MHC Class I Downregulation in Cancer: Underlying Mechanisms and Potential Targets for Cancer Immunotherapy


Abstract
:1. Introduction
2. Dysregulation of MHC-I Expression in Cancer
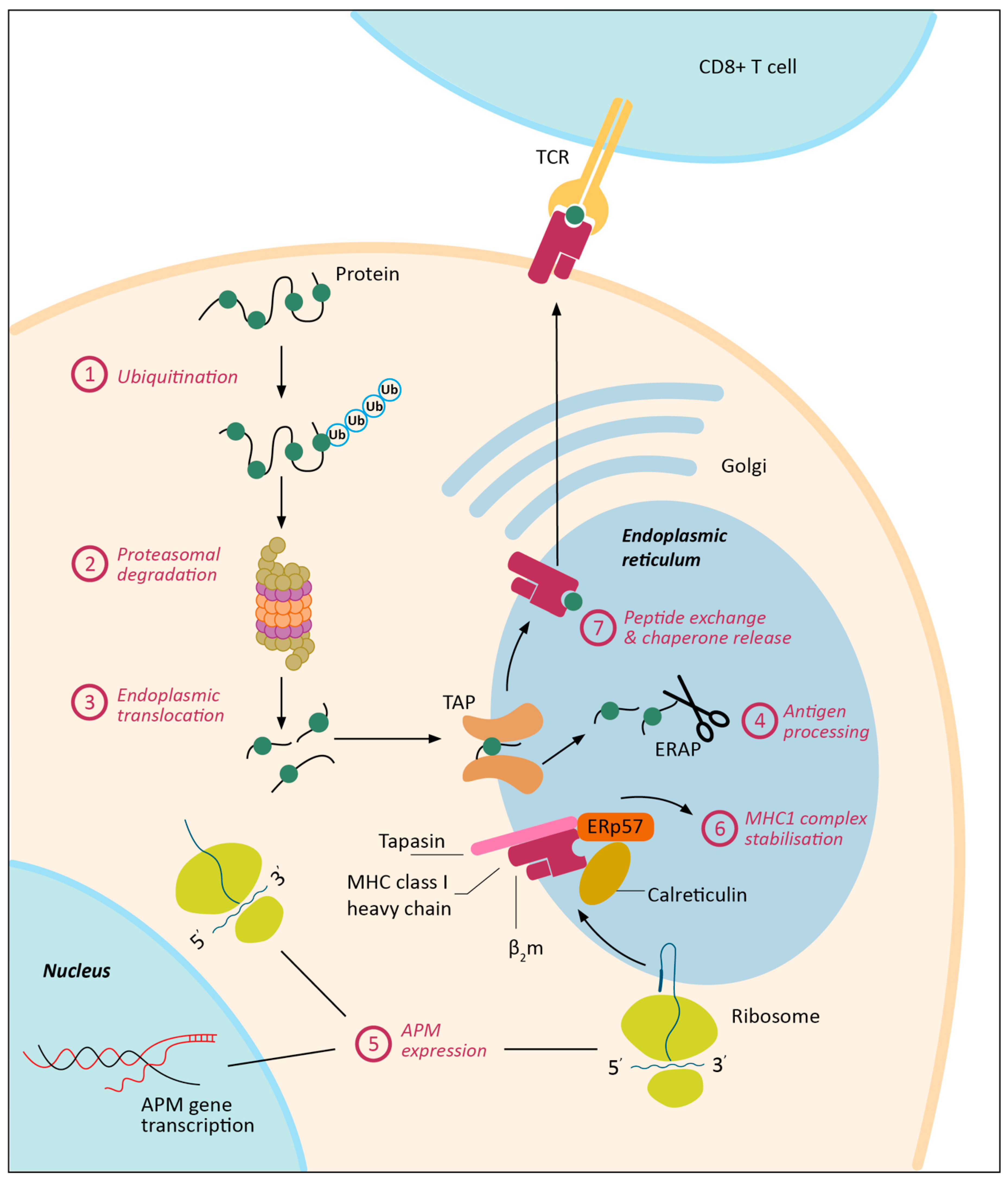
3. MHC-I Expression Regulation
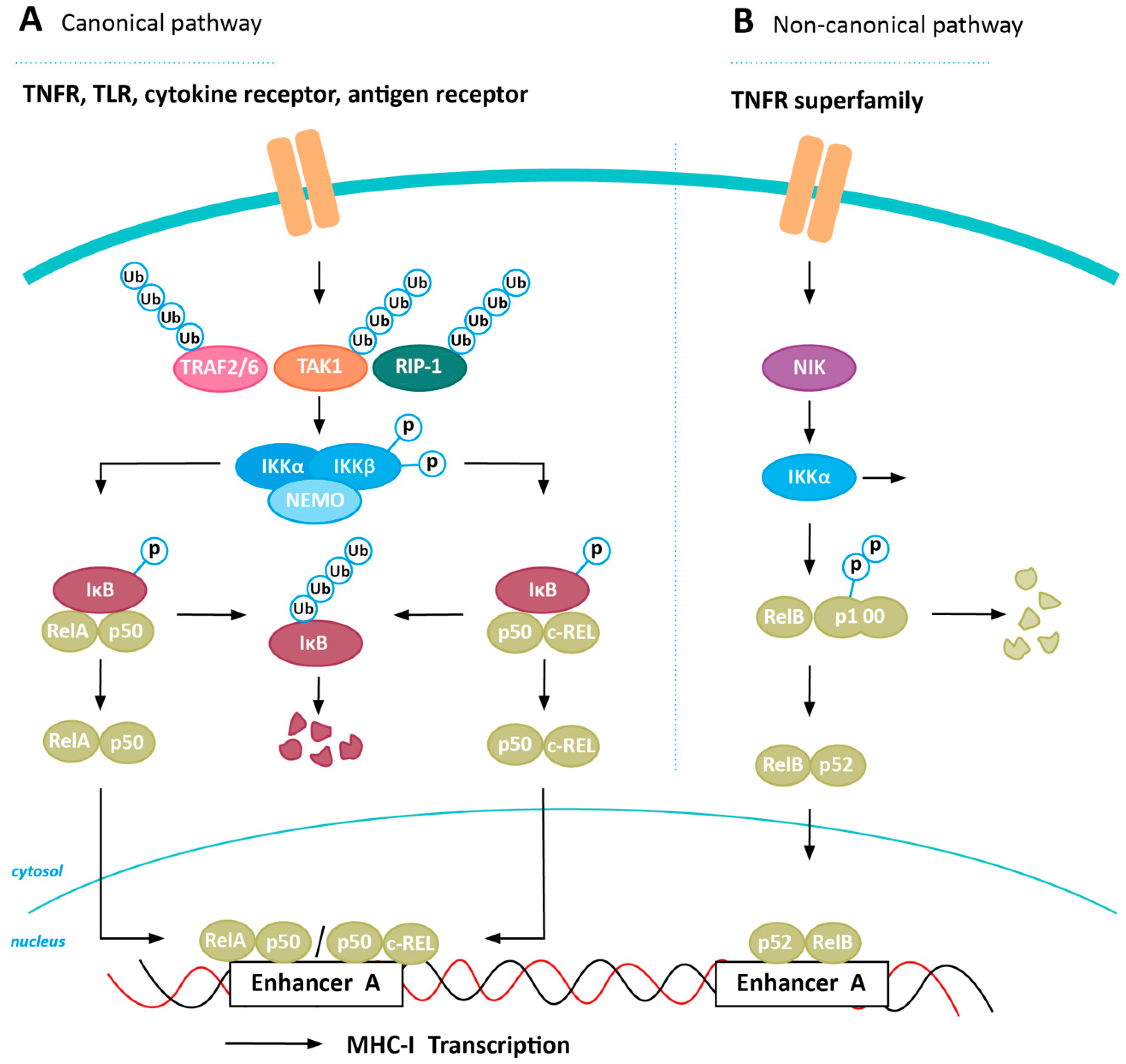
4. Inducing MHC-I Expression in Cancer via NFkB Stabilization
4.1. Positive Regulators of NFkB Expression
4.2. Negative Regulators of NFkB Expression
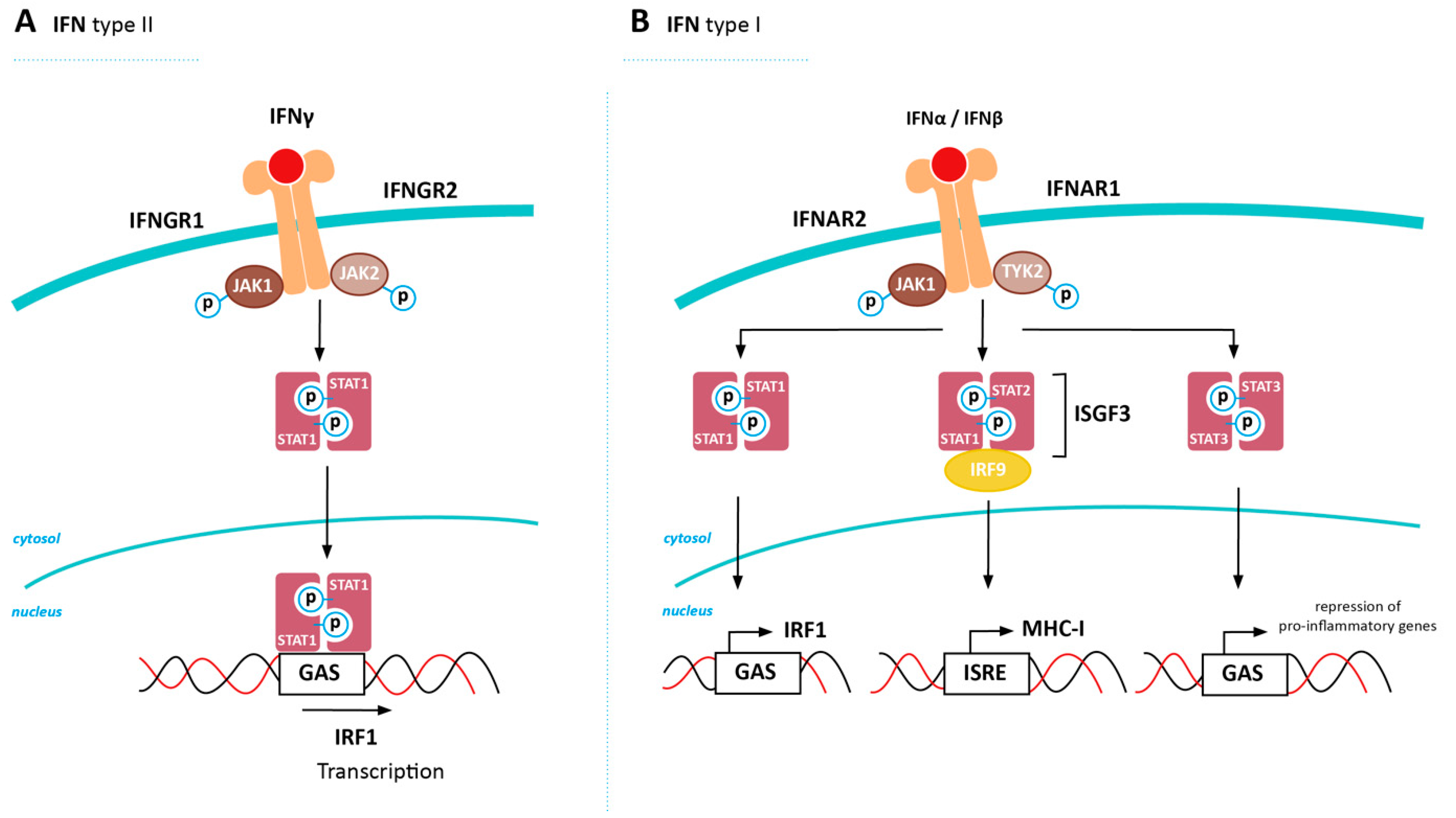
5. Inducing MHC-I Expression in Cancer via Restored IFN Signaling
5.1. Positive Regulators of IFN Signalling
5.2. Negative Regulators of IFN Signaling
6. NLRC5-Mediated Upregulation of MHC-I
7. Inducing MHC-I Expression in Cancer via STAT3 Inhibition
8. Inducing MHC-I Expression in Cancer via STING Induction
9. Well-Known Oncogenic Pathways Affect MHC-I Expression
10. Chemotherapy- and Radiation-Induced MHC-I Expression
11. Epigenetic Silencing Affecting MHC-I Expression
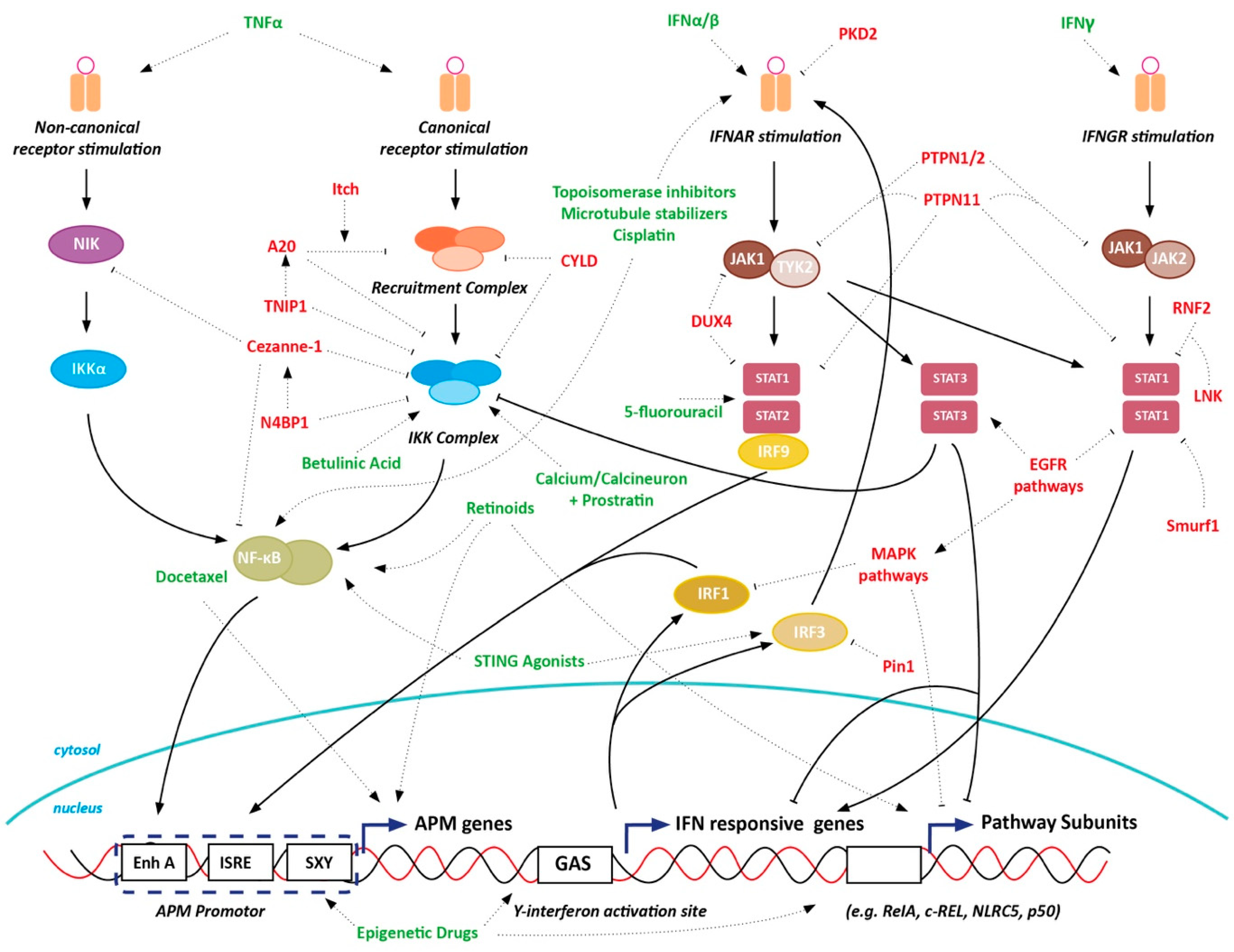
12. Discussion
13. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Sharma, P.; Hu-Lieskovan, S.; Wargo, J.A.; Ribas, A. Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell 2017, 168, 707–723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Angell, T.E.; Lechner, M.G.; Jang, J.K.; LoPresti, J.S.; Epstein, A.L. MHC class I loss is a frequent mechanism of immune escape in papillary thyroid cancer that is reversed by interferon and selumetinib treatment in Vitro. Clin. Cancer Res. 2014, 20, 6034–6044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mina, M.; Boldrini, R.; Citti, A.; Romania, P.; D’Alicandro, V.; De Ioris, M.; Castellano, A.; Furlanello, C.; Locatelli, F.; Fruci, D. Tumor-infiltrating T lymphocytes improve clinical outcome of therapy-resistant neuroblastoma. Oncoimmunology 2015, 4, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, A.H.; Ho, L.-L.; Levine, S.; Yadav, V.; Cheah, J.; Soule, C.; Frederick, D.T.; Liu, D.; Boland, G.; Kellis, M. Epigenomic correlates of checkpoint blockade immunotherapy resistance. Proc. Am. Assoc. Cancer Res. Annu. Meet. 2019, 79. [Google Scholar] [CrossRef]
- Gettinger, S.; Choi, J.; Hastings, K.; Truini, A.; Datar, I.; Sowell, R.; Wurtz, A.; Dong, W.; Cai, G.; Melnick, M.A.; et al. Impaired HLA class I antigen processing and presentation as a mechanism of acquired resistance to immune checkpoint inhibitors in lung cancer. Cancer Discov. 2017, 7, 1420–1435. [Google Scholar] [CrossRef] [Green Version]
- Sade-Feldman, M.; Jiao, Y.J.; Chen, J.H.; Rooney, M.S.; Barzily-Rokni, M.; Eliane, J.P.; Bjorgaard, S.L.; Hammond, M.R.; Vitzthum, H.; Blackmon, S.M.; et al. Resistance to checkpoint blockade therapy through inactivation of antigen presentation. Nat. Commun. 2017, 8. [Google Scholar] [CrossRef]
- Chowell, D.; Morris, L.G.T.; Grigg, C.M.; Weber, J.K.; Samstein, R.M.; Makarov, V.; Kuo, F.; Kendall, S.M.; Requena, D.; Riaz, N.; et al. Patient HLA class I genotype influences cancer response to checkpoint blockade immunotherapy. Science 2018, 359, 582–587. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.H.; Shklovskaya, E.; Lim, S.Y.; Carlino, M.S.; Menzies, A.M.; Stewart, A.; Pedersen, B.; Irvine, M.; Alavi, S.; Yang, J.Y.H.; et al. Transcriptional downregulation of MHC class I and melanoma de- differentiation in resistance to PD-1 inhibition. Nat. Commun. 2020, 11, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benitez, R.; Godelaine, D.; Lopez-Nevot, M.A.; Brasseur, F.; Jiménez, P.; Marchand, M.; Oliva, M.R.; Van Baren, N.; Cabrera, T.; Andry, G.; et al. Mutations of the β2-microglobulin gene result in a lack of HLA class I molecules on melanoma cells of two patients immunized with MAGE peptides. Tissue Antigens 1998, 52, 520–529. [Google Scholar] [CrossRef]
- Lauss, M.; Donia, M.; Harbst, K.; Andersen, R.; Mitra, S.; Rosengren, F.; Salim, M.; Vallon-Christersson, J.; Törngren, T.; Kvist, A.; et al. Mutational and putative neoantigen load predict clinical benefit of adoptive T cell therapy in melanoma. Nat. Commun. 2017, 8, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Khong, H.T.; Wang, Q.J.; Rosenberg, S.A. Identification of multiple antigens recognized by tumor-infiltrating lymphocytes from a single patient: Tumor escape by antigen loss and loss of MHC expression. J. Immunother. 2004, 27, 184–190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maleno, I.; López-Nevot, M.; Cabrera, T.; Salinero, J.; Garrido, F. Multiple mechanisms generate HLA class I altered phenotypes in laryngeal carcinomas: High frequency of HLA haplotype loss associated with loss of heterozygosity in chromosome region 6p21. Cancer Immunol. Immunother. 2002, 51, 389–396. [Google Scholar] [CrossRef]
- Maleno, I.; Cabrera, C.M.; Cabrera, T.; Paco, L.; López-Nevot, M.A.; Collado, A.; Ferrón, A.; Garrido, F. Distribution of HLA class I altered phenotypes in colorectal carcinomas: High frequency of HLA haplotype loss associated with loss of heterozygosity in chromosome region 6p21. Immunogenetics 2004, 56, 244–253. [Google Scholar] [CrossRef] [PubMed]
