-onset breast cancer cases with and without BRCA1 and BRCA2 mutations. J. Urol. 2011, 185. [Google Scholar] [CrossRef]
- De Bock, G.H.; Jacobi, C.E.; Seynaeve, C.; Krol-Warmerdam, E.M.; Blom, J.; van Asperen, C.J.; Cornelisse, C.J.; Klijn, J.G.; Devilee, P.; Tollenaar, R.A.; et al. A family history of breast cancer will not predict female early onset breast cancer in a population-based setting. BMC Cancer 2008, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Razavi, P.; Chang, M.T.; Xu, G.; Bandlamudi, C.; Ross, D.S.; Vasan, N.; Cai, Y.; Bielski, C.M.; Donoghue, M.T.A.; Jonsson, P.; et al. The genomic landscape of endocrine-resistant advanced breast cancers. Cancer Cell 2018, 34, 427–438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Curtis, C.; Shah, S.P.; Chin, S.F.; Turashvili, G.; Rueda, O.M.; Dunning, M.J.; Speed, D.; Lynch, A.G.; Samarajiwa, S.; Yuan, Y.; et al. The genomic and transcriptomic architecture of 2000 breast tumours reveals novel subgroups. Nature 2012, 486, 346–352. [Google Scholar] [CrossRef]
- Pereira, B.; Chin, S.F.; Rueda, O.M.; Vollan, H.K.; Provenzano, E.; Bardwell, H.A.; Pugh, M.; Jones, L.; Russell, R.; Sammut, S.J.; et al. The somatic mutation profiles of 2433 breast cancers refines their genomic and transcriptomic landscapes. Nat. Commun. 2016, 7. [Google Scholar] [CrossRef] [Green Version]
- Stephens, P.J.; Tarpey, P.S.; Davies, H.; Van Loo, P.; Greenman, C.; Wedge, D.C.; Nik-Zainal, S.; Martin, S.; Varela, I.; Bignell, G.R.; et al. The landscape of cancer genes and mutational processes in breast cancer. Nature 2012, 486, 400–404. [Google Scholar] [CrossRef]
- Encinas, G.; Sabelnykova, V.Y.; de Lyra, E.C.; Hirata Katayama, M.L.; Maistro, S.; de Vasconcellos Valle, P.W.M.; de Lima Pereira, G.F.; Rodrigues, L.M.; de Menezes Pacheco Serio, P.A.; de Gouvea, A.; et al. Somatic mutations in early onset luminal breast cancer. Oncotarget 2018, 9, 22460–22479. [Google Scholar] [CrossRef] [Green Version]
- Kadalayil, L.; Khan, S.; Nevanlinna, H.; Fasching, P.A.; Couch, F.J.; Hopper, J.L.; Liu, J.; Maishman, T.; Durcan, L.; Gerty, S.; et al. Germline variation in ADAMTSL1 is associated with prognosis following breast cancer treatment in young women. Nat. Commun. 2017, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, M.; Yang, L.; Lei, T.; Xiao, L.; Li, L.; Zhang, P.; Feng, W.; Ye, F.; Bu, H. BRCA1/2 mutation spectrum in Chinese early-onset breast cancer. Transl. Cancer Res. 2019, 8, 483–490. [Google Scholar] [CrossRef]
- Zhang, Y.; Cai, Q.; Shu, X.O.; Gao, Y.T.; Li, C.; Zheng, W.; Long, J. Whole-Exome Sequencing Identifies Novel Somatic Mutations in Chinese Breast Cancer Patients. J. Mol. Genet. Med. 2015, 9. [Google Scholar] [CrossRef] [PubMed]
- Ahsan, H.; Halpern, J.; Kibriya, M.G.; Pierce, B.L.; Tong, L.; Gamazon, E.; McGuire, V.; Felberg, A.; Shi, J.; Jasmine, F.; et al. A genome-wide association study of early-onset breast cancer identifies PFKM as a novel breast cancer gene and supports a common genetic spectrum for breast cancer at any age. Cancer Epidemiol. Biomark. Prev. 2014, 23, 658–669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marcos-Silva, L.; Narimatsu, Y.; Halim, A.; Campos, D.; Yang, Z.; Tarp, M.A.; Pereira, P.J.; Mandel, U.; Bennett, E.P.; Vakhrushev, S.Y.; et al. Characterization of binding epitopes of CA125 monoclonal antibodies. J. Proteome Res. 2014, 13, 3349–3359. [Google Scholar] [CrossRef] [Green Version]
