The CCL5/CCR5 Axis in Cancer Progression


Abstract
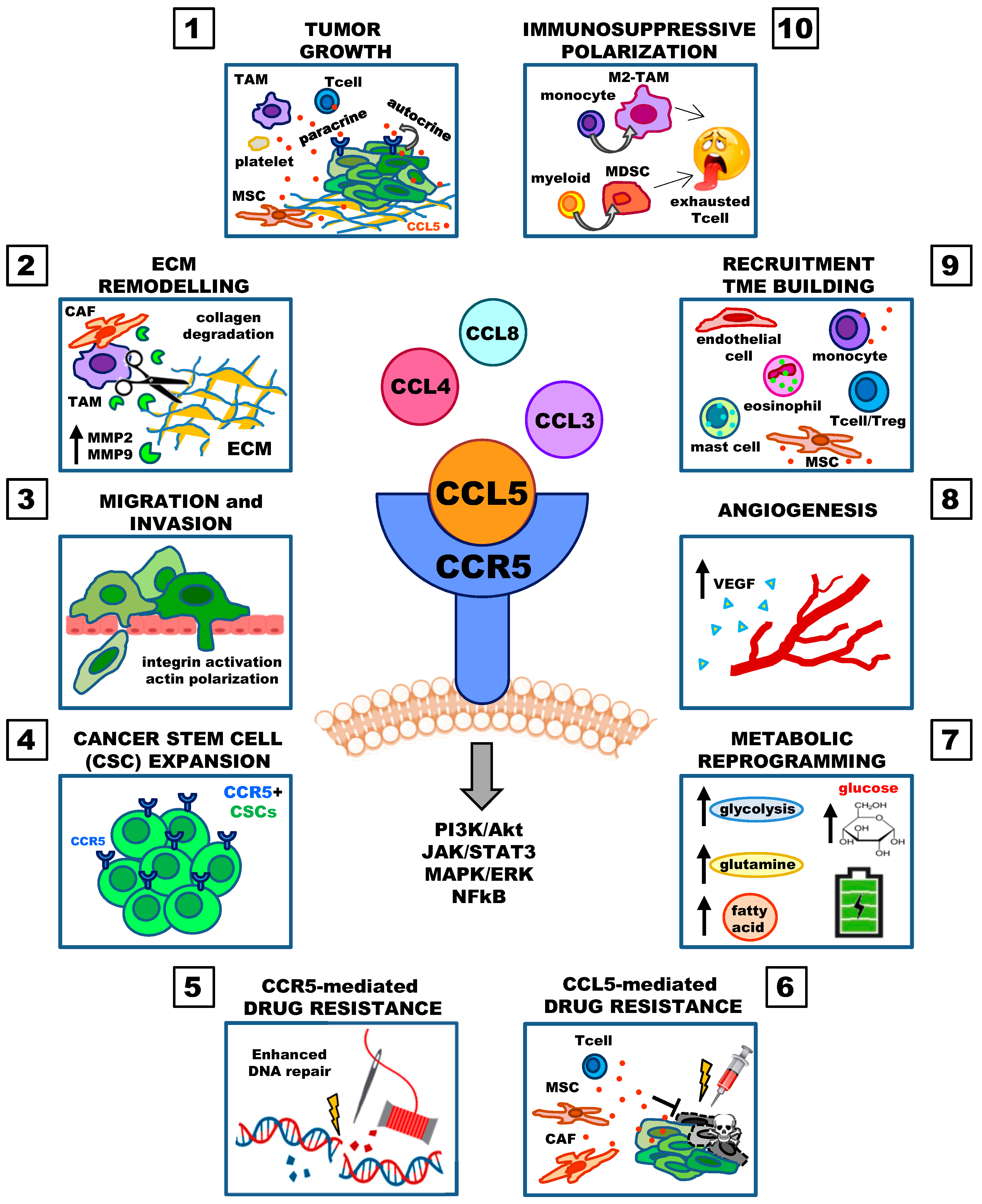
:1. Introduction
2. Involvement of the CCL5/CCR5 Axis in Cancer Progression
2.1. Tumor Growth
2.2. Extracellular Matrix Remodeling and Migration
2.3. Cancer Stem Cell Expansion, Decreased Cytotoxicity of DNA-Damaging Agents, Drug Resistance
2.4. Deregulated Cellular Energetics (Metabolic Reprogramming)
2.5. Angiogenesis
2.6. Recruitment of Immune and Stromal Cells and Immunosuppressive Polarization
3. The Tumor-Promoting Role of the CCL5/CCR5 Axis
3.1. The CCL5/CCR5 Axis in Hematological Malignancies
3.1.1. Acute Myeloid Leukemia
3.1.2. Acute Lymphoblastic Leukemia and Chronic Lymphoblastic Leukemia
3.1.3. Hodgkin Lymphoma
3.1.4. Multiple Myeloma
3.1.5. Other Lymphomas
3.2. The CCL5/CCR5 Axis in Solid Tumors
3.2.1. Breast Cancer
3.2.2. Breast Phyllodes
3.2.3. Cholangiocarcinoma
3.2.4. Colorectal Cancer
3.2.5. Esophageal Squamous Cell Carcinoma
3.2.6. Gastric Cancer
3.2.7. Glioblastoma
3.2.8. Head and Neck Cancer and Squamous Cell Carcinoma of Tongue and Floor of the Mouth
3.2.9. Lung Cancer
3.2.10. Melanoma
3.2.11. Osteosarcoma
3.2.12. Ovarian Cancer
3.2.13. Pancreatic Cancer
3.2.14. Pituitary Tumors
3.2.15. Prostate Cancer
3.2.16. Thyroid Carcinoma
4. Counteracting the CCL5/CCR5 Axis: Clinical Applications
4.1. Inhibition of CCR5/CCL5 Interactions in Clinical Trials
4.2. Pharmacological Inhibition of CCL5 Secretion by Cancer Cells and the TME
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Nagarsheth, N.; Wicha, M.S.; Zou, W. Chemokines in the cancer microenvironment and their relevance in cancer immunotherapy. Nat. Rev. Immunol. 2017, 17, 559–572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Do, H.T.T.; Lee, C.H.; Cho, J. Chemokines and their Receptors: Multifaceted Roles in Cancer Progression and Potential Value as Cancer Prognostic Markers. Cancers 2020, 12, 287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aldinucci, D.; Borghese, C.; Casagrande, N. Formation of the Immunosuppressive Microenvironment of Classic Hodgkin Lymphoma and Therapeutic Approaches to Counter It. Int. J. Mol. Sci. 2019, 20, 2416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, L.-Y.; Lin, Y.-C.; Mahalingam, J.; Huang, C.-T.; Chen, T.-W.; Kang, C.-W.; Peng, H.-M.; Chu, Y.-Y.; Chiang, J.-M.; Dutta, A.; et al. Tumor-Derived Chemokine CCL5 Enhances TGF-Mediated Killing of CD8+ T Cells in Colon Cancer by T-Regulatory Cells. Cancer Res. 2012, 72, 1092–1102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cook, J.; Hagemann, T. Tumour-associated macrophages and cancer. Curr. Opin. Pharmacol. 2013, 13, 595–601. [Google Scholar] [CrossRef] [PubMed]
- Bronte, V.; Brandau, S.; Chen, S.-H.; Colombo, M.P.; Frey, A.; Greten, T.F.; Mandruzzato, S.; Murray, P.J.; Ochoa, A.; Ostrand-Rosenberg, S.; et al. Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards. Nat. Commun. 2016, 7, 12150. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Hou, J.; Han, Z.; Wang, Y.; Hao, C.; Wei, L.; Shi, Y. One cell, multiple roles: Contribution of mesenchymal stem cells to tumor development in tumor microenvironment. Cell Biosci. 2013, 3, 5. [Google Scholar] [CrossRef] [Green Version]
- Wu, Q.; Li, B.; Li, Z.; Li, J.; Sun, S.; Sun, S. Cancer-associated adipocytes: Key players in breast cancer progression. J. Hematol. Oncol. 2019, 12, 95–115. [Google Scholar] [CrossRef]
- Schlecker, E.; Stojanovic, A.; Eisen, C.; Quack, C.; Falk, C.S.; Umansky, V.; Cerwenka, A.; Xu, M.; Hadinoto, V.; Appanna, R.; et al. Tumor-Infiltrating Monocytic Myeloid-Derived Suppressor Cells Mediate CCR5-Dependent Recruitment of Regulatory T Cells Favoring Tumor Growth. J. Immunol. 2012, 189, 5602–5611. [Google Scholar] [CrossRef] [Green Version]
- Karnoub, A.E.; Dash, A.B.; Vo, A.P.; Sullivan, A.; Brooks, M.W.; Bell, G.W.; Richardson, A.L.; Polyak, K.; Tubo, R.; Weinberg, R.A. Mesenchymal stem cells within tumour stroma promote breast cancer metastasis. Nature 2007, 449, 557–563. [Google Scholar] [CrossRef]
- Ben-Baruch, A. The Tumor-Promoting Flow of Cells Into, Within and Out of the Tumor Site: Regulation by the Inflammatory Axis of TNFα and Chemokines. Cancer Microenviron. 2011, 5, 151–164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marcuzzi, E.; Angioni, R.; Molon, B.; Calì, B. Chemokines and Chemokine Receptors: Orchestrating Tumor Metastasization. Int. J. Mol. Sci. 2018, 20, 96. [Google Scholar] [CrossRef] [Green Version]
- Pallegar, N.K.; Christian, S.L. Adipocytes in the Tumour Microenvironment. Adv. Exp. Med. Biol. 2020, 1234, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Teleanu, R.I.; Chircov, C.; Grumezescu, A.M.; Teleanu, D.M. Tumor Angiogenesis and Anti-Angiogenic Strategies for Cancer Treatment. J. Clin. Med. 2019, 9, 84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pérez-Velázquez, J.; Rejniak, K.A. Drug-Induced Resistance in Micrometastases: Analysis of Spatio-Temporal Cell Lineages. Front. Physiol. 2020, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gentles, A.J.; Newman, A.M.; Liu, C.L.; Bratman, S.V.; Feng, W.; Kim, N.; Nair, V.S.; Xu, Y.; Khuong, A.; Hoang, C.D.; et al. The prognostic landscape of genes and infiltrating immune cells across human cancers. Nat. Med. 2015, 21, 938–945. [Google Scholar] [CrossRef] [PubMed]
- Miao, M.; De Clercq, E.; Li, G. Clinical significance of chemokine receptor antagonists. Expert Opin. Drug Metab. Toxicol. 2020, 16, 11–30. [Google Scholar] [CrossRef]
- Atretkhany, K.-S.; Drutskaya, M.; Nedospasov, S.; Grivennikov, S.; Kuprash, D.V. Chemokines, cytokines and exosomes help tumors to shape inflammatory microenvironment. Pharmacol. Ther. 2016, 168, 98–112. [Google Scholar] [CrossRef] [PubMed]
- Aldinucci, D.; Colombatti, A. The Inflammatory Chemokine CCL5 and Cancer Progression. Mediat. Inflamm. 2014, 2014, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiao, X.; Nawab, O.; Patel, T.; Kossenkov, A.V.; Halama, N.; Jaeger, D.; Pestell, R.G. Recent Advances Targeting CCR5 for Cancer and Its Role in Immuno-Oncology. Cancer Res. 2019, 79, 4801–4807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dangaj, D.; Bruand, M.; Grimm, A.J.; Ronet, C.; Barras, D.; Duttagupta, P.A.; Lanitis, E.; Duraiswamy, J.; Tanyi, J.L.; Benencia, F.; et al. Cooperation between Constitutive and Inducible Chemokines Enables T Cell Engraftment and Immune Attack in Solid Tumors. Cancer Cell 2019, 35, 885–900.e10. [Google Scholar] [CrossRef] [PubMed]
- De Galarreta, M.R.; Bresnahan, E.; Molina-Sanchez, P.; Lindblad, K.E.; Maier, B.; Sia, D.; Puigvehi, M.; Miguela, V.; Casanova-Acebes, M.; Dhainaut, M.; et al. β-catenin activation promotes immune escape and resistance to anti-PD-1 therapy in hepatocellular carcinoma. Cancer Discov. 2019, 9, 1124–1141. [Google Scholar] [CrossRef]
- Huffman, A.P.; Lin, J.H.; Kim, S.I.; Byrne, K.T.; Vonderheide, R.H. CCL5 mediates CD40-driven CD4+ T cell tumor infiltration and immunity. JCI Insight 2020, 5, 5. [Google Scholar] [CrossRef] [PubMed]
- Böttcher, J.P.; Bonavita, E.; Chakravarty, P.; Blees, H.; Cabeza-Cabrerizo, M.; Sammicheli, S.; Rogers, N.C.; Sahai, E.; Zelenay, S.; Sousa, C.R.E. NK Cells Stimulate Recruitment of cDC1 into the Tumor Microenvironment Promoting Cancer Immune Control. Cell 2018, 172, 1022–1037.e14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seo, W.; Shimizu, K.; Kojo, S.; Okeke, A.; Kohwi-Shigematsu, T.; Fujii, S.-I.; Taniuchi, I. Runx-mediated regulation of CCL5 via antagonizing two enhancers influences immune cell function and anti-tumor immunity. Nat. Commun. 2020, 11, 1–16. [Google Scholar] [CrossRef]
- Soria, G.; Ben-Baruch, A. The inflammatory chemokines CCL2 and CCL5 in breast cancer. Cancer Lett. 2008, 267, 271–285. [Google Scholar] [CrossRef]