- Maleno, I.; Romero, J.M.; Cabrera, T.; Paco, L.; Aptsiauri, N.; Cozar, J.M.; Tallada, M.; López-Nevot, M.A.; Garrido, F. LOH at 6p21.3 region and HLA class altered phenotypes in bladder carcinomas. Immunogenetics 2006, 58, 503–510. [Google Scholar] [CrossRef]
- Feenstra, M.; Veltkamp, M.; Van Kuik, J.; Wiertsema, S.; Slootweg, P.; Van den Tweel, J.; De Weger, R.; Tilanus, M. HLA class 1 expression and chromosomal deletions at 6p and 15q in head and neck squamous cell carcinomas. Tissue Antigens 1999, 54, 235–245. [Google Scholar] [CrossRef] [PubMed]
- Garrido, M.A.; Rodriguez, T.; Zinchenko, S.; Maleno, I.; Ruiz-Cabello, F.; Concha, Á.; Olea, N.; Garrido, F.; Aptsiauri, N. HLA class I alterations in breast carcinoma are associated with a high frequency of the loss of heterozygosity at chromosomes 6 and 15. Immunogenetics 2018, 70, 647–659. [Google Scholar] [CrossRef]
- Seliger, B.; Ritz, U.; Bock, M.; Huber, C.; Abele, R.; Tampé, R.; Sutter, G.; Sutter, G.; Drexler, I.; Ferrone, S. Immune escape of melanoma: First evidence of structural alterations in two distinct components of the MHC class I antigen processing pathway. Cancer Res. 2001, 61, 8647–8650. [Google Scholar]
- Meissner, M.; Reichert, T.E.; Kunkel, M.; Gooding, W.; Whiteside, T.L.; Ferrone, S.; Seliger, B. Defects in the human leukocyte antigen class I antigen-processing machinery in head and neck squamous cell carcinoma: Association with clinical outcome. Clin. Cancer Res. 2005, 11, 2552–2560. [Google Scholar] [CrossRef] [Green Version]
- Spel, L.; Boelens, J.J.; Van Der Steen, D.M.; Blokland, N.J.G.; van Noesel, M.M.; Molenaar, J.J.; Heemskerk, M.H.M.; Boes, M.; Nierkens, S. Natural killer cells facilitate PRAME-specific T-cell reactivity against neuroblastoma. Oncotarget 2015, 6, 35770–35781. [Google Scholar] [CrossRef] [Green Version]
- Romero, J.M.; Jiménez, P.; Cabrera, T.; Cózar, J.M.; Pedrinaci, S.; Tallada, M.; Garrido, F.; Ruiz-Cabello, F. Coordinated downregulation of the antigen presentation machinery and HLA class I/β2-microglobulin complex is responsible for HLA-ABC loss in bladder cancer. Int. J. Cancer 2005, 113, 605–610. [Google Scholar] [CrossRef]
- Squire, R.; Fowler, C.L.; Brooks, S.P.; Rich, G.A.; Cooney, D.R. The relationship of class I MHC antigen expression to stage IV-S disease and survival in neuroblastoma. J. Pediatr. Surg. 1990, 25, 381–386. [Google Scholar] [CrossRef]
- Watson, N.F.S.; Ramage, J.M.; Madjd, Z.; Spendlove, I.; Ellis, I.O.; Scholefield, J.H.; Durrant, L.G. Immunosurveillance is active in colorectal cancer as downregulation but not complete loss of MHC class I expression correlates with a poor prognosis. Int. J. Cancer 2006, 118, 6–10. [Google Scholar] [CrossRef] [PubMed]
- Turcotte, S.; Katz, S.C.; Shia, J.; Jarnagin, W.R.; Kingham, T.P.; Allen, P.J.; Fong, Y.; D’Angelica, M.I.; DeMatteo, R.P. Tumor MHC class I expression improves the prognostic value of T-cell density in resected colorectal liver metastases. Cancer Immunol. Res. 2014, 2, 530–537. [Google Scholar] [CrossRef] [Green Version]
- Andersson, E.; Villabona, L.; Bergfeldt, K.; Carlson, J.W.; Ferrone, S.; Kiessling, R.; Seliger, B.; Masucci, G.V. Correlation of HLA-A02* genotype and HLA class I antigen down-regulation with the prognosis of epithelial ovarian cancer. Cancer Immunol. Immunother. 2012, 61, 1243–1253. [Google Scholar] [CrossRef] [PubMed]
- Roemer, M.G.M.; Advani, R.H.; Redd, R.A.; Pinkus, G.S.; Natkunam, Y.; Ligon, A.H.; Connelly, C.F.; Pak, C.J.; Carey, C.D.; Daadi, S.E.; et al. Classical Hodgkin lymphoma with reduced β2M/MHC class i expression is associated with inferior outcome independent of 9p24.1 status. Cancer Immunol. Res. 2016, 4, 910–916. [Google Scholar] [CrossRef] [Green Version]
- Van Houdt, I.S.; Sluijter, B.J.R.; Moesbergen, L.M.; Vos, W.M.; De Gruijl, T.D.; Molenkamp, B.G.; Van Den Eertwegh, A.J.M.; Hooijberg, E.; Van Leeuwen, P.A.M.; Meijer, C.J.L.M.; et al. Favorable outcome in clinically stage II melanoma patients is associated with the presence of activated tumor infiltrating T-lymphocytes and preserved MHC class I antigen expression. Int. J. Cancer 2008, 123, 609–615. [Google Scholar] [CrossRef]
- Simpson, J.A.D.; Al-Attar, A.; Watson, N.F.S.; Scholefield, J.H.; Ilyas, M.; Durrant, L.G. Intratumoral T cell infiltration, MHC class I and STAT1 as biomarkers of good prognosis in colorectal cancer. Gut 2010, 59, 926–933. [Google Scholar] [CrossRef] [PubMed]
- Inoue, M.; Mimura, K.; Izawa, S.; Shiraishi, K.; Inoue, A.; Shiba, S.; Watanabe, M.; Maruyama, T.; Kawaguchi, Y.; Inoue, S.; et al. Expression of mhc class i on breast cancer cells correlates inversely with her2 expression. Oncoimmunology 2012, 1, 1104–1110. [Google Scholar] [CrossRef] [Green Version]
- Hanagiri, T.; Shigematsu, Y.; Kuroda, K.; Baba, T.; Shiota, H.; Ichiki, Y.; Nagata, Y.; Yasuda, M.; Uramoto, H.; So, T.; et al. Prognostic implications of human leukocyte antigen class i expression in patients who underwent surgical resection for non-small-cell lung cancer. J. Surg. Res. 2013, 181, e57–e63. [Google Scholar] [CrossRef]
- Spel, L.; Schiepers, A.; Boes, M. NFκB and MHC-1 interplay in neuroblastoma and immunotherapy. Trends Cancer 2018, 4, 715–717. [Google Scholar] [CrossRef]