- Kanwal, M.; Ding, X.J.; Song, X.; Zhou, G.B.; Cao, Y. MUC16 overexpression induced by gene mutations promotes lung cancer cell growth and invasion. Oncotarget 2018, 9, 12226–12239. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Pasche, B.; Zhang, W.; Chen, K. Association of MUC16 mutation with tumor mutation load and outcomes in patients with gastric cancer. JAMA Oncol. 2018, 4, 1691–1698. [Google Scholar] [CrossRef]
- Patel, J.S.; Callahan, B.M.; Chobrutskiy, B.I.; Blanck, G. Matrix-Metalloprotease resistant mucin-16 (muc16) peptide mutants represent a worse lung adenocarcinoma outcome. proteomics Clin. Appl. 2019, 13. [Google Scholar] [CrossRef]
- Taniguchi, T.; Woodward, A.M.; Magnelli, P.; McColgan, N.M.; Lehoux, S.; Jacobo, S.M.P.; Mauris, J.; Argueso, P. N-Glycosylation affects the stability and barrier function of the MUC16 mucin. J. Biol. Chem. 2017, 292, 11079–11090. [Google Scholar] [CrossRef] [Green Version]
- Williams, K.A.; Terry, K.L.; Tworoger, S.S.; Vitonis, A.F.; Titus, L.J.; Cramer, D.W. Polymorphisms of MUC16 (CA125) and MUC1 (CA15.3) in relation to ovarian cancer risk and survival. PLoS ONE 2014, 9, e88334. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Hu, S.; Li, Y. KRT18 is correlated with the malignant status and acts as an oncogene in colorectal cancer. Biosci. Rep. 2019, 39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sporikova, Z.; Koudelakova, V.; Trojanec, R.; Hajduch, M. Genetic Markers in Triple-Negative Breast Cancer. Clin. Breast Cancer 2018, 18. [Google Scholar] [CrossRef] [PubMed]
- Hamdi, Y.; Boujemaa, M.; Ben Rekaya, M.; Ben Hamda, C.; Mighri, N.; El Benna, H.; Mejri, N.; Labidi, S.; Daoud, N.; Naouali, C.; et al. Family specific genetic predisposition to breast cancer: Results from Tunisian whole exome sequenced breast cancer cases. J. Transl. Med. 2018, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O′Regan, G.M.; Sandilands, A.; McLean, W.H.I.; Irvine, A.D. Filaggrin in atopic dermatitis. J. Allergy Clin. Immunol. 2008, 122, 689–693. [Google Scholar] [CrossRef]
- Bisgaard, H.; Simpson, A.; Palmer, C.N.; Bonnelykke, K.; McLean, I.; Mukhopadhyay, S.; Pipper, C.B.; Halkjaer, L.B.; Lipworth, B.; Hankinson, J.; et al. Gene-environment interaction in the onset of eczema in infancy: Filaggrin loss-of-function mutations enhanced by neonatal cat exposure. PLoS Med. 2008, 5. [Google Scholar] [CrossRef] [Green Version]
- Henderson, J.; Northstone, K.; Lee, S.P.; Liao, H.; Zhao, Y.; Pembrey, M.; Mukhopadhyay, S.; Smith, G.D.; Palmer, C.N.; McLean, W.H.; et al. The burden of disease associated with filaggrin mutations: A population-based, longitudinal birth cohort study. J. Allergy Clin. Immunol. 2008, 121, 872–877. [Google Scholar] [CrossRef]