- Udi, J.; Schüler, J.; Wider, D.; Ihorst, G.; Catusse, J.; Waldschmidt, J.; Schnerch, D.; Follo, M.; Wäsch, R.; Engelhardt, M. Potentin vitroandin vivoactivity of sorafenib in multiple myeloma: Induction of cell death, CD138-downregulation and inhibition of migration through actin depolymerization. Br. J. Haematol. 2013, 161, 104–116. [Google Scholar] [CrossRef]
- Appay, V.; Rowland-Jones, S.L. RANTES: A versatile and controversial chemokine. Trends Immunol. 2001, 22, 83–87. [Google Scholar] [CrossRef]
- Roscic-Mrkic, B.; Fischer, M.; Leemann, C.; Manrique, A.; Gordon, C.J.; Moore, J.P.; Proudfoot, A.E.I.; Trkola, A.; Balta, G.; Gümrük, F.; et al. RANTES (CCL5) uses the proteoglycan CD44 as an auxiliary receptor to mediate cellular activation signals and HIV-1 enhancement. Blood 2003, 102, 1169–1177. [Google Scholar] [CrossRef]
- Dedoni, S.; Campbell, L.; Harvey, B.K.; Avdoshina, V.; Mocchetti, I. The orphan G-protein-coupled receptor 75 signaling is activated by the chemokine CCL 5. J. Neurochem. 2018, 146, 526–539. [Google Scholar] [CrossRef]
- Aldinucci, D.; Gloghini, A.; Pinto, A.; Colombatti, A.; Carbone, A. The role of CD40/CD40L and interferon regulatory factor 4 in Hodgkin lymphoma microenvironment. Leuk. Lymphoma 2011, 53, 195–201. [Google Scholar] [CrossRef] [PubMed]
- Ridley, A.J. Cell Migration: Integrating Signals from Front to Back. Science 2003, 302, 1704–1709. [Google Scholar] [CrossRef] [Green Version]
- Oppermann, M. Chemokine receptor CCR5: Insights into structure, function, and regulation. Cell. Signal. 2004, 16, 1201–1210. [Google Scholar] [CrossRef] [PubMed]
- Vangelista, L.; Vento, S. The Expanding Therapeutic Perspective of CCR5 Blockade. Front. Immunol. 2018, 8. [Google Scholar] [CrossRef] [PubMed]
- Gavegnano, C.; Savarino, A.; Owanikoko, T.; Marconi, V.C. Crossroads of Cancer and HIV-1: Pathways to a Cure for HIV. Front. Immunol. 2019, 10, 2267. [Google Scholar] [CrossRef]
- Kulmann-Leal, B.; Ellwanger, J.H.; Chies, J.A.B. A functional interaction between the CCR5 and CD34 molecules expressed in hematopoietic cells can support (or even promote) the development of cancer. Hematol. Transfus. Cell Ther. 2020, 42, 70–76. [Google Scholar] [CrossRef] [PubMed]
- Velasco-Velázquez, M.A.; Jiao, X.; De La Fuente-Granada, M.; Pestell, T.G.; Ertel, A.; Lisanti, M.P.; Pestell, R.G. CCR5 Antagonist Blocks Metastasis of Basal Breast Cancer Cells. Cancer Res. 2012, 72, 3839–3850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casagrande, N.; Borghese, C.; Visser, L.; Mongiat, M.; Colombatti, A.; Aldinucci, D. CCR5 antagonism by maraviroc inhibits Hodgkin lymphoma microenvironment interactions and xenograft growth. Haematologica 2018, 104, 564–575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zi, J.; Yuan, S.; Qiao, J.; Zhao, K.; Xu, L.; Qi, K.; Xu, K.; Zeng, L. Treatment with the C-C chemokine receptor type 5 (CCR5)-inhibitor maraviroc suppresses growth and induces apoptosis of acute lymphoblastic leukemia cells. Am. J. Cancer Res. 2017, 7, 869–880. [Google Scholar]
- Aldinucci, D.; Lorenzon, D.; Cattaruzza, L.; Pinto, A.; Gloghini, A.; Carbone, A.; Colombatti, A. Expression of CCR5 receptors on Reed–Sternberg cells and Hodgkin lymphoma cell lines: Involvement of CCL5/Rantes in tumor cell growth and microenvironmental interactions. Int. J. Cancer 2008, 122, 769–776. [Google Scholar] [CrossRef]
- Menu, E.; De Leenheer, E.; De Raeve, H.; Coulton, L.; Imanishi, T.; Miyashita, K.; Van Valckenborgh, E.; Van Riet, I.; Van Camp, B.; Horuk, R.; et al. Role of CCR1 and CCR5 in homing and growth of multiple myeloma and in the development of osteolytic lesions: A study in the 5TMM model. Clin. Exp. Metastasis 2006, 23, 291–300. [Google Scholar] [CrossRef] [PubMed]
- Trentin, L.; Miorin, M.; Facco, M.; Baesso, I.; Carraro, S.; Cabrelle, A.; Maschio, N.; Bortoli, M.; Binotto, G.; Piazza, F.; et al. Multiple myeloma plasma cells show different chemokine receptor profiles at sites of disease activity. Br. J. Haematol. 2007, 138, 594–602. [Google Scholar] [CrossRef] [PubMed]
- Jiao, X.; Velasco-Velázquez, M.A.; Wang, M.; Li, Z.; Rui, H.; Peck, A.R.; Korkola, J.; Chen, X.; Xu, S.; DuHadaway, J.; et al. CCR5 Governs DNA Damage Repair and Breast Cancer Stem Cell Expansion. Cancer Res. 2018, 78, 1657–1671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halama, N.; Zoernig, I.; Berthel, A.; Kahlert, C.; Klupp, F.; Suarez-Carmona, M.; Suetterlin, T.; Brand, K.; Krauss, J.; Lasitschka, F.; et al. Tumoral Immune Cell Exploitation in Colorectal Cancer Metastases Can Be Targeted Effectively by Anti-CCR5 Therapy in Cancer Patients. Cancer Cell 2016, 29, 587–601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pervaiz, A.; Ansari, S.; Berger, M.R.; Adwan, H. CCR5 blockage by maraviroc induces cytotoxic and apoptotic effects in colorectal cancer cells. Med Oncol. 2015, 32, 158. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.-C.; Shen, Y.-C.; Chang, J.W.-C.; Hsieh, J.-J.; Chu, Y.; Wang, C.-H. Autocrine CCL5 promotes tumor progression in esophageal squamous cell carcinoma in vitro. Cytokine 2018, 110, 94–103. [Google Scholar] [CrossRef]
- Aldinucci, D.; Casagrande, N. Inhibition of the CCL5/CCR5 Axis against the Progression of Gastric Cancer. Int. J. Mol. Sci. 2018, 19, 1477. [Google Scholar] [CrossRef] [Green Version]
- Ryu, H.; Baek, S.W.; Moon, J.Y.; Jo, I.; Kim, N.; Lee, H.J. C-C motif chemokine receptors in gastric cancer. Mol. Clin. Oncol. 2017, 8, 3–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalez-Arriagada, W.A.; Lozano-Burgos, C.; Zúñiga-Moreta, R.; González-Díaz, P.; Della Coletta, R. Clinicopathological significance of chemokine receptor (CCR1, CCR3, CCR4, CCR5, CCR7 and CXCR4) expression in head and neck squamous cell carcinomas. J. Oral Pathol. Med. 2018, 47, 755–763. [Google Scholar] [CrossRef]
- Liu, J.; Wang, C.; Ma, X.; Tian, Y.; Wang, C.; Fu, Y.; Luo, Y. High expression of CCR5 in melanoma enhances epithelial-mesenchymal transition and metastasis via TGFβ1. J. Pathol. 2019, 247, 481–493. [Google Scholar] [CrossRef]
- Singh, S.K.; Mishra, M.K.; Eltoum, I.-E.A.; Bae, S.; Lillard, J.W.; Singh, R. CCR5/CCL5 axis interaction promotes migratory and invasiveness of pancreatic cancer cells. Sci. Rep. 2018, 8, 1323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaday, G.G.; Peehl, D.M.; Kadam, P.A.; Lawrence, D.M. Expression of CCL5 (RANTES) and CCR5 in prostate cancer. Prostate 2006, 66, 124–134. [Google Scholar] [CrossRef] [PubMed]
- Murooka, T.T.; Rahbar, R.; Fish, E.N. CCL5 promotes proliferation of MCF-7 cells through mTOR-dependent mRNA translation. Biochem. Biophys. Res. Commun. 2009, 387, 381–386. [Google Scholar] [CrossRef] [PubMed]
- Ding, H.; Zhao, L.; Dai, S.; Li, L.; Wang, F.; Shan, B. CCL5 secreted by tumor associated macrophages may be a new target in treatment of gastric cancer. Biomed. Pharmacother. 2016, 77, 142–149. [Google Scholar] [CrossRef]
- Gao, D.; Fish, E.N. Chemokines in breast cancer: Regulating metabolism. Cytokine 2018, 109, 57–64. [Google Scholar] [CrossRef]
- Huang, C.-Y.; Fong, Y.-C.; Lee, C.-Y.; Chen, M.-Y.; Tsai, H.-C.; Hsu, H.-C.; Tang, C.-H. CCL5 increases lung cancer migration via PI3K, Akt and NF-κB pathways. Biochem. Pharmacol. 2009, 77, 794–803. [Google Scholar] [CrossRef]
- Wang, S.-W.; Wu, H.-H.; Liu, S.-C.; Wang, P.-C.; Ou, W.-C.; Chou, W.-Y.; Shen, Y.-S.; Tang, C.-H. CCL5 and CCR5 Interaction Promotes Cell Motility in Human Osteosarcoma. PLoS ONE 2012, 7, e35101. [Google Scholar] [CrossRef] [Green Version]
- Agere, S.A.; Akhtar, N.; Watson, J.M.; Ahmed, S. RANTES/CCL5 Induces Collagen Degradation by Activating MMP-1 and MMP-13 Expression in Human Rheumatoid Arthritis Synovial Fibroblasts. Front. Immunol. 2017, 8. [Google Scholar] [CrossRef] [Green Version]
- Long, H.; Xie, R.; Xiang, T.; Zhao, Z.; Lin, S.; Liang, Z.; Chen, Z.; Zhu, B. Autocrine CCL5 Signaling Promotes Invasion and Migration of CD133 + Ovarian Cancer Stem-Like Cells via NF-κB-Mediated MMP-9 Upregulation. Stem Cells 2012, 30, 2309–2319. [Google Scholar] [CrossRef]