- Anfossi, N.; André, P.; Guia, S.; Falk, C.S.; Roetynck, S.; Stewart, C.A.; Breso, V.; Frassati, C.; Reviron, D.; Middleton, D.; et al. Human NK Cell Education by Inhibitory Receptors for MHC Class, I. Immunity 2006, 25, 331–342. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-C.; Lee, K.-M.; Kim, D.-W.; Heo, D.S. Elevated TGF-β1 Secretion and Down-Modulation of NKG2D Underlies Impaired NK Cytotoxicity in Cancer Patients. J. Immunol. 2004, 172, 7335–7340. [Google Scholar] [CrossRef] [PubMed]
- Jonges, L.E.; Giezeman-Smits, K.M.; Van Vlierberghe, R.L.E.; Ensink, N.G.; Hagenaars, M.; Joly, É.; Eggermont, A.M.M.; Van de Velde, C.J.H.; Fleuren, G.J.; Kuppen, P.J.K. NK cells modulate MHC class I expression on tumor cells and their susceptibility to lysis. Immunobiology 2000, 202, 326–338. [Google Scholar] [CrossRef]
- Jongsma, M.L.M.; Guarda, G.; Spaapen, R.M. The regulatory network behind MHC class I expression. Mol. Immunol. 2019, 113, 16–21. [Google Scholar] [CrossRef] [PubMed]
- Lorenzi, S.; Forloni, M.; Cifaldi, L.; Antonucci, C.; Citti, A.; Boldrini, R.; Pezzullo, M.; Castellano, A.; Russo, V.; van der Bruggen, P.; et al. IRF1 and NF-kB Restore MHC Class I-Restricted Tumor Antigen Processing and Presentation to Cytotoxic T Cells in Aggressive Neuroblastoma. PLoS ONE 2012, 7, 1–8. [Google Scholar] [CrossRef]
- Sun, S.C. The non-canonical NF-κB pathway in immunity and inflammation. Nat. Rev. Immunol. 2017, 17, 545–558. [Google Scholar] [CrossRef]
- Chaturvedi, M.M.; Sung, B.; Yadav, V.R.; Kannappan, R.; Aggarwal, B.B. NF-κB addiction and its role in cancer: One size does not fit all. Oncogene 2011, 30, 1615–1630. [Google Scholar] [CrossRef] [Green Version]
- Wells, K.; Hintzsche, J.; Amato, C.M.; Tobin, R.; Vorwald, V.; McCarter, M.; Shellman, Y.; Tan, A.C.; Robinson, W. Investigating the role of NF- κB signaling and immune checkpoint blockade therapy in melanoma. Clin. Res. 2019, 90, abstract 5002. [Google Scholar]
- Ea, C.K.; Deng, L.; Xia, Z.P.; Pineda, G.; Chen, Z.J. Activation of IKK by TNFα Requires Site-Specific Ubiquitination of RIP1 and Polyubiquitin Binding by NEMO. Mol. Cell 2006, 22, 245–257. [Google Scholar] [CrossRef]
- Cai, W.; Kerner, Z.J.; Sun, J. Targeted Cancer Therapy with Tumor Necrosis Factor-Alpha. Biochem. Insights 2008, 15–21. [Google Scholar] [CrossRef] [Green Version]
- Segars, J.H.; Nagata, T.; Bours, V.; Medin, J.A.; Franzoso, G.; Blanco, J.C.; Drew, P.D.; Becker, K.G.; An, J.; Tang, T. Retinoic acid induction of major histocompatibility complex class I genes in NTera-2 embryonal carcinoma cells involves induction of NF-kappa B (p50-p65) and retinoic acid receptor beta-retinoid X receptor beta heterodimers. Mol. Cell. Biol. 1993, 13, 6157–6169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farina, A.R.; Masciulli, M.P.; Tacconelli, A.; Cappabianca, L.; De Santis, G.; Gulino, A.; Mackay, A.R. All-trans-retinoic acid induces nuclear factor κB activation and matrix metalloproteinase-9 expression and enhances basement membrane invasivity of differentiation-resistant human SK-N-BE 9N neuroblastoma cells. Cell Growth Differ. 2002, 13, 343–354. [Google Scholar] [PubMed]
- Vertuani, S.; De Geer, A.; Levitsky, V.; Kogner, P.; Kiessling, R.; Levitskaya, J. Retinoids Act as Multistep Modulators of the Major Histocompatibility Class I Presentation Pathway and Sensitize Neuroblastomas to Cytotoxic Lymphocytes. Cancer Res. 2003, 63, 8006–8013. [Google Scholar]
- Matthay, K.K.; Reynolds, C.P.; Seeger, R.C.; Shimada, H.; Adkins, E.S.; Haas-Kogan, D.; Gerbing, R.B.; London, W.B.; Villablanca, J.G. Long-term results for children with high-risk neuroblastoma treated on a randomized trial of myeloablative therapy followed by 13-cis-retinoic acid: A children’s oncology group study. J. Clin. Oncol. 2009, 27, 1007–1013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kasperczyk, H.; La Ferla-Brühl, K.; Westhoff, M.A.; Behrend, L.; Zwacka, R.M.; Debatin, K.M.; Fulda, S. Betulinic acid as new activator of NF-κB: Molecular mechanisms and implications for cancer therapy. Oncogene 2005, 24, 6945–6956. [Google Scholar] [CrossRef] [Green Version]
- Selzer, E.; Pimentel, E.; Wacheck, V.; Schlegel, W.; Pehamberger, H.; Jansen, B.; Kodym, R. Effects of betulinic acid alone and in combination with irradiation in human melanoma cells. J. Invest. Dermatol. 2000, 114, 935–940. [Google Scholar] [CrossRef] [Green Version]
- Wick, W.; Grimmel, C.; Wagenknecht, B.; Dichgans, J.; Weller, M. Betulinic acid-induced apoptosis in glioma cells: A sequential requirement for new protein synthesis, formation of reactive oxygen species, and caspase processing. J. Pharmacol. Exp. Ther. 1999, 289, 1306–1312. [Google Scholar]
- Fulda, S.; Friesen, C.; Los, M.; Scaffidi, C.; Mier, W.; Benedict, M.; Nuñez, G.; Krammer, P.H.; Peter, M.E.; Debatin, K.M. Betulinic acid triggers CD95 (APO-1/Fas)- and p53-independent apoptosis via activation of caspases in neuroectodermal tumors. Cancer Res. 1997, 57, 4956–4964. [Google Scholar] [PubMed]
- Chowdhury, A.R.; Suparna, M.; Mittra, B.; Sharma, S.; Mukhopadhyay, S.; Majumder, H.K. Betulinic acid, a potent inhibitor of eukaryotic topoisomerase I: Identification of the inhibitory step, the major functional group responsible and development of more potent derivatives. Med. Sci. Monit. 2002, 8, 254–261. [Google Scholar]