- Machado, C.; Sunkel, C.E.; Andrew, D.J. Human autoantibodies reveal titin as a chromosomal protein. J. Cell Biol. 1998, 141, 321–333. [Google Scholar] [CrossRef] [Green Version]
- Tan, H.; Bao, J.; Zhou, X. Genome-wide mutational spectra analysis reveals significant cancer-specific heterogeneity. Sci. Rep. 2015, 5. [Google Scholar] [CrossRef] [Green Version]
- Greenman, C.; Stephens, P.; Smith, R.; Dalgliesh, G.L.; Hunter, C.; Bignell, G.; Davies, H.; Teague, J.; Butler, A.; Stevens, C.; et al. Patterns of somatic mutation in human cancer genomes. Nature 2007, 446, 153–158. [Google Scholar] [CrossRef] [Green Version]
- Kufe, D.W. Mucins in cancer: Function, prognosis and therapy. Nat. Rev. Cancer 2009, 9, 874–885. [Google Scholar] [CrossRef] [Green Version]
- Yang, B.; Wu, A.; Hu, Y.; Tao, C.; Wang, J.M.; Lu, Y.; Xing, R. Mucin 17 inhibits the progression of human gastric cancer by limiting inflammatory responses through a MYH9-p53-RhoA regulatory feedback loop. J. Exp. Clin. Cancer Res. 2019, 38. [Google Scholar] [CrossRef] [PubMed]
- Vasconcelos, I.; Hussainzada, A.; Berger, S.; Fietze, E.; Linke, J.; Siedentopf, F.; Schoenegg, W. The St. Gallen surrogate classification for breast cancer subtypes successfully predicts tumor presenting features, nodal involvement, recurrence patterns and disease free survival. Breast 2016, 29, 181–185. [Google Scholar] [CrossRef] [PubMed]
- Kondov, B.; Milenkovikj, Z.; Kondov, G.; Petrushevska, G.; Basheska, N.; Bogdanovska-Todorovska, M.; Tolevska, N.; Ivkovski, L. Presentation of the molecular subtypes of breast cancer detected by immunohistochemistry in surgically treated patients. Open Access Maced. J. Med. Sci. 2018, 6, 961–967. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, R.K.; Jain, M. NGS QC Toolkit: A toolkit for quality control of next generation sequencing data. PLoS ONE 2012, 7, e30619. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DePristo, M.A.; Banks, E.; Poplin, R.; Garimella, K.V.; Maguire, J.R.; Hartl, C.; Philippakis, A.A.; del Angel, G.; Rivas, M.A.; Hanna, M.; et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 2011, 43, 491–498. [Google Scholar] [CrossRef]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38. [Google Scholar] [CrossRef]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [Green Version]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013, 6. [Google Scholar] [CrossRef] [Green Version]
- Mermel, C.H.; Schumacher, S.E.; Hill, B.; Meyerson, M.L.; Beroukhim, R.; Getz, G. GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome Biol. 2011, 12. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Category | Description | Number of Patients | WES | WGS |
|---|---|---|---|---|
| Subtype | Her2+ | 9 | 8 | 1 |
| Luminal A | 34 | 34 | 0 | |
| Luminal B/Her2+ | 20 | 18 | 2 | |
| Luminal B/Her2- | 14 | 13 | 1 | |
| Triple negative | 13 | 13 | 0 | |
| Age group | < 37 (median) | 39 | 37 | 2 |
| ≥ 37 | 51 | 49 | 2 | |
| Stage group | Ia, Ib | 15 | 13 | 2 |
| IIa, IIb | 42 | 41 | 1 | |
| IIIa, IIIb, IIIc | 24 | 23 | 1 | |
| IVa, IVb | 5 | 5 | 0 | |
| Unknown | 4 | 4 | 0 | |
| Family history | No family history | 79 | 75 | 4 |
| With family history | 11 | 11 | 0 |