- Kato, T.; Fujita, Y.; Nakane, K.; Mizutani, K.; Terazawa, R.; Ehara, H.; Kanimoto, Y.; Kojima, T.; Nozawa, Y.; Deguchi, T.; et al. CCR1/CCL5 interaction promotes invasion of taxane-resistant PC3 prostate cancer cells by increasing secretion of MMPs 2/9 and by activating ERK and Rac signaling. Cytokine 2013, 64, 251–257. [Google Scholar] [CrossRef]
- Xiang, P.; Jin, S.; Yang, Y.; Sheng, J.; He, Q.; Song, Y.; Yu, W.; Hu, S.; Jin, J. Infiltrating CD4+ T cells attenuate chemotherapy sensitivity in prostate cancer via CCL5 signaling. Prostate 2019, 79, 1018–1031. [Google Scholar] [CrossRef]
- Zhou, B.; Sun, C.; Li, N.; Shan, W.; Lu, H.; Guo, L.; Guo, E.; Xia, M.; Weng, D.; Meng, L.; et al. Cisplatin-induced CCL5 secretion from CAFs promotes cisplatin-resistance in ovarian cancer via regulation of the STAT3 and PI3K/Akt signaling pathways. Int. J. Oncol. 2016, 48, 2087–2097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yi, E.H.; Lee, C.S.; Lee, J.-K.; Lee, Y.J.; Shin, M.K.; Cho, C.-H.; Kang, K.W.; Lee, J.W.; Han, W.; Noh, D.-Y.; et al. STAT3-RANTES Autocrine Signaling Is Essential for Tamoxifen Resistance in Human Breast Cancer Cells. Mol. Cancer Res. 2012, 11, 31–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faubert, B.; Solmonson, A.; DeBerardinis, R.J. Metabolic reprogramming and cancer progression. Science 2020, 368, eaaw5473. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Xiao, Z.; Chen, T.; Liang, S.H.; Guo, H. Glucose Metabolism on Tumor Plasticity, Diagnosis, and Treatment. Front. Oncol. 2020, 10, 317. [Google Scholar] [CrossRef] [PubMed]
- Bose, S.; Le, A. Glucose Metabolism in Cancer. Plant Promot. Transcr. Factors 2018, 3–12. [Google Scholar] [CrossRef]
- Suffee, N.; Hlawaty, H.; Meddahi-Pelle, A.; Maillard, L.; Louedec, L.; Haddad, O.; Martin, L.; Laguillier-Morizot, C.; Richard, B.; Oudar, O.; et al. RANTES/CCL5-induced pro-angiogenic effects depend on CCR1, CCR5 and glycosaminoglycans. Angiogenesis 2012, 15, 727–744. [Google Scholar] [CrossRef]
- Wang, S.-W.; Liu, S.-C.; Sun, H.-L.; Huang, T.-Y.; Chan, C.-H.; Yang, C.-Y.; Yeh, H.-I.; Chou, W.-Y.; Lin, Y.-M.; Tang, C.-H. CCL5/CCR5 axis induces vascular endothelial growth factor-mediated tumor angiogenesis in human osteosarcoma microenvironment. Carcinogenesis 2014, 36, 104–114. [Google Scholar] [CrossRef]
- Wang, L.-H.; Lin, C.-Y.; Liu, S.-C.; Liu, G.-T.; Chen, Y.-L.; Chen, J.-J.; Chan, C.-H.; Lin, T.-Y.; Chen, C.-K.; Xu, G.-H.; et al. CCL5 promotes VEGF-C production and induces lymphangiogenesis by suppressing miR-507 in human chondrosarcoma cells. Oncotarget 2016, 7, 36896–36908. [Google Scholar] [CrossRef] [Green Version]
- Mantovani, A.; Marchesi, F.; Malesci, A.; Laghi, L.; Allavena, P. Tumour-associated macrophages as treatment targets in oncology. Nat. Rev. Clin. Oncol. 2017, 14, 399–416. [Google Scholar] [CrossRef]
- Deng, G. Tumor-infiltrating regulatory T cells: Origins and features. Am. J. Clin. Exp. Immunol. 2018, 7, 81–87. [Google Scholar] [PubMed]
- Barcellos-De-Souza, P.; Comito, G.; Pons-Segura, C.; Taddei, M.L.; Gori, V.; Becherucci, V.; Bambi, F.; Margheri, F.; Laurenzana, A.; Del Rosso, M.; et al. Mesenchymal Stem Cells are Recruited and Activated into Carcinoma-Associated Fibroblasts by Prostate Cancer Microenvironment-Derived TGF-β1. Stem Cells 2016, 34, 2536–2547. [Google Scholar] [CrossRef] [PubMed]
- Bankov, K.; Döring, C.; Ustaszewski, A.; Giefing, M.; Herling, M.; Cencioni, C.; Spallotta, F.; Gaetano, C.; Küppers, R.; Hansmann, M.-L.; et al. Fibroblasts in Nodular Sclerosing Classical Hodgkin Lymphoma Are Defined by a Specific Phenotype and Protect Tumor Cells from Brentuximab-Vedotin Induced Injury. Cancers 2019, 11, 1687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- LeBleu, V.S.; Kalluri, R. A peek into cancer-associated fibroblasts: Origins, functions and translational impact. Dis. Model. Mech. 2018, 11, dmm029447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanahan, D.; Coussens, L.M. Accessories to the Crime: Functions of Cells Recruited to the Tumor Microenvironment. Cancer Cell 2012, 21, 309–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ansems, M.; Span, P.N. The tumor microenvironment and radiotherapy response; a central role for cancer-associated fibroblasts. Clin. Transl. Radiat. Oncol. 2020, 22, 90–97. [Google Scholar] [CrossRef]
- Cammarota, F.; Laukkanen, M.O. Mesenchymal Stem/Stromal Cells in Stromal Evolution and Cancer Progression. Stem Cells Int. 2015, 2016, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Ban, Y.; Mai, J.; Li, X.; Mitchell-Flack, M.; Zhang, T.; Zhang, L.; Chouchane, L.; Ferrari, M.; Shen, H.; Ma, X. Targeting Autocrine CCL5–CCR5 Axis Reprograms Immunosuppressive Myeloid Cells and Reinvigorates Antitumor Immunity. Cancer Res. 2017, 77, 2857–2868. [Google Scholar] [CrossRef] [Green Version]
- Maeda, K.; Das, D.; Nakata, H.; Mitsuya, H. CCR5 inhibitors: Emergence, success, and challenges. Expert Opin. Emerg. Drugs 2012, 17, 135–145. [Google Scholar] [CrossRef]
- Tupova, L.; Ceckova, M.; Ambrus, C.; Sorf, A.; Ptackova, Z.; Gaborik, Z.; Staud, F. Interactions between Maraviroc and the ABCB1, ABCG2, and ABCC2 Transporters: An Important Role in Transplacental Pharmacokinetics. Drug Metab. Dispos. 2019, 47, 954–960. [Google Scholar] [CrossRef]
- Zhang, X.; Haney, K.M.; Richardson, A.C.; Wilson, E.; Gewirtz, D.A.; Ware, J.L.; Zehner, Z.E.; Zhang, Y. Anibamine, a natural product CCR5 antagonist, as a novel lead for the development of anti-prostate cancer agents. Bioorganic Med. Chem. Lett. 2010, 20, 4627–4630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, F.; Arnatt, C.K.; Haney, K.M.; Fang, H.C.; Bajacan, J.E.; Richardson, A.C.; Ware, J.L.; Zhang, Y. Structure activity relationship studies of natural product chemokine receptor CCR5 antagonist anibamine toward the development of novel anti prostate cancer agents. Eur. J. Med. Chem. 2012, 55, 395–408. [Google Scholar] [CrossRef]
- Mukaida, N.; Tanabe, Y.; Baba, T. Chemokines as a Conductor of Bone Marrow Microenvironment in Chronic Myeloid Leukemia. Int. J. Mol. Sci. 2017, 18, 1824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abe, M.; Hiura, K.; Wilde, J.; Moriyama, K.; Hashimoto, T.; Ozaki, S.; Wakatsuki, S.; Kosaka, M.; Kido, S.; Inoue, D.; et al. Role for macrophage inflammatory protein (MIP)-1alpha and MIP-1beta in the development of osteolytic lesions in multiple myeloma. Blood 2002, 100, 2195–2202. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Feng, W.; Wang, H.; Wang, L.; Yang, X.; Yang, F.; Zhang, Y.; Liu, X.; Zhang, D.; Ren, Q.; et al. Blocking migration of regulatory T cells to leukemic hematopoietic microenvironment delays disease progression in mouse leukemia model. Cancer Lett. 2019, 469, 151–161. [Google Scholar] [CrossRef]
- Oba, Y.; Lee, J.W.; Ehrlich, L.A.; Chung, H.Y.; Jelinek, D.F.; Callander, N.; Horuk, R.; Choi, S.J.; Roodman, G.D. MIP-1α utilizes both CCR1 and CCR5 to induce osteoclast formation and increase adhesion of myeloma cells to marrow stromal cells. Exp. Hematol. 2005, 33, 272–278. [Google Scholar] [CrossRef]
- Binder, S.; Luciano, M.; Horejs-Höck, J. The cytokine network in acute myeloid leukemia (AML): A focus on pro- and anti-inflammatory mediators. Cytokine Growth Factor Rev. 2018, 43, 8–15. [Google Scholar] [CrossRef]
- Cignetti, A.; Vallario, A.; Roato, I.; Circosta, P.; Strola, G.; Scielzo, C.; Allione, B.; Garetto, L.; Caligaris-Cappio, F.; Ghia, P. The characterization of chemokine production and chemokine receptor expression reveals possible functional cross-talks in AML blasts with monocytic differentiation. Exp. Hematol. 2003, 31, 495–503. [Google Scholar] [CrossRef]