- Kwon, H.J.; Shim, J.S.; Kim, J.H.; Cho, H.Y.; Yum, Y.N.; Kim, S.H.; Yu, J. Betulinic acid inhibits growth factor-induced in vitro angiogenesis via the modulation of mitochondrial function in endothelial cells. Japanese J. Cancer Res. 2002, 93, 417–425. [Google Scholar] [CrossRef]
- Chan, J.K.; Bhattacharyya, D.; Lassen, K.G.; Ruelas, D.; Greene, W.C. Calcium/calcineurin synergizes with prostratin to promote NF-κB dependent activation of latent HIV. PLoS ONE 2013, 8, e77749. [Google Scholar] [CrossRef] [PubMed]
- Fang, D.F.; He, K.; Wang, N.; Sang, Z.H.; Qiu, X.; Xu, G.; Jian, Z.; Liang, B.; Li, T.; Li, H.Y.; et al. NEDD4 ubiquitinates TRAF3 to promote CD40-mediated AKT activation. Nat. Commun. 2014, 5, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Oberst, A.; Malatesta, M.; Aqeilan, R.I.; Rossi, M.; Salomoni, P.; Murillas, R.; Sharma, P.; Kuehn, M.R.; Oren, M.; Croce, C.M.; et al. The Nedd4-binding partner 1 (N4BP1) protein is an inhibitor of the E3 ligase Itch. Proc. Natl. Acad. Sci. USA 2007, 104, 11280–11285. [Google Scholar] [CrossRef] [Green Version]
- Spel, L.; Nieuwenhuis, J.; Haarsma, R.; Stickel, E.; Bleijerveld, O.B.; Altelaar, M.; Boelens, J.J.; Brummelkamp, T.R.; Nierkens, S.; Boes, M. Nedd4-binding protein 1 and TNFAIP3-interacting protein 1 control MHC-1 display in neuroblastoma. Cancer Res. 2018, 78, 6621–6631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fenner, B.J.; Scannell, M.; Prehn, J.H.M. Identification of polyubiquitin binding proteins involved in NF-κB signaling using protein arrays. Biochim. Biophys. Acta Proteins Proteomics 2009, 1794, 1010–1016. [Google Scholar] [CrossRef] [PubMed]
- Nepravishta, R.; Ferrentino, F.; Mandaliti, W.; Mattioni, A.; Weber, J.; Polo, S.; Castagnoli, L.; Cesareni, G.; Paci, M.; Santonico, E. CoCUN, a novel ubiquitin binding domain identified in N4BP1. Biomolecules 2019, 9, 284. [Google Scholar] [CrossRef] [Green Version]
- Hu, H.; Brittain, G.C.; Chang, J.H.; Puebla-Osorio, N.; Jin, J.; Zal, A.; Xiao, Y.; Cheng, X.; Chang, M.; Fu, Y.X.; et al. OTUD7B controls non-canonical NF-κB activation through deubiquitination of TRAF3. Nature 2013, 494, 371–374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, X.D.; Sun, S.C. Targeting signaling factors for degradation, an emerging mechanism for TRAF functions. Immunol. Rev. 2015, 266, 56–71. [Google Scholar] [CrossRef] [Green Version]
- G’Sell, R.T.; Gaffney, P.M.; Powell, D.W. A20-binding inhibitor of NF-κB activation 1 is a physiologic inhibitor of NF-κB: A molecular switch for inflammation and autoimmunity. Arthritis Rheumatol. 2015, 67, 2292–2302. [Google Scholar] [CrossRef] [Green Version]
- Wagner, S.; Carpentier, I.; Rogov, V.; Kreike, M.; Ikeda, F.; Löhr, F.; Wu, C.J.; Ashwell, J.D.; Dötsch, V.; Dikic, I.; et al. Ubiquitin binding mediates the NF-κB inhibitory potential of ABIN proteins. Oncogene 2008, 27, 3739–3745. [Google Scholar] [CrossRef] [Green Version]
- Cohen, S.; Ciechanover, A.; Kravtsova-Ivantsiv, Y.; Lapid, D.; Lahav-Baratz, S. ABIN-1 negatively regulates NF-κB by inhibiting processing of the p105 precursor. Biochem. Biophys. Res. Commun. 2009, 389, 205–210. [Google Scholar] [CrossRef] [PubMed]
- Mauro, C.; Pacifico, F.; Lavorgna, A.; Mellone, S.; Iannetti, A.; Acquaviva, R.; Formisano, S.; Vito, P.; Leonardi, A. ABIN-1 binds to NEMO/IKKγ and co-operates with A20 in inhibiting NF-κB. J. Biol. Chem. 2006, 281, 18482–18488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cruz, J.A.; Childs, E.E.; Amatya, N.; Garg, A.V.; Beyaert, R.; Kane, L.P.; Aneskievich, B.J.; Ma, A.; Gaffen, S.L. IL-17 Signaling Triggers Degradation of the Constitutive NF-κB Inhibitor ABIN-1. ImmunoHorizons 2017, 1, 133–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, B.; Kang, H.; Fung, A.; Zhao, H.; Wang, T.; Ma, D. The role of interleukin 17 in tumour proliferation, angiogenesis, and metastasis. Mediators Inflamm. 2014, 2014. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; He, K.; Guo, Q.; Chen, J.; Zhang, M.; Huang, K.; Yang, D.; Wu, L.; Deng, Y.; Luo, X.; et al. SSRP1 promotes colorectal cancer progression and is negatively regulated by miR-28-5p. J. Cell. Mol. Med. 2019, 23, 3118–3129. [Google Scholar] [CrossRef]
- Wang, C.; Wu, C.; Yang, Q.; Ding, M.; Zhong, J.; Zhang, C.Y.; Ge, J.; Wang, J.; Zhang, C. miR-28-5p acts as a tumor suppressor in renal cell carcinoma for multiple antitumor effects by targeting RAP1B. Oncotarget 2016, 7, 73888–73902. [Google Scholar] [CrossRef] [Green Version]
- Shi, X.; Teng, F. Down-regulated miR-28-5p in human hepatocellular carcinoma correlated with tumor proliferation and migration by targeting insulin-like growth factor-1 (IGF-1). Mol. Cell. Biochem. 2015, 408, 283–293. [Google Scholar] [CrossRef]
- Song, L.; Lin, C.; Gong, H.; Wang, C.; Liu, L.; Wu, J.; Tao, S.; Hu, B.; Cheng, S.Y.; Li, M.; et al. MiR-486 sustains NF-κB activity by disrupting multiple NF-κB-negative feedback loops. Cell Res. 2013, 23, 274–289. [Google Scholar] [CrossRef]