| Pathway ID | Pathway | Her2+ | Luminal_A | Luminal B Her2+ | Luminal B Her 2- | Triple Negative | Whole Cohort |
|---|---|---|---|---|---|---|---|
| * hsa05222 | Small cell lung cancer | 0.001 | 0.009 | 0.032 | 0.006 | 0.020 | 0.014 |
| hsa04380 | Osteoclast differentiation | 0.006 | – | – | – | – | – |
| * hsa05146 | Amoebiasis | 0.021 | 0.018 | – | – | – | – |
| * hsa05200 | Pathways in cancer | 0.022 | – | – | 0.012 | – | – |
| hsa04919 | Thyroid hormone-signaling pathway | 0.026 | – | – | – | – | – |
| hsa04510 | Focal adhesion | 0.028 | 0.013 | 0.009 | 0.0001 | 0.051 | 0.004 |
| hsa04071 | Sphingolipid-signaling pathway | 0.030 | – | – | – | – | – |
| hsa04512 | ECM–receptor interaction | – | 0.001 | 0.008 | 0.007 | – | 0.001 |
| * hsa05016 | Huntington’s disease | – | 0.010 | – | – | – | 0.031 |
| hsa04151 | PI3K–Akt-signaling pathway | – | 0.016 | 0.009 | 0.001 | – | 0.032 |
| * hsa05213 | Endometrial cancer | – | – | 0.006 | 0.021 | 0.048 | – |
| hsa02010 | ABC transporters | – | – | 0.024 | 0.030 | – | 0.00004 |
| hsa03460 | Fanconi anemia pathway | – | – | 0.039 | – | – | – |
| hsa04015 | Rap1-signaling pathway | – | – | – | 0.0004 | – | – |
| hsa05230 | Central carbon metabolism in cancer | – | – | – | 0.002 | – | – |
| * hsa05218 | Melanoma | – | – | – | 0.014 | – | – |
| * hsa05215 | Prostate cancer | – | – | – | 0.020 | – | – |
| hsa04060 | Cytokine–cytokine receptor interaction | – | – | – | 0.024 | – | – |
| hsa04923 | Regulation of lipolysis in adipocytes | – | – | – | 0.030 | – | – |
| * hsa05412 | Arrhythmogenic right ventricular cardiomyopathy (ARVC) | – | – | – | 0.037 | – | – |
| hsa04520 | Adherens junction | – | – | – | 0.037 | – | – |
| hsa05205 | Proteoglycans in cancer | – | – | – | 0.043 | – | – |
| * hsa04930 | Type II diabetes mellitus | – | – | – | 0.043 | – | – |
| hsa04611 | Platelet activation | – | – | – | 0.048 | – | – |
| * hsa05210 | Colorectal cancer | – | – | – | 0.049 | – | – |
| hsa04630 | Jak–STAT-signaling pathway | – | – | – | 0.050 | – | – |
| hsa04530 | Tight junction | – | – | – | – | – | 0.008 |
| hsa04974 | Protein digestion and absorption | – | – | – | – | – | 0.017 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Midha, M.K.; Huang, Y.-F.; Yang, H.-H.; Fan, T.-C.; Chang, N.-C.; Chen, T.-H.; Wang, Y.-T.; Kuo, W.-H.; Chang, K.-J.; Shen, C.-Y.; et al. Comprehensive Cohort Analysis of Mutational Spectrum in Early Onset Breast Cancer Patients. Cancers 2020, 12, 2089. https://doi.org/10.3390/cancers12082089
Midha MK, Huang Y-F, Yang H-H, Fan T-C, Chang N-C, Chen T-H, Wang Y-T, Kuo W-H, Chang K-J, Shen C-Y, et al. Comprehensive Cohort Analysis of Mutational Spectrum in Early Onset Breast Cancer Patients. Cancers. 2020; 12(8):2089. https://doi.org/10.3390/cancers12082089
Chicago/Turabian StyleMidha, Mohit K., Yu-Feng Huang, Hsiao-Hsiang Yang, Tan-Chi Fan, Nai-Chuan Chang, Tzu-Han Chen, Yu-Tai Wang, Wen-Hung Kuo, King-Jen Chang, Chen-Yang Shen, and et al. 2020. "Comprehensive Cohort Analysis of Mutational Spectrum in Early Onset Breast Cancer Patients" Cancers 12, no. 8: 2089. https://doi.org/10.3390/cancers12082089