- Bruserud, Ø.; Ryningen, A.; Olsnes, A.M.; Stordrange, L.; Øyan, A.M.; Kalland, K.H.; Gjertsen, B.T. Subclassification of patients with acute myelogenous leukemia based on chemokine responsiveness and constitutive chemokine release by their leukemic cells. Haematologica 2007, 92, 332–341. [Google Scholar] [CrossRef] [Green Version]
- Yazdani, Z.; Mousavi, Z.; Ghasemimehr, N.; Khandany, B.K.; Nikbakht, R.; Jafari, E.; Fatemi, A.; Hassanshahi, G. Differential regulatory effects of chemotherapeutic protocol on CCL3_CCL4_CCL5/CCR5 axes in acute myeloid leukemia patients with monocytic lineage. Life Sci. 2019, 240, 117071. [Google Scholar] [CrossRef]
- Waldeck, S.; Rassner, M.; Keye, P.; Follo, M.; Herchenbach, D.; Endres, C.; Charlet, A.; Andrieux, G.; Salzer, U.; Boerries, M.; et al. CCL5 mediates target-kinase independent resistance to FLT3 inhibitors in FLT3-ITD-positive AML. Mol. Oncol. 2020, 14, 779–794. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Philip, M.; Ferrell, P.B. Alterations of T-cell-mediated immunity in acute myeloid leukemia. Oncogene 2020, 39, 3611–3619. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.; Qazi, S.; Ozer, Z.; Gaynon, P.; Reaman, G.H.; Uckun, F.M. CD22 Exon 12 deletion is a characteristic genetic defect of therapy-refractory clones in paediatric acute lymphoblastic leukaemia. Br. J. Haematol. 2011, 156, 89–98. [Google Scholar] [CrossRef]
- Zucchetto, A.; Benedetti, D.; Tripodo, C.; Bomben, R.; Bo, M.D.; Marconi, D.; Bossi, F.; Lorenzon, D.; Degan, M.; Rossi, F.M.; et al. CD38/CD31, the CCL3 and CCL4 Chemokines, and CD49d/Vascular Cell Adhesion Molecule-1 Are Interchained by Sequential Events Sustaining Chronic Lymphocytic Leukemia Cell Survival. Cancer Res. 2009, 69, 4001–4009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zucchetto, A.; Tripodo, C.; Benedetti, D.; Deaglio, S.; Gaïdano, G.; Del Poeta, G.; Gattei, V. Monocytes/macrophages but not T lymphocytes are the major targets of the CCL3/CCL4 chemokines produced by CD38+CD49d+chronic lymphocytic leukaemia cells. Br. J. Haematol. 2010, 150, 111–113. [Google Scholar] [CrossRef]
- Aldinucci, D.; Celegato, M.; Casagrande, N.; Information, P.E.K.F.C. Microenvironmental interactions in classical Hodgkin lymphoma and their role in promoting tumor growth, immune escape and drug resistance. Cancer Lett. 2016, 380, 243–252. [Google Scholar] [CrossRef] [PubMed]
- Fischer, M.; Juremalm, M.; Olsson, N.; Backlin, C.; Sundström, C.; Nilsson, K.; Enblad, G.; Nilsson, G. Expression of CCL5/RANTES by Hodgkin and Reed-Sternberg cells and its possible role in the recruitment of mast cells into lymphomatous tissue. Int. J. Cancer 2003, 107, 197–201. [Google Scholar] [CrossRef] [PubMed]
- Buri, C.; Körner, M.; Schärli, P.; Cefai, D.; Uguccioni, M.; Mueller, C.; Laissue, J.A.; Mazzucchelli, L.; Ben-Smith, A.; Dove, S.K.; et al. CC chemokines and the receptors CCR3 and CCR5 are differentially expressed in the nonneoplastic leukocytic infiltrates of Hodgkin disease. Blood 2001, 97, 1543–1548. [Google Scholar] [CrossRef] [PubMed]
- Maggio, E.; Berg, A.V.D.; Diepstra, A.; Kluiver, J.; Visser, L.; Poppema, S. Chemokines, cytokines and their receptors in Hodgkin’s lymphoma cell lines and tissues. Ann. Oncol. 2002, 13, 52–56. [Google Scholar] [CrossRef]
- Uchihara, J.-N.; Krensky, A.M.; Matsuda, T.; Kawakami, H.; Okudaira, T.; Masuda, M.; Ohta, T.; Takasu, N.; Mori, N. Retracted: Transactivation of the CCL5/RANTES gene by Epstein-Barr virus latent membrane protein 1. Int. J. Cancer 2004, 114, 747–755. [Google Scholar] [CrossRef]
- Aldinucci, D.; Pinto, A.; Gloghini, A.; Carbone, A. Chemokine receptors as therapeutic tools in Hodgkin lymphoma: CCR4 and beyond. Blood 2010, 115, 746–747. [Google Scholar] [CrossRef] [Green Version]
- Aldinucci, D.; Gloghini, A.; Pinto, A.; De Filippi, R.; Carbone, A. The classical Hodgkin’s lymphoma microenvironment and its role in promoting tumour growth and immune escape. J. Pathol. 2010, 221, 248–263. [Google Scholar] [CrossRef] [PubMed]
- Carbone, A.; Gloghini, A.; Aldinucci, D.; Gattei, V.; Dalla-Favera, R.; Gaidano, G. Expression pattern of MUM1/IRF4 in the spectrum of pathology of Hodgkin’s disease. Br. J. Haematol. 2002, 117, 366–372. [Google Scholar] [CrossRef] [PubMed]
- Aldinucci, D.; Celegato, M.; Borghese, C.; Colombatti, A.; Carbone, A. IRF4 silencing inhibits Hodgkin lymphoma cell proliferation, survival and CCL5 secretion. Br. J. Haematol. 2010, 152, 182–190. [Google Scholar] [CrossRef] [PubMed]
- Lentzsch, S.; Gries, M.; Janz, M.; Bargou, R.; Dörken, B.; Mapara, M.Y. Macrophage inflammatory protein 1-alpha (MIP-1α) triggers migration and signaling cascades mediating survival and proliferation in multiple myeloma (MM) cells. Blood 2003, 101, 3568–3573. [Google Scholar] [CrossRef] [Green Version]
- Vallet, S.; Anderson, K. CCR1 as a target for multiple myeloma. Expert Opin. Ther. Targets 2011, 15, 1037–1047. [Google Scholar] [CrossRef]
- Borrello, I.M. Can we change the disease biology of multiple myeloma? Leuk. Res. 2012, 36, S3–S12. [Google Scholar] [CrossRef] [Green Version]
- Abe, M.; Hiura, K.; Wilde, J.; Shioyasono, A.; Moriyama, K.; Hashimoto, T.; Kido, S.; Oshima, T.; Shibata, H.; Ozaki, S.; et al. Osteoclasts enhance myeloma cell growth and survival via cell-cell contact: A vicious cycle between bone destruction and myeloma expansion. Blood 2004, 104, 2484–2491. [Google Scholar] [CrossRef]
- Lee, J.-W.; Hoshino, A.; Inoue, K.; Saitou, T.; Uehara, S.; Kobayashi, Y.; Ueha, S.; Matsushima, K.; Yamaguchi, A.; Imai, Y.; et al. The HIV co-receptor CCR5 regulates osteoclast function. Nat. Commun. 2017, 8, 2226. [Google Scholar] [CrossRef]
- Vallet, S.; Pozzi, S.; Patel, K.; Vaghela, N.; Fulciniti, M.T.; Veiby, P.; Hideshima, T.; Santo, L.; Cirstea, D.; Scadden, D.T.; et al. A novel role for CCL3 (MIP-1α) in myeloma-induced bone disease via osteocalcin downregulation and inhibition of osteoblast function. Leukemia 2011, 25, 1174–1181. [Google Scholar] [CrossRef]
- Terpos, E.; Politou, M.; Szydlo, R.; Goldman, J.M.; Apperley, J.F.; Rahemtulla, A. Serum levels of macrophage inflammatory protein-1 alpha (MIP-1α) correlate with the extent of bone disease and survival in patients with multiple myeloma. Br. J. Haematol. 2003, 123, 106–109. [Google Scholar] [CrossRef] [PubMed]
- Roussou, M.; Tasidou, A.; Dimopoulos, M.A.; Kastritis, E.; Migkou, M.; Christoulas, D.; Gavriatopoulou, M.; Zagouri, F.; Matsouka, C.; Anagnostou, D.; et al. Increased expression of macrophage inflammatory protein-1α on trephine biopsies correlates with extensive bone disease, increased angiogenesis and advanced stage in newly diagnosed patients with multiple myeloma. Leukemia 2009, 23, 2177–2181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hashimoto, T.; Abe, M.; Oshima, T.; Shibata, H.; Ozaki, S.; Inoue, D.; Matsumoto, T. Ability of myeloma cells to secrete macrophage inflammatory protein (MIP)-1alpha and MIP-1beta correlates with lytic bone lesions in patients with multiple myeloma. Br. J. Haematol. 2004, 125, 38–41. [Google Scholar] [CrossRef] [PubMed]
- Dairaghi, D.J.; Oyajobi, B.O.; Gupta, A.; McCluskey, B.; Miao, S.; Powers, J.P.; Seitz, L.C.; Wang, Y.; Zeng, Y.; Zhang, P.; et al. CCR1 blockade reduces tumor burden and osteolysis in vivo in a mouse model of myeloma bone disease. Blood 2012, 120, 1449–1457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.S.; Ryu, K.J.; Ko, Y.H. Macrophage inflammatory protein 1 alpha (MIP-1?) may be associated with poor outcome in patients with extranodal NK/T-cell lymphoma. Hematol. Oncol. 2016, 35, 310–316. [Google Scholar] [CrossRef]
- Butrym, A.; Kryczek, I.; Dlubek, D.; Jaskula, E.; Lange, A.; Jurczyszyn, A.; Mazur, G. High expression of CC chemokine receptor 5 (CCR5) promotes disease progression in patients with B-cell non-Hodgkin lymphomas. Curr. Probl. Cancer 2018, 42, 268–275. [Google Scholar] [CrossRef] [PubMed]