- Tan, G.; Wu, L.; Tan, J.; Zhang, B.; Tai, W.C.S.; Xiong, S.; Chen, W.; Yang, J.; Li, H. MIR-1180 promotes apoptotic resistance to human hepatocellular carcinoma via activation of NF-κB signaling pathway. Sci. Rep. 2016, 6, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Jiang, N.; Shi, H.; Zhao, S.; Yao, S.; Shen, H. MiR-28-5p promotes the development and progression of ovarian cancer through inhibition of N4BP1. Int. J. Oncol. 2017, 50, 1383–1391. [Google Scholar] [CrossRef] [Green Version]
- Zhu, G.; Wang, Z.; Mijiti, M.; Du, G.; Li, Y.; Dangmurenjiafu, G. MiR-28-5p promotes human glioblastoma cell growth through inactivation of FOXO1. Int. J. Clin. Exp. Pathol. 2019, 12, 2972–2980. [Google Scholar] [PubMed]
- Shembade, N.; Harhaj, E.W. Regulation of NF-κB signaling by the A20 deubiquitinase. Cell. Mol. Immunol. 2012, 9, 123–130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wertz, I.E.; Rourke, K.M.O.; Zhou, H.; Eby, M.; Aravind, L.; Seshagiri, S.; Wu, P.; Wiesmann, C.; Dixit, V.M. ligase domains of A20 downregulate NF- k B signalling. Nature 2004, 430, 1–6. [Google Scholar] [CrossRef]
- Shembade, N.; Harhaj, N.S.; Parvatiyar, K.; Copeland, N.G.; Jenkins, N.A.; Matesic, L.E.; Harhaj, E.W. The E3 ligase Itch negatively regulates inflammatory signaling pathways by controlling the function of the ubiquitin-editing enzyme A20. Nat. Immunol. 2008, 9, 254–262. [Google Scholar] [CrossRef]
- Ishihara, T.; Tsuda, H.; Hotta, A.; Kozaki, K.I.; Yoshida, A.; Noh, J.Y.; Ito, K.; Imoto, I.; Inazawa, J. ITCH is a putative target for a novel 20q11.22 amplification detected in anaplastic thyroid carcinoma cells by array-based comparative genomic hybridization. Cancer Sci. 2008, 99, 1940–1949. [Google Scholar] [CrossRef] [PubMed]
- Ho, K.C.; Zhou, Z.; She, Y.M.; Chun, A.; Cyr, T.D.; Yang, X. Itch E3 ubiquitin ligase regulates large tumor suppressor 1 tumor-suppressor stability. Proc. Natl. Acad. Sci. USA 2011, 108, 4870–4875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salah, Z.; Melino, G.; Aqeilan, R.I. Negative regulation of the Hippo pathway by E3 ubiquitin ligase ITCH is sufficient to promote tumorigenicity. Cancer Res. 2011, 71, 2010–2020. [Google Scholar] [CrossRef] [Green Version]
- Salah, Z.; Itzhaki, E.; Aqeilan, R.I. The ubiquitin E3 ligase ITCH enhances breast tumor progression by inhibiting the Hippo tumor suppressor pathway. Oncotarget 2014, 5, 10886–10900. [Google Scholar] [CrossRef] [Green Version]
- Meng, J.; Tagalakis, A.D.; Hart, S.L. Silencing E3 Ubiqutin ligase ITCH as a potential therapy to enhance chemotherapy efficacy in p53 mutant neuroblastoma cells. Sci. Rep. 2020, 10, 1–12. [Google Scholar] [CrossRef]
- Rossi, M.; Rotblat, B.; Ansell, K.; Amelio, I.; Caraglia, M.; Misso, G.; Bernassola, F.; Cavasotto, C.N.; Knight, R.A.; Ciechanover, A.; et al. High throughput screening for inhibitors of the HECT ubiquitin E3 ligase ITCH identifies antidepressant drugs as regulators of autophagy. Cell Death Dis. 2014, 5, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.M.; HuangFu, W.C.; Huang, H.L.; Wu, W.C.; Chen, Y.L.; Yen, Y.; Huang, H.L.; Nien, C.Y.; Lai, M.J.; Pan, S.L.; et al. 1,4-Naphthoquinones as inhibitors of Itch, a HECT domain-E3 ligase, and tumor growth suppressors in multiple myeloma. Eur. J. Med. Chem. 2017, 140, 84–91. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Bengtsson, B.O.; Mix, E.; Thorell, L.H.; Olsson, T.; Link, H. Effect of monoamine reuptake inhibiting antidepressants on major histocompatibility complex expression on macrophages in normal rats and rats with experimental allergic neuritis (EAN). Immunopharmacology 1994, 27, 225–244. [Google Scholar] [CrossRef]
- Kovalenko, A.; Chable-Bessia, C.; Cantarella, G.; Israël, A.; Wallach, D.; Courtois, G. The tumour suppressor CYLD negatively regulates NF-κB signalling by deubiquitination. Nature 2003, 424, 801–805. [Google Scholar] [CrossRef] [PubMed]
- Yin, J.; Weng, C.; Ma, J.; Chen, F.; Huang, Y.; Feng, M. MicroRNA-1288 promotes cell proliferation of human glioblastoma cells by repressing ubiquitin carboxyl-terminal hydrolase CYLD expression. Mol. Med. Rep. 2017, 16, 6764–6770. [Google Scholar] [CrossRef] [PubMed]
- Qiu, H.; Yuan, S.; Lu, X. miR-186 suppressed CYLD expression and promoted cell proliferation in human melanoma. Oncol. Lett. 2016, 12, 2301–2306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ni, F.; Zhao, H.; Cui, H.; Wu, Z.; Chen, L.; Hu, Z.; Guo, C.; Liu, Y.; Chen, Z.; Wang, X.; et al. MicroRNA-362-5p promotes tumor growth and metastasis by targeting CYLD in hepatocellular carcinoma. Cancer Lett. 2015, 356, 809–818. [Google Scholar] [CrossRef] [PubMed]
- Farshi, P.; Deshmukh, R.R.; Nwankwo, J.O.; Arkwright, R.T.; Cvek, B.; Liu, J.; Dou, Q.P. Deubiquitinases (DUBs) and DUB inhibitors: A patent review. Expert Opin. Ther. Pat. 2015, 25, 1191–1208. [Google Scholar] [CrossRef] [Green Version]
- Sun, S.C. CYLD: A tumor suppressor deubiquitinase regulating NF-B activation and diverse biological processes. Cell Death Differ. 2010, 17, 25–34. [Google Scholar] [CrossRef]