- Halvorsen, E.C.; Hamilton, M.J.; Young, A.; Wadsworth, B.J.; LePard, N.E.; Lee, H.N.; Firmino, N.; Collier, J.L.; Bennewith, K.L. Maraviroc decreases CCL8-mediated migration of CCR5+ regulatory T cells and reduces metastatic tumor growth in the lungs. OncoImmunology 2016, 5, e1150398. [Google Scholar] [CrossRef] [Green Version]
- Sax, M.J.; Gasch, C.; Athota, V.R.; Freeman, R.; Rasighaemi, P.; Westcott, D.E.; Day, C.J.; Nikolic, I.; Elsworth, B.L.; Wei, M.; et al. Cancer cell CCL5 mediates bone marrow independent angiogenesis in breast cancer. Oncotarget 2016, 7, 85437–85449. [Google Scholar] [CrossRef] [Green Version]
- Jin, K.; Pandey, N.B.; Popel, A.S. Simultaneous blockade of IL-6 and CCL5 signaling for synergistic inhibition of triple-negative breast cancer growth and metastasis. Breast Cancer Res. 2018, 20, 54. [Google Scholar] [CrossRef] [Green Version]
- Zazo, S.; González-Alonso, P.; Martin-Aparicio, E.; Chamizo, C.; Luque, M.; Sanz-Alvarez, M.; Mínguez, P.; Gómez-López, G.; Cristobal, I.; Caramés, C.; et al. Autocrine CCL5 effect mediates trastuzumab resistance by ERK pathway activation in HER2-positive breast cancer. Mol. Cancer Ther. 2020. [Google Scholar] [CrossRef]
- Brett, E.; Sauter, M.; Timmins, É.; Azimzadeh, O.; Rosemann, M.; Merl-Pham, J.; Hauck, S.M.; Nelson, P.J.; Becker, K.F.; Schunn, I.; et al. Oncogenic Linear Collagen VI of Invasive Breast Cancer Is Induced by CCL5. J. Clin. Med. 2020, 9, 991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nie, Y.; Huang, H.; Guo, M.; Chen, J.; Wu, W.; Li, W.; Xu, X.; Lin, X.; Fu, W.; Yao, Y.-D.; et al. Breast Phyllodes Tumors Recruit and Repolarize Tumor-Associated Macrophages via Secreting CCL5 to Promote Malignant Progression, Which Can Be Inhibited by CCR5 Inhibition Therapy. Clin. Cancer Res. 2019, 25, 3873–3886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, W.; Tong, Y.; Li, Y.; Yuan, J.; Hu, S.; Hu, T.; Song, G. Mesenchymal stem cells in inflammatory microenvironment potently promote metastatic growth of cholangiocarcinoma via activating Akt/NF-κB signaling by paracrine CCL5. Oncotarget 2017, 8, 73693–73704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanabe, Y.; Sasaki, S.; Mukaida, N.; Baba, T. Blockade of the chemokine receptor, CCR5, reduces the growth of orthotopically injected colon cancer cells via limiting cancerassociated fibroblast accumulation. Oncotarget 2016, 7, 48335–48345. [Google Scholar] [CrossRef] [Green Version]
- Nishikawa, G.; Kawada, K.; Nakagawa, J.; Toda, K.; Ogawa, R.; Inamoto, S.; Mizuno, R.; Itatani, Y.; Sakai, Y. Bone marrow-derived mesenchymal stem cells promote colorectal cancer progression via CCR5. Cell Death Dis. 2019, 10, 264. [Google Scholar] [CrossRef] [Green Version]
- Cambien, B.; Richard-Fiardo, P.; Karimdjee, B.F.; Martini, V.; Ferrua, B.; Pitard, B.; Schmid-Antomarchi, H.; Schmid-Alliana, A. CCL5 Neutralization Restricts Cancer Growth and Potentiates the Targeting of PDGFRβ in Colorectal Carcinoma. PLoS ONE 2011, 6, e28842. [Google Scholar] [CrossRef]
- Liu, C.; Yao, Z.; Wang, J.; Zhang, W.; Yang, Y.; Zhang, Y.; Qu, X.; Zhu, Y.; Zou, J.; Peng, S.; et al. Macrophage-derived CCL5 facilitates immune escape of colorectal cancer cells via the p65/STAT3-CSN5-PD-L1 pathway. Cell Death Differ. 2019, 27, 1765–1781. [Google Scholar] [CrossRef]
- Zhang, S.; Zhong, M.; Wang, C.; Xu, Y.; Gao, W.-Q.; Zhang, Y. CCL5-deficiency enhances intratumoral infiltration of CD8+ T cells in colorectal cancer. Cell Death Dis. 2018, 9, 766. [Google Scholar] [CrossRef] [Green Version]
- Okita, K.; Furuhata, T.; Kimura, Y.; Kawakami, M.; Yamaguchi, K.; Tsuruma, T.; Zembutsu, H.; Hirata, K. The interplay between gastric cancer cell lines and PBMCs mediated by the CC chemokine RANTES plays an important role in tumor progression. J. Exp. Clin. Cancer Res. 2005, 24, 439–446. [Google Scholar] [PubMed]
- Sugasawa, H.; Ichikura, T.; Kinoshita, M.; Ono, S.; Majima, T.; Tsujimoto, H.; Chochi, K.; Hiroi, S.; Takayama, E.; Saitoh, D.; et al. Gastric cancer cells exploit CD4+ cell-derived CCL5 for their growth and prevention of CD8+ cell-involved tumor elimination. Int. J. Cancer 2008, 122, 2535–2541. [Google Scholar] [CrossRef]
- Laudati, E.; Currò, D.; Navarra, P.; Lisi, L. Blockade of CCR5 receptor prevents M2 microglia phenotype in a microglia-glioma paradigm. Neurochem. Int. 2017, 108, 100–108. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Wang, Y.; Xue, Y.; Lv, W.; Zhang, Y.; He, S. Critical roles of chemokine receptor CCR5 in regulating glioblastoma proliferation and invasion. Acta Biochim. et Biophys. Sin. 2015, 47, 890–898. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Chen, L.; Dang, W.-Q.; Cao, M.-F.; Xiao, J.-F.; Lv, S.-Q.; Jiang, W.-J.; Yao, X.-H.; Lu, H.-M.; Miao, J.-Y.; et al. CCL8 secreted by tumor-associated macrophages promotes invasion and stemness of glioblastoma cells via ERK1/2 signaling. Lab. Investig. 2019, 100, 619–629. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Luan, X.R. miR-147a suppresses the metastasis of non-small-cell lung cancer by targeting CCL5. J. Int. Med Res. 2019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, L.; Zhu, X.; Zhang, L.; Xu, Y.; Chen, G.; Luo, J. EZH2 enhances expression of CCL5 to promote recruitment of macrophages and invasion in lung cancer. Biotechnol. Appl. Biochem. 2020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Umansky, V.; Blattner, C.; Gebhardt, C.; Utikal, J.S. CCR5 in recruitment and activation of myeloid-derived suppressor cells in melanoma. Cancer Immunol. Immunother. 2017, 66, 1015–1023. [Google Scholar] [CrossRef]
- Blattner, C.; Fleming, V.; Weber, R.; Himmelhan, B.; Altevogt, P.; Gebhardt, C.; Schulze, T.J.; Razon, H.; Hawila, E.; Wildbaum, G.; et al. CCR5+Myeloid-Derived Suppressor Cells Are Enriched and Activated in Melanoma Lesions. Cancer Res. 2017, 78, 157–167. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Arnatt, C.K.; Zhang, F.; Wang, J.; Haney, K.M.; Fang, X. The potential role of anibamine, a natural product CCR5 antagonist, and its analogues as leads toward development of anti-ovarian cancer agents. Bioorganic Med. Chem. Lett. 2012, 22, 5093–5097. [Google Scholar] [CrossRef]
- Mitra, A.K.; Zillhardt, M.; Hua, Y.; Tiwari, P.; Murmann, A.E.; Peter, M.E.; Lengyel, E. MicroRNAs reprogram normal fibroblasts into cancer-associated fibroblasts in ovarian cancer. Cancer Discov. 2012, 2, 1100–1108. [Google Scholar] [CrossRef] [Green Version]
- Long, H.; Xiang, T.; Qi, W.; Huang, J.; Chen, J.; He, L.; Liang, Z.; Guo, B.; Li, Y.; Xie, R.; et al. CD133+ ovarian cancer stem-like cells promote non-stem cancer cell metastasis via CCL5 induced epithelial-mesenchymal transition. Oncotarget 2015, 6, 5846–5859. [Google Scholar] [CrossRef] [Green Version]
- Tang, S.; Xiang, T.; Huang, S.; Zhou, J.; Wang, Z.; Xie, R.; Long, H.; Zhu, B. Ovarian cancer stem-like cells differentiate into endothelial cells and participate in tumor angiogenesis through autocrine CCL5 signaling. Cancer Lett. 2016, 376, 137–147. [Google Scholar] [CrossRef]
- Huang, H.; Zepp, M.; Georges, R.B.; Jarahian, M.; Kazemi, M.; Eyol, E.; Berger, M.R. The CCR5 antagonist maraviroc causes remission of pancreatic cancer liver metastasis in nude rats based on cell cycle inhibition and apoptosis induction. Cancer Lett. 2020, 474, 82–93. [Google Scholar] [CrossRef] [PubMed]
- Barry, S.; Carlsen, E.; Marques, P.; Stiles, C.E.; Gadaleta, E.; Berney, D.M.; Roncaroli, F.; Chelala, C.; Solomou, A.; Herincs, M.; et al. Tumor microenvironment defines the invasive phenotype of AIP-mutation-positive pituitary tumors. Oncogene 2019, 38, 5381–5395. [Google Scholar] [CrossRef] [PubMed]
- Zhao, R.; Bei, X.; Yang, B.; Wang, X.; Jiang, C.; Shi, F.; Wang, X.; Zhu, Y.; Jing, Y.; Han, B.; et al. Endothelial cells promote metastasis of prostate cancer by enhancing autophagy. J. Exp. Clin. Cancer Res. 2018, 37, 221. [Google Scholar] [CrossRef] [Green Version]