- Wang, J.H.; Wei, W.; Guo, Z.X.; Shi, M.; Guo, R. Decreased Cezanne expression is associated with the progression and poor prognosis in hepatocellular carcinoma. J. Transl. Med. 2015, 13, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Tilborghs, S.; Corthouts, J.; Verhoeven, Y.; Arias, D.; Rolfo, C.; Trinh, X.B.; van Dam, P.A. The role of Nuclear Factor-kappa B signaling in human cervical cancer. Crit. Rev. Oncol. Hematol. 2017, 120, 141–150. [Google Scholar] [CrossRef]
- Musella, M.; Manic, G.; De Maria, R.; Vitale, I.; Sistigu, A. Type-I-interferons in infection and cancer: Unanticipated dynamics with therapeutic implications. Oncoimmunology 2017, 6, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Castro, F.; Cardoso, A.P.; Gonçalves, R.M.; Serre, K.; Oliveira, M.J. Interferon-gamma at the crossroads of tumor immune surveillance or evasion. Front. Immunol. 2018, 9, 1–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaretsky, J.M.; Garcia-Diaz, A.; Shin, D.S.; Escuin-Ordinas, H.; Hugo, W.; Hu-Lieskovan, S.; Torrejon, D.Y.; Abril-Rodriguez, G.; Sandoval, S.; Barthly, L.; et al. Mutations associated with acquired resistance to PD-1 blockade in melanoma. N. Engl. J. Med. 2016, 375, 819–829. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Shi, L.Z.; Zhao, H.; Chen, J.; Xiong, L.; He, Q.; Chen, T.; Roszik, J.; Bernatchez, C.; Woodman, S.E.; et al. Loss of IFN-γ Pathway Genes in Tumor Cells as a Mechanism of Resistance to Anti-CTLA-4 Therapy. Cell 2016, 167, 397–404.e9. [Google Scholar] [CrossRef] [PubMed]
- Stelloo, E.; Versluis, M.A.; Nijman, H.W.; De Bruyn, M.; Plat, A.; Osse, E.M.; Van Dijk, R.H.; Nout, R.A.; Creutzberg, C.L.; De Bock, G.H.; et al. Microsatellite instability derived JAK1 frameshift mutations are associated with tumor immune evasion in endometrioid endometrial cancer. Oncotarget 2016, 7, 39885–39893. [Google Scholar] [CrossRef] [Green Version]
- Smithy, J.W.; Moore, L.M.; Pelekanou, V.; Rehman, J.; Gaule, P.; Wong, P.F.; Neumeister, V.M.; Sznol, M.; Kluger, H.M.; Rimm, D.L. Nuclear IRF-1 expression as a mechanism to assess “Capability” to express PD-L1 and response to PD-1 therapy in metastatic melanoma. J. Immunother. Cancer 2017, 5, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Bhat, M.Y.; Solanki, H.S.; Advani, J.; Khan, A.A.; Keshava Prasad, T.S.; Gowda, H.; Thiyagarajan, S.; Chatterjee, A. Comprehensive network map of interferon gamma signaling. J. Cell Commun. Signal. 2018, 12, 745–751. [Google Scholar] [CrossRef]
- Ivashkiv, L.B.; Donlin, L.T. Regulation of type i interferon responses. Nat. Rev. Immunol. 2014, 14, 36–49. [Google Scholar] [CrossRef] [Green Version]
- Propper, D.J.; Chao, D.; Braybrooke, J.P.; Bahl, P.; Thavasu, P.; Balkwill, F.; Turley, H.; Dobbs, N.; Gatter, K.; Talbot, D.C.; et al. Low-Dose IFN-γ Induces Tumor MHC Expression in Metastatic Malignant Melanoma. Clin. Cancer Res. 2003, 9, 84–92. [Google Scholar]
- Yang, I.; Kremen, T.J.; Giovannone, A.J.; Paik, E.; Odesa, S.K.; Prins, R.M.; Liau, L.M. Modulation of major histocompatibility complex Class I molecules and major histocompatibility complex-bound immunogenic peptides induced by interferon-α and interferon-γ treatment of human glioblastoma multiforme. J. Neurosurg. 2004, 100, 310–319. [Google Scholar] [CrossRef]
- George, P.M.; Badiger, R.; Alazawi, W.; Foster, G.R.; Mitchell, J.A. Pharmacology and therapeutic potential of interferons. Pharmacol. Ther. 2012, 135, 44–53. [Google Scholar] [CrossRef] [PubMed]
- Hemmerle, T.; Neri, D. The dose-dependent tumor targeting of antibody-IFNγ fusion proteins reveals an unexpected receptor-trapping mechanism in vivo. Cancer Immunol. Res. 2014, 2, 559–567. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Lim, O.; Kim, T.M.; Ahn, Y.O.; Choi, H.; Chung, H.; Min, B.; Her, J.H.; Cho, S.Y.; Keam, B.; et al. Phase I study of random healthy donor-derived allogeneic natural killer cell therapy in patients with malignant lymphoma or advanced solid tumors. Cancer Immunol. Res. 2016, 4, 215–224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, Y.-H.; Choi, M.G.; Kim, D.H.; Choi, Y.J.; Kim, S.Y.; Sung, K.J.; Lee, J.C.; Kim, S.-Y.; Rho, J.K.; Choi, C.M. Natural Killer Cells as a Potential Biomarker for Predicting Immunotherapy Efficacy in Patients with Non-Small Cell Lung Cancer. Target. Oncol. 2020, 15, 241–247. [Google Scholar] [CrossRef] [PubMed]
- Mazzaschi, G.; Facchinetti, F.; Missale, G.; Canetti, D.; Madeddu, D.; Zecca, A.; Veneziani, M.; Gelsomino, F.; Goldoni, M.; Buti, S.; et al. The circulating pool of functionally competent NK and CD8+ cells predicts the outcome of anti-PD1 treatment in advanced NSCLC. Lung Cancer 2019, 127, 153–163. [Google Scholar] [CrossRef] [PubMed]
- Castriconi, R.; Dondero, A.; Augugliaro, R.; Cantoni, C.; Carnemolla, B.; Sementa, A.R.; Negri, F.; Conte, R.; Corrias, M.V.; Moretta, L.; et al. Identification of 4Ig-B7-H3 as a neuroblastoma-associated molecule that exerts a protective role from an NK cell-mediated lysis. Proc. Natl. Acad. Sci. USA 2004, 101, 12640–12645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.H.; Martin-Orozco, N.; Zheng, P.; Li, J.; Zhang, P.; Tan, H.; Park, H.J.; Jeong, M.; Chang, S.H.; Kim, B.S.; et al. Inhibition of the B7-H3 immune checkpoint limits tumor growth by enhancing cytotoxic lymphocyte function. Cell Res. 2017, 27, 1034–1045. [Google Scholar] [CrossRef]