- Xing, Q.; Xie, H.; Zhu, B.; Sun, Z.; Huang, Y. MiR-455-5p Suppresses the Progression of Prostate Cancer by Targeting CCR5. BioMed Res. Int. 2019, 2019, 6394784–6394788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luboshits, G.; Shina, S.; Kaplan, O.; Engelberg, S.; Nass, D.; Lifshitz-Mercer, B.; Chaitchik, S.; Keydar, I.; Ben-Baruch, A. Elevated expression of the CC chemokine regulated on activation, normal T cell expressed and secreted (RANTES) in advanced breast carcinoma. Cancer Res. 1999, 59. [Google Scholar]
- Mi, Z.; Bhattacharya, S.D.; Kim, V.M.; Guo, H.; Talbot, L.J.; Kuo, P.C. Osteopontin promotes CCL5-mesenchymal stromal cell-mediated breast cancer metastasis. Carcinogenesis 2011, 32, 477–487. [Google Scholar] [CrossRef]
- Lv, D.; Zhang, Y.; Kim, H.-J.; Zhang, L.; Ma, X. CCL5 as a potential immunotherapeutic target in triple-negative breast cancer. Cell. Mol. Immunol. 2013, 10, 303–310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smeets, A.; Brouwers, B.; Hatse, S.; Laenen, A.; Paridaens, R.; Floris, G.; Wildiers, H.; Christiaens, M.-R. Circulating CCL5 Levels in Patients with Breast Cancer: Is There a Correlation with Lymph Node Metastasis? ISRN Immunol. 2013, 2013, 1–5. [Google Scholar] [CrossRef]
- Li, S.; Wan, S.; Hu, J.; Fang, D.; Zhao, R.; Yuan, S.; Sun, L.; Zhang, L. Chemokine C-C motif receptor 5 and C-C motif ligand 5 promote cancer cell migration under hypoxia. Cancer Sci. 2012, 103, 904–912. [Google Scholar] [CrossRef]
- Swamydas, M.; Ricci, K.; Rego, S.L.; Dréau, D. Mesenchymal stem cell-derived CCL-9 and CCL-5 promote mammary tumor cell invasion and the activation of matrix metalloproteinases. Cell Adhes. Migr. 2013, 7, 315–324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Lv, D.; Kim, H.-J.; Kurt, R.A.; Bu, W.; Li, Y.; Ma, X. A novel role of hematopoietic CCL5 in promoting triple-negative mammary tumor progression by regulating generation of myeloid-derived suppressor cells. Cell Res. 2012, 23, 394–408. [Google Scholar] [CrossRef] [PubMed]
- Walens, A.; DiMarco, A.V.; Lupo, R.; Kroger, B.R.; Damrauer, J.S.; Alvarez, J.V. CCL5 promotes breast cancer recurrence through macrophage recruitment in residual tumors. eLife 2019, 8, 43653. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.; Fertig, E.J.; Jin, K.; Sukumar, S.; Pandey, N.B.; Popel, A.S. Breast cancer cells condition lymphatic endothelial cells within pre-metastatic niches to promote metastasis. Nat. Commun. 2014, 5, 4715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Limaiem, F.; Kashyap, S. Cancer, Phyllodes Tumor Of The Breast (Cystosarcoma). In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2019; Available online: https://www.ncbi.nlm.nih.gov/books/NBK541138/PMID:31082182 (accessed on 21 April 2020).
- Musha, H.; Ohtani, H.; Mizoi, T.; Kinouchi, M.; Nakayama, T.; Shiiba, K.; Miyagawa, K.; Nagura, H.; Yoshie, O.; Sasaki, I. Selective infiltration of CCR5+CXCR3+ T lymphocytes in human colorectal carcinoma. Int. J. Cancer 2005, 116, 949–956. [Google Scholar] [CrossRef] [PubMed]
- Üçüncü, M.; Serilmez, M.; Sarı, M.; Bademler, S.; Karabulut, S. The Diagnostic Significance of PDGF, EphA7, CCR5, and CCL5 Levels in Colorectal Cancer. Biomolecules 2019, 9, 464. [Google Scholar] [CrossRef] [Green Version]
- Sasaki, S.; Baba, T.; Shinagawa, K.; Matsushima, K.; Mukaida, N. Crucial involvement of the CCL3-CCR5 axis-mediated fibroblast accumulation in colitis-associated carcinogenesis in mice. Int. J. Cancer 2014, 135, 1297–1306. [Google Scholar] [CrossRef] [Green Version]
- Suarez-Carmona, M.; Chaorentong, P.; Kather, J.N.; Rothenheber, R.; Ahmed, A.; Berthel, A.; Heinzelmann, A.; Moraleda, R.; Valous, N.A.; Koşaloğlu-Yalçın, Z.; et al. CCR5 status and metastatic progression in colorectal cancer. OncoImmunology 2019, 8, e1626193. [Google Scholar] [CrossRef]
- Sima, A.R.; Sima, H.R.; Rafatpanah, H.; Hosseinnezhad, H.; Ghaffarzadehgan, K.; Valizadeh, N.; Bahar, M.M.; Hakimi, H.R.; Masoom, A.; Noorbakhsh, A.; et al. Serum Chemokine Ligand 5 (CCL5/RANTES) Level Might be Utilized as a Predictive Marker of Tumor Behavior and Disease Prognosis in Patients with Gastric Adenocarcinoma. J. Gastrointest. Cancer 2014, 45, 476–480. [Google Scholar] [CrossRef]
- Wang, T.; Wei, Y.; Tian, L.; Song, H.; Ma, Y.; Yao, Q.; Feng, M.; Wang, Y.; Gao, M.; Xue, Y. C-C motif chemokine ligand 5 (CCL5) levels in gastric cancer patient sera predict occult peritoneal metastasis and a poorer prognosis. Int. J. Surg. 2016, 32, 136–142. [Google Scholar] [CrossRef]
- Cao, Z.; Xu, X.; Luo, X.; Li, L.; Huang, B.; Li, X.; Tao, D.; Hu, J.; Gong, J. Role of RANTES and its receptor in gastric cancer metastasis. Acta Acad. Med. Wuhan 2011, 31, 342–347. [Google Scholar] [CrossRef] [PubMed]
- Fukui, R.; Nishimori, H.; Hata, F.; Yasoshima, T.; Ohno, K.; Nomura, H.; Yanai, Y.; Tanaka, H.; Kamiguchi, K.; Denno, R.; et al. Metastases-Related Genes in the Classification of Liver and Peritoneal Metastasis in Human Gastric Cancer. J. Surg. Res. 2005, 129, 94–100. [Google Scholar] [CrossRef]
- Mencarelli, A.; Graziosi, L.; Renga, B.; Cipriani, S.; D’Amore, C.; Francisci, D.; Bruno, A.; Baldelli, F.; Donini, A.; Fiorucci, S. CCR5 Antagonism by Maraviroc Reduces the Potential for Gastric Cancer Cell Dissemination. Transl. Oncol. 2013, 6, 784–793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, S.-S.; Fan, W.; Sun, L.; Li, S.; Zhao, R.-P.; Zhang, L.; Yu, B.-Y.; Yuan, S.-T. The saponin DT-13 inhibits gastric cancer cell migration through down-regulation of CCR5-CCL5 axis. Chin. J. Nat. Med. 2014, 12, 833–840. [Google Scholar] [CrossRef]
- Sugasawa, H.; Ichikura, T.; Tsujimoto, H.; Kinoshita, M.; Morita, D.; Ono, S.; Chochi, K.; Tsuda, H.; Seki, S.; Mochizuki, H. Prognostic significance of expression of CCL5/RANTES receptors in patients with gastric cancer. J. Surg. Oncol. 2008, 97, 445–450. [Google Scholar] [CrossRef]
- Yang, T.; Chen, M.; Yang, X.; Zhang, X.; Zhang, Z.; Sun, Y.; Xu, B.; Hua, J.; He, Z.; Song, Z. Down-regulation of KLF5 in cancer-associated fibroblasts inhibit gastric cancer cells progression by CCL5/CCR5 axis. Cancer Biol. Ther. 2017, 18, 806–815. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Qu, B. Expression of chemotactic factor CCL5 in gastric cancer tissue and its correlation with macrophage marker CD86. Biomed. Res. 2017, 28, 6388–6391. [Google Scholar]
- Kranjc, M.K.; Novak, M.; Pestell, R.G.; Lah, T.T. Cytokine CCL5 and receptor CCR5 axis in glioblastoma multiforme. Radiol. Oncol. 2019, 53, 397–406. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Wang, Y.; Liu, T.; Yang, N.; Xu, S.; Li, X. Hypoxia and macrophages promote glioblastoma invasion by the CCL4-CCR5 axis. Oncol. Rep. 2016, 36, 3522–3528. [Google Scholar] [CrossRef]
- Solga, A.C.; Pong, W.W.; Kim, K.-Y.; Cimino, P.J.; Toonen, J.A.; Walker, J.; Wylie, T.; Magrini, V.; Griffith, M.; Griffith, O.L.; et al. RNA Sequencing of Tumor-Associated Microglia Reveals Ccl5 as a Stromal Chemokine Critical for Neurofibromatosis-1 Glioma Growth. Neoplasia 2015, 17, 776–788. [Google Scholar] [CrossRef] [Green Version]
- Pan, Y.; Smithson, L.J.; Ma, Y.; Hambardzumyan, D.; Gutmann, D.H. Ccl5 establishes an autocrine high-grade glioma growth regulatory circuit critical for mesenchymal glioblastoma survival. Oncotarget 2017, 8, 32977–32989. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blaszczyk, J.; Van Coillie, E.; Proost, P.; Van Damme, J.; Opdenakker, G.; Bujacz, G.D.; Wang, J.M.; Ji, X. Complete Crystal Structure of Monocyte Chemotactic Protein-2, a CC Chemokine that Interacts with Multiple Receptors. Biochemistry 2000, 39, 14075–14081. [Google Scholar] [CrossRef] [PubMed]
- Domingueti, C.-B.; Janini, J.-B.-M.; Paranaíba, L.-M.-R.; Lozano-Burgos, C.; Olivero, P.; González-Arriagada, W.-A. Prognostic value of immunoexpression of CCR4, CCR5, CCR7 and CXCR4 in squamous cell carcinoma of tongue and floor of the mouth. Med. Oral Patol. Oral Cir. Bucal 2019, 24, e354–e363. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Wang, J.; Qian, Z.; Han, Y. CCR5 is Associated With Immune Cell Infiltration and Prognosis of Lung Cancer. J. Thorac. Oncol. 2019, 14, e102–e103. [Google Scholar] [CrossRef]
- Brehm, J.E.; Bedogni, B. Blockade of CCR5 in melanoma: An alternative immune checkpoint modulator. Exp. Dermatol. 2019, 29, 196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, K.; Gong, C.; Peng, H.; Fang, H.; Zhou, J.; Li, J.; Chen, S.; Zheng, H. High CCL5 expression is associated with osteosarcoma metastasis and poor prognosis of patients with osteosarcoma. Mol. Med. Rep. 2017, 16, 6953–6957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farsinejad, S.; Cattabiani, T.; Muranen, T.; Iwanicki, M. Ovarian Cancer Dissemination-A Cell Biologist’s Perspective. Cancers 2019, 11, 1957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milliken, D.; Scotton, C.; Raju, S.; Balkwill, F.; Wilson, J. Analysis of chemokines and chemokine receptor expression in ovarian cancer ascites. Clin. Cancer Res. 2002, 8, 1108–1114. [Google Scholar] [PubMed]
- Tsukishiro, S.; Suzumori, N.; Nishikawa, H.; Arakawa, A.; Suzumori, K. Elevated serum RANTES levels in patients with ovarian cancer correlate with the extent of the disorder. Gynecol. Oncol. 2006, 102, 542–545. [Google Scholar] [CrossRef] [PubMed]
- You, Y.; Li, Y.; Li, M.; Lei, M.; Wu, M.; Qu, Y.; Yuan, Y.; Chen, T.; Jiang, H. Ovarian cancer stem cells promote tumour immune privilege and invasion via CCL5 and regulatory T cells. Clin. Exp. Immunol. 2017, 191, 60–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schickel, R.; Boyerinas, B.; Park, S.-M.; Peter, M.E. MicroRNAs: Key players in the immune system, differentiation, tumorigenesis and cell death. Oncogene 2008, 27, 5959–5974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pokhriyal, R.; Hariprasad, R.; Kumar, L.; Hariprasad, G. Chemotherapy Resistance in Advanced Ovarian Cancer Patients. Biomark. Cancer 2019, 11. [Google Scholar] [CrossRef] [PubMed]
- Pasquier, J.; Gosset, M.; Geyl, C.; Hoarau-Véchot, J.; Chevrot, A.; Pocard, M.; Mirshahi, M.; Lis, R.; Rafii, A.; Touboul, C. CCL2/CCL5 secreted by the stroma induce IL-6/PYK2 dependent chemoresistance in ovarian cancer. Mol. Cancer 2018, 17, 47. [Google Scholar] [CrossRef]
- Cui, Z.Y.; Park, S.J.; Jo, E.; Hwang, I.-H.; Lee, K.-B.; Kim, S.-W.; Kim, D.J.; Joo, J.C.; Hong, S.H.; Lee, M.-G.; et al. Cordycepin induces apoptosis of human ovarian cancer cells by inhibiting CCL5-mediated Akt/NF-κB signaling pathway. Cell Death Discov. 2018, 4, 62. [Google Scholar] [CrossRef] [Green Version]
- Daly, A.F.; Tichomirowa, M.A.; Petrossians, P.; Heliövaara, E.; Jaffrain-Rea, M.L.; Barlier, A.; Naves, L.A.; Ebeling, T.; Karhu, A.; Raappana, A.; et al. Clinical Characteristics and Therapeutic Responses in Patients with Germ-Line AIP Mutations and Pituitary Adenomas: An International Collaborative Study. J. Clin. Endocrinol. Metab. 2010, 95, E373–E383. [Google Scholar] [CrossRef] [Green Version]
- Huang, R.; Wang, S.; Wang, N.; Zheng, Y.; Zhou, J.; Yang, B.; Wang, X.; Zhang, J.; Guo, L.; Wang, S.; et al. CCL5 derived from tumor-associated macrophages promotes prostate cancer stem cells and metastasis via activating β-catenin/STAT3 signaling. Cell Death Dis. 2020, 11, 1–20. [Google Scholar] [CrossRef]
- Silver, D.A.; Pellicer, I.; Fair, W.R.; Heston, W.D.; Cordon-Cardo, C. Prostate-specific membrane antigen expression in normal and malignant human tissues. Clin. Cancer Res. 1997, 3, 81–85. [Google Scholar]
- Colombatti, M.; Grasso, S.; Porzia, A.; Fracasso, G.; Scupoli, M.T.; Cingarlini, S.; Poffe, O.; Naim, H.Y.; Heine, M.; Tridente, G.; et al. The Prostate Specific Membrane Antigen Regulates the Expression of IL-6 and CCL5 in Prostate Tumour Cells by Activating the MAPK Pathways1. PLoS ONE 2009, 4, e4608. [Google Scholar] [CrossRef]
- Luo, J.; Lee, S.O.; Cui, Y.; Yang, R.; Li, L.; Chang, C. Infiltrating bone marrow mesenchymal stem cells (BM-MSCs) increase prostate cancer cell invasion via altering the CCL5/HIF2α/androgen receptor signals. Oncotarget 2015, 6, 27555–27565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borghese, C.; Cattaruzza, L.; Pivetta, E.; Normanno, N.; De Luca, A.; Mazzucato, M.; Celegato, M.; Colombatti, A.; Aldinucci, D. Gefitinib inhibits the cross-talk between mesenchymal stem cells and prostate cancer cells leading to tumor cell proliferation and inhibition of docetaxel activity. J. Cell. Biochem. 2013, 114, 1135–1144. [Google Scholar] [CrossRef]
- Luo, J.; Lee, S.O.; Liang, L.; Huang, C.-K.; Li, L.; Wen, S.; Chang, C. Infiltrating bone marrow mesenchymal stem cells increase prostate cancer stem cell population and metastatic ability via secreting cytokines to suppress androgen receptor signaling. Oncogene 2013, 33, 2768–2778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Machlus, K.R.; Johnson, K.E.; Kulenthirarajan, R.; Forward, J.A.; Tippy, M.D.; Soussou, T.S.; El-Husayni, S.H.; Wu, S.K.; Wang, S.; Watnick, R.S.; et al. CCL5 derived from platelets increases megakaryocyte proplatelet formation. Blood 2016, 127, 921–926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haemmerle, M.; Stone, R.L.; Menter, D.G.; Afshar-Kharghan, V.; Sood, A.K. The Platelet Lifeline to Cancer: Challenges and Opportunities. Cancer Cell 2018, 33, 965–983. [Google Scholar] [CrossRef] [PubMed]
- Schlesinger, M. Role of platelets and platelet receptors in cancer metastasis. J. Hematol. Oncol. 2018, 11, 125. [Google Scholar] [CrossRef]
- Sun, C.; Li, Q.; Hu, Z.; He, J.; Li, C.; Li, G.; Tao, X.; Yang, A. Treatment and Prognosis of Anaplastic Thyroid Carcinoma: Experience from a Single Institution in China. PLoS ONE 2013, 8, e80011. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Chu, H.-Y.; Zhong, Z.-M.; Qi, X.; Cheng, R.; Qin, R.-J.; Liang, J.; Zhu, X.-F.; Zeng, M.; Sun, C. Platelet-secreted CCL3 and its receptor CCR5 promote invasive and migratory abilities of anaplastic thyroid carcinoma cells via MMP-1. Cell. Signal. 2019, 63, 109363. [Google Scholar] [CrossRef]
- Hinshaw, D.C.; Shevde, L.A. The Tumor Microenvironment Innately Modulates Cancer Progression. Cancer Res. 2019, 79, 4557–4566. [Google Scholar] [CrossRef] [Green Version]
- Germano, G.; Frapolli, R.; Simone, M.; Tavecchio, M.; Erba, E.; Pesce, S.; Pasqualini, F.; Grosso, F.; Sanfilippo, R.; Casali, P.G.; et al. Antitumor and Anti-inflammatory Effects of Trabectedin on Human Myxoid Liposarcoma Cells. Cancer Res. 2010, 70, 2235–2244. [Google Scholar] [CrossRef] [Green Version]
- Gallo, M.; De Luca, A.; Lamura, L.; Normanno, N. Zoledronic acid blocks the interaction between mesenchymal stem cells and breast cancer cells: Implications for adjuvant therapy of breast cancer. Ann. Oncol. 2012, 23, 597–604. [Google Scholar] [CrossRef]
- Liu, H.; Wang, S.H.; Chen, S.-C.; Chen, C.-Y.; Lin, T. Zoledronic acid blocks the interaction between breast cancer cells and regulatory T-cells. BMC Cancer 2019, 19, 176. [Google Scholar] [CrossRef] [Green Version]
- Song, X.; Zhou, X.; Qin, Y.; Yang, J.; Wang, Y.; Sun, Z.; Yu, K.; Zhang, S.; Liu, S. Emodin inhibits epithelial-mesenchymal transition and metastasis of triple negative breast cancer via antagonism of CC-chemokine ligand 5 secreted from adipocytes. Int. J. Mol. Med. 2018, 42, 579–588. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
| Tumor Type | Molecules | Related Studies/Effects | Ref. |
|---|---|---|---|
| Acute lymphoblastic leukemia | Maraviroc | Induced tumor cell apoptosis in vitro, and decreased tumor xenograft growth. | [39] |