- Hu, Y.; Tian, Z.; Zhang, C. Natural Killer Cell-Based Immunotherapy for Cancer: Advances and Prospects. Engineering 2019, 5, 106–114. [Google Scholar] [CrossRef]
- Bernson, E.; Hallner, A.; E Sander, F.; Wilsson, O.; Werlenius, O.; Rydström, A.; Kiffin, R.; Brune, M.; Foà, R.; Aurelius, J.; et al. Impact of killer-immunoglobulin-like receptor and human leukocyte antigen genotypes on the efficacy of immunotherapy in acute myeloid leukemia. Leuk. 2017, 31, 2552–2559. [Google Scholar] [CrossRef] [Green Version]
- Boudreau, J.E.; Giglio, F.; Gooley, T.A.; Stevenson, P.A.; Le Luduec, J.-B.; Shaffer, B.C.; Rajalingam, R.; Hou, L.; Hurley, C.K.; Noreen, H.; et al. KIR3DL1/HLA-B Subtypes Govern Acute Myelogenous Leukemia Relapse After Hematopoietic Cell Transplantation. J. Clin. Oncol. 2017, 35, 2268–2278. [Google Scholar] [CrossRef]
- Lupo, K.B.; Matosevic, S. Natural Killer Cells as Allogeneic Effectors in Adoptive Cancer Immunotherapy. Cancers 2019, 11, 769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dondero, A.; Pastorino, F.; Della Chiesa, M.; Corrias, M.V.; Morandi, F.; Pistoia, V.; Olive, D.; Bellora, F.; Locatelli, F.; Castellano, A.; et al. PD-L1 expression in metastatic neuroblastoma as an additional mechanism for limiting immune surveillance. Oncoimmunology 2016, 5, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.; Crabill, G.A.; Pritchard, T.S.; McMiller, T.L.; Wei, P.; Pardoll, D.M.; Pan, F.; Topalian, S.L. Mechanisms regulating PD-L1 expression on tumor and immune cells. J. Immunother. Cancer 2019, 7, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Chew, G.-L.; Campbell, A.E.; De Neef, E.; Sutliff, N.A.; Shadle, S.C.; Tapscott, S.J.; Bradley, R.K. DUX4 Suppresses MHC Class I to Promote Cancer Immune Evasion and Resistance to Checkpoint Blockade. Dev. Cell 2019, 50, 658–671.e7. [Google Scholar] [CrossRef]
- Bosnakovski, D.; da Silva, M.T.; Sunny, S.T.; Ener, E.T.; Toso, E.A.; Yuan, C.; Cui, Z.; Walters, M.A.; Jadhav, A.; Kyba, M. A novel P300 inhibitor reverses DUX4-mediated global histone H3 hyperacetylation, target gene expression, and cell death. Sci. Adv. 2019, 5, eaaw7781. [Google Scholar] [CrossRef] [Green Version]
- Lasko, L.M.; Jakob, C.G.; Edalji, R.P.; Qiu, W.; Montgomery, D.; Digiammarino, E.L.; Hansen, T.M.; Risi, R.M.; Frey, R.; Manaves, V.; et al. Discovery of a selective catalytic p300/CBP inhibitor that targets lineage-specific tumours. Nature 2017, 550, 128–132. [Google Scholar] [CrossRef]
- Oliva, J.; Galasinski, S.; Richey, A.; Campbell, A.E.; Meyers, M.J.; Modi, N.; Zhong, J.W.; Tawil, R.; Tapscott, S.J.; Sverdrup, F.M. Clinically advanced p38 inhibitors suppress DUX4 expression in cellular and animal models of facioscapulohumeral muscular dystrophys. J. Pharmacol. Exp. Ther. 2019, 370, 219–230. [Google Scholar] [CrossRef] [PubMed]
- Yong, H.Y.; Koh, M.S.; Moon, A. The p38 MAPK inhibitors for the treatment of inflammatory diseases and cancer. Expert Opin. Investig. Drugs 2009, 18, 1893–1905. [Google Scholar] [CrossRef] [PubMed]
- Ding, L.W.; Sun, Q.Y.; Lin, D.C.; Chien, W.; Hattori, N.; Dong, X.M.; Gery, S.; Garg, M.; Doan, N.B.; Said, J.W.; et al. LNK (SH2B3): Paradoxical effects in ovarian cancer. Oncogene 2015, 34, 1463–1474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, L.W.; Sun, Q.Y.; Edwards, J.J.; Fernández, L.T.; Ran, X.B.; Zhou, S.Q.; Scolyer, R.A.; Wilmott, J.S.; Thompson, J.F.; Doan, N.; et al. LNK suppresses interferon signaling in melanoma. Nat. Commun. 2019, 10. [Google Scholar] [CrossRef]
- Kriegel, A.J.; Baker, M.A.; Liu, Y.; Liu, P.; Cowley, A.W.; Liang, M. Endogenous MicroRNAs in human microvascular endothelial cells regulate mRNAs encoded by hypertension-related genes. Hypertension 2015, 66, 793–799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Jin, B.J.; Chen, Q.; Yan, B.J.; Liu, Z.L. MicroRNA-29b upregulation improves myocardial fibrosis and cardiac function in myocardial infarction rats through targeting SH2B3. Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 10115–10122. [Google Scholar] [CrossRef]
- Saitoh, T.; Tun-Kyi, A.; Ryo, A.; Yamamoto, M.; Finn, G.; Fujita, T.; Akira, S.; Yamamoto, N.; Lu, K.P.; Yamaoka, S. Negative regulation of interferon-regulatory factor 3-dependent innate antiviral response by the prolyl isomerase Pin1. Nat. Immunol. 2006, 7, 598–605. [Google Scholar] [CrossRef]
- Chen, Y.; Wu, Y.; Yang, H.; Li, X.; Jie, M.; Hu, C.; Wu, Y.; Yang, S.; Yang, Y. Prolyl isomerase Pin1: A promoter of cancer and a target for therapy. Cell Death Dis. 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Hennig, L.; Christner, C.; Kipping, M.; Schelbert, B.; Rücknagel, K.P.; Grabley, S.; Küllertz, G.; Fischer, G. Selective inactivation of parvulin-like peptidyl-prolyl cis/trans isomerases by juglone. Biochemistry 1998, 37, 5953–5960. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhang, B.; Gao, J.; Wang, X.; Liu, Z. Regulation of the MicroRNA 200b (miRNA-200b) by transcriptional regulators PEA3 and ELK-1 protein affects expression of Pin1 protein to control anoikis. J. Biol. Chem. 2013, 288, 32742–32752. [Google Scholar] [CrossRef] [Green Version]
- Luo, M.L.; Gong, C.; Chen, C.H.; Lee, D.Y.; Hu, H.; Huang, P.; Yao, Y.; Guo, W.; Reinhardt, F.; Wulf, G.; et al. Prolyl isomerase pin1 acts downstream of mir200c to promote cancer stem-like cell traits in breast cancer. Cancer Res. 2014, 74, 3603–3616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, K.H.; Lin, F.C.; Hsu, T.I.; Lin, J.T.; Guo, J.H.; Tsai, C.H.; Lee, Y.C.; Lee, Y.C.; Chen, C.L.; Hsiao, M.; et al. MicroRNA-296-5p (miR-296-5p) functions as a tumor suppressor in prostate cancer by directly targeting Pin1. Biochim. Biophys. Acta Mol. Cell Res. 2014, 1843, 2055–2066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, S.; Kozono, S.; Kats, L.; Nechama, M.; Li, W.; Guarnerio, J.; Luo, M.; You, M.H.; Yao, Y.; Kondo, A.; et al. Active Pin1 is a key target of all-trans retinoic acid in acute promyelocytic leukemia and breast cancer. Nat. Med. 2015, 21, 457–466. [Google Scholar] [CrossRef] [PubMed]
- Campaner, E.; Rustighi, A.; Zannini, A.; Cristiani, A.; Piazza, S.; Ciani, Y.; Kalid, O.; Golan, G.; Baloglu, E.; Shacham, S.; et al. A covalent PIN1 inhibitor selectively targets cancer cells by a dual mechanism of action. Nat. Commun. 2017, 8. [Google Scholar] [CrossRef]
- Myers, M.P.; Andersen, J.N.; Cheng, A.; Tremblay, M.L.; Horvath, C.M.; Parisien, J.P.; Salmeen, A.; Barford, D.; Tonks, N.K. TYK2 and JAK2 Are Substrates of Protein-tyrosine Phosphatase 1B. J. Biol. Chem. 2001, 276, 47771–47774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- You, M.; Yu, D.-H.; Feng, G.-S. Shp-2 Tyrosine Phosphatase Functions as a Negative Regulator of the Interferon-Stimulated Jak/STAT Pathway. Mol. Cell. Biol. 1999, 19, 2416–2424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bollu, L.R.; Mazumdar, A.; Savage, M.I.; Brown, P.H. Molecular pathways: Targeting protein tyrosine phosphatases in cancer. Clin. Cancer Res. 2017, 23, 2136–2142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yi, T.; Pathak, M.K.; Lindner, D.J.; Ketterer, M.E.; Farver, C.; Borden, E.C. Anticancer Activity of Sodium Stibogluconate in Synergy with IFNs. J. Immunol. 2002, 169, 5978–5985. [Google Scholar] [CrossRef] [Green Version]
- Lantz, K.A.; Hart, S.G.E.; Planey, S.L.; Roitman, M.F.; Ruiz-White, I.A.; Wolfe, H.R.; McLane, M.P. Inhibition of PTP1B by trodusquemine (MSI-1436) causes fat-specific weight loss in diet-induced obese mice. Obesity 2010, 18, 1516–1523. [Google Scholar] [CrossRef]
- Huffaker, T.B.; Lee, S.H.; Tang, W.W.; Wallace, J.A.; Alexander, M.; Runtsch, M.C.; Larsen, D.K.; Thompson, J.; Ramstead, A.G.; Voth, W.P.; et al. Antitumor immunity is defective in T cell–specific microRNA-155– deficient mice and is rescued by immune checkpoint blockade. J. Biol. Chem. 2017, 292, 18530–18541. [Google Scholar] [CrossRef] [Green Version]
- Higgs, G.; Slack, F. The multiple roles of microRNA-155 in oncogenesis. J. Clin. Bioinforma. 2013, 3, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Liu, B.; Mink, S.; Wong, K.A.; Stein, N.; Getman, C.; Dempsey, P.W.; Wu, H.; Shuai, K. PIAS1 selectively inhibits interferon-inducible genes and is important in innate immunity. Nat. Immunol. 2004, 5, 891–898. [Google Scholar] [CrossRef]
- Yuan, C.; Qi, J.; Zhao, X.; Gao, C. Smurf1 protein negatively regulates interferon-γ signaling through promoting STAT1 protein ubiquitination and degradation. J. Biol. Chem. 2012, 287, 17006–17015. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Jiang, M.; Wang, W.; Liu, W.; Song, X.; Ma, Z.; Zhang, S.; Liu, L.; Liu, Y.; Cao, X. Nuclear RNF2 inhibits interferon function by promoting K33-linked STAT1 disassociation from DNA article. Nat. Immunol. 2018, 19, 41–50. [Google Scholar] [CrossRef]
- Sánchez-Beato, M.; Sánchez, E.; González-Carreró, J.; Morente, M.; Díez, A.; Sánchez-Verde, L.; Martín, M.C.; Cigudosa, J.C.; Vidal, M.; Piris, M.A. Variability in the expression of polycomb proteins in different normal and tumoral tissues. A pilot study using tissue microarrays. Mod. Pathol. 2006, 19, 684–694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romero, I.; Martinez, M.; Garrido, C.; Collado, A.; Algarra, I.; Garrido, F.; Garcia-Lora, A.M. The tumour suppressor Fhit positively regulates MHC class I expression on cancer cells. J. Pathol. 2012, 227, 367–379. [Google Scholar] [CrossRef] [PubMed]
- Rai, K.; Akdemir, K.C.; Kwong, L.N.; Fiziev, P.; Wu, C.J.; Keung, E.Z.; Sharma, S.; Samant, N.S.; Williams, M.; Axelrad, J.B.; et al. Dual roles of RNF2 in melanoma progression. Cancer Discov. 2015, 5, 1314–1327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rabellino, A.; Andreani, C.; Scaglioni, P.P. The Role of PIAS SUMO E3-Ligases in Cancer. Cancer Res. 2017, 77, 1542–1547. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Yu, N.; Xu, J.; Ding, X.; Deng, W.; Wu, G.; Li, X.; Hou, Y.; Liu, Z.; Zhao, Y.; et al. SMURF1 facilitates estrogen receptor a signaling in breast cancer cells. J. Exp. Clin. Cancer Res. 2018, 37, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Chen, S.; Li, Y.; Gao, Y.; Huang, S.; Li, H.; Zhu, Y. SMURF1-mediated ubiquitination of ARHGAP26 promotes ovarian cancer cell invasion and migration. Exp. Mol. Med. 2019, 51. [Google Scholar] [CrossRef] [Green Version]
- Nair, S.; Bist, P.; Dikshit, N.; Krishnan, M.N. Global functional profiling