| Acute myeloid leukemia | Maraviroc | Inhibited T-reg accumulation and increased antileukemic effects of CD8+ T cells in tumor xenografts. | [85] |
| Hodgkin lymphoma | Anti-CCL5 antibody | Inhibited autocrine tumor growth; reduced tumor cell recruitment of CD4+T cells, eosinophils, monocytes, and MSCs; Inhibited tumor cell clonogenic growth induced by CCL5 secreted by tumor-educated MSCs. | [38,40] |
| Maraviroc | Inhibited: clonogenic growth of tumor cells; monocyte migration induced by tumor-educated MSCs; heterospheroids formation and viability; tumor xenograft growth, and monocyte accumulation in tumor tissues. Enhanced doxorubicin and brentuximab cytotoxic activity. | [38] | |
| Multiple myeloma | TAK779 | Inhibited bone marrow homing of tumor cells, osteoclastogenesis and osteoclastic resorption in tumor xenografts. | [41] |
| Anti-CCR5 antibody | Inhibited osteoclast formation and myeloma cell adhesion to stromal cells. | [86] |
| Tumor | Treatment | Effects | Reference |
|---|---|---|---|
| Breast cancer | Maraviroc | Decreased Treg migration, reduced lung metastasis in mouse models | [117] |
| Inhibited angiogenesis, reduced metastasis and improved survival in mouse models | [118] | ||
| Reduced immune-suppressive myeloid cells, increased antitumor immunity and reduced tumor growth in mouse models (in presence of CCL5-targeting siRNA) | [78] | ||
| Reduced mice tumor growth and thoracic metastasis (in presence of anti-IL-6R) | [119] | ||
| Reduced metastasis in mouse models Enhanced cell killing by doxorubicin in vitro | [43] | ||
| Reduced resistance to trastuzumab due to CCL5 overexpression | [120] | ||
| Anti-CCL5 antibodies | Counteracted the ability of MSCs to induce metastasis in tumor xenografts | [10] | |
| Inhibited collagen VI production by fibroblasts in response to the paracrine coculture of adipose-derived stem cells and tumor cells | [121] | ||
| Breast phyllodes | Maraviroc | Inhibited monocyte recruitment and tumor growth in patient-derived mouse xenografts | [122] |
| Cholangio- carcinoma | Anti-CCL5 antibody, maraviroc | Decreased cell invasion and CCR5 expression induced by MSCs | [123] |
| Colon cancer | Maraviroc | Decreased CAF and tumor xenograft growth | [124] |
| Repolarized immunosuppressive TAMs towards an anti-tumoral M1-like state in organoids from patients with metastatic CRC. Generated objective clinical responses in CRC patients (MARACON-001 phase I trial) | [44] | ||
| Reduced MSC-induced tumor xenograft growth in vivo | [125] | ||
| Anti-CCL5 antibodies | Decreased in vitro growth and migration of tumor cells, xenograft growth, liver metastases, sensitized to PDGFRβ therapy | [126] | |
| Inhibited the effects of macrophage CCL5: PDL-1 stabilization and inhibition of tumor killing by T cells | [127] | ||
| RNAi-CCL5 silencing | Decreased tumor growth in immunocompetent syngeneic mice and reduced Treg infiltration | [4] | |
| CCL5 knockout | Decreased tumor growth and metastasis facilitating CD8+ T cell accumulation in the tumor site in CRC xenografts. Decreased resistance to anti-PD-1 therapy | [128] | |
| Esophageal carcinoma | Maraviroc, siRNA-CCL5 silencing | Blocked tumor cell migration and invasion in vitro. Decreased cell growth rate, migration and invasion, and induced apoptosis in vitro | [46] |
| Gastric cancer | Anti-CCL5 antibodies | Decreased tumor invasion induced by CCL5 secreted by PBMCs educated by tumor cells | [129] |
| Decreased in vivo growth of tumor cells coinjected with PBMCs | [130] | ||
| Glioblastoma | Maraviroc, siRNA-CCR5 silencing | Induced M1 polarization of microglia and reduced migration | [131] |
| Inhibited tumor growth, invasion, and xenograft growth | [132] | ||
| Anti-CCL8 antibody | Reduced invasion and stemness of tumor cells | [133] | |
| Lung cancer | Anti-CCL5 antibody | Decreased tumor cell migration, invasion, and colony formation; inhibited growth and metastasis in xenografts | [134] |
| shRNA-EZH2 silencing siRNA-CCL5 silencing | Reduced tumor cell migration and invasion, suppressed metastasis and macrophage infiltration in xenografts | [135] | |
| Melanoma | mCCR5-Ig | Increased survival of tumor-bearing mice and decreased migration and immunosuppressive potential of MDSCs | [136,137] |
| Ovarian cancer | Anibamine | Inhibited tumor cell growth and intracellular Ca2+ flux | [138] |
| shRNA-CCL5 silencing, anti-CCR5 antibody | Inhibited the invasive capacity of CSLCs, inhibited invasion of ovarian cancer stem-like cells in vitro and in tumor xenografts | [59] | |
| Anti-CCL5 antibody | Decreased tumor growth and invasion induced by CAFs | [139] | |
| Reduced epithelial-mesenchymal transition-like process of non-stem ovarian cancer cells, metastasis in xenografts | [140] | ||
| Anti-CCL5 antibody, shRNA-CCL5 silencing | Inhibited endothelial cell differentiation and tube formation of cancer stem-like cells in vitro and in xenografts | [141] | |
| Pancreatic cancer | Maraviroc | Decreased tumor cell growth and invasion | [51] |
| Decreased tumor cell Inhibited, induced apoptosis in vitro, reduced liver metastasis from xenografts | [142] | ||
| Pituitary tumor | Maraviroc | Decreased macrophage migration induced by tumor cells | [143] |
| Prostate cancer | Anibamine | Reduced tumor xenograft growth | [82] |
| Maraviroc | Reduced metastasis and improved overall survival (in presence of chloroquine) | [144] | |
| miR-455-5p | Inducted apoptosis, suppressed cellular proliferation, and tumor xenograft growth | [145] |
| Treatment | Tumor | Name (Recruitment Status) | ClinicalTrials.gov Identifier |
|---|---|---|---|
| Leronlimab + carboplatin | Triple-negative breast cancer | A Phase Ib/II Study of Leronlimab (PRO 140) Combined With Carboplatin in Patients With CCR5+ Metastatic Triple Negative Breast Cancer (recruiting) | NCT03838367 |
| Leronlimab | Triple-negative breast cancer | A Compassionate Use Study of Leronlimab (PRO 140) in Combination With Treatment of Physician’s Choice in Patients With CCR5+ Metastatic Triple Negative Breast Cancer (available) | NCT04313075 |
| Maraviroc | Colorectal cancer | Treatment of Advanced Colorectal Cancer Patients With Hepatic Liver Metastases Using the CCR5-Antagonist Maraviroc (Phase I Maracon Trial) (completed) | NCT01736813 |
| Maraviroc + pembrolizumab | Colorectal cancer | A Phase I Trial of Combined PD-1 Inhibition (Pembrolizumab) and CCR5 Inhibition (Maraviroc) for the Treatment of Refractory Microsatellite Stable (MSS) Metastatic Colorectal Cancer (PICCASSO) (completed March 2020) | NCT03274804 |
| Vicriviroc + pembrolizumab | Microsatellite stable colorectal cancer | A Phase 2 Trial to Evaluate the safety and Efficacy of Vicriviroc (MK-7690) in Combination With Pembrolizumab (MK-3475) in Participants With Advanced/Metastatic Microsatellite Stable Colorectal Cancer (active, not recruiting) | NCT03631407 |
| Maraviroc | Kaposi’s sarcoma | Effects of Maraviroc on HIV-related Kaposi’s Sarcoma (completed) | NCT01276236 |
| BMS-813160 + nivolumab | Advanced pancreatic ductal adenocarcinoma | A Phase I/II Trial of Combination Immunotherapy With Nivolumab and a CCR2/CCR5 Dual Antagonist (BMS-813160) With or Without GVAX Following Chemotherapy and Radiotherapy for Locally Advanced Pancreatic Ductal Adenocarcinomas (recruiting) | NCT03767582 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aldinucci, D.; Borghese, C.; Casagrande, N. The CCL5/CCR5 Axis in Cancer Progression. Cancers 2020, 12, 1765. https://doi.org/10.3390/cancers12071765
Aldinucci D, Borghese C, Casagrande N. The CCL5/CCR5 Axis in Cancer Progression. Cancers. 2020; 12(7):1765. https://doi.org/10.3390/cancers12071765
Chicago/Turabian StyleAldinucci, Donatella, Cinzia Borghese, and Naike Casagrande. 2020. "The CCL5/CCR5 Axis in Cancer Progression" Cancers 12, no. 7: 1765. https://doi.org/10.3390/cancers12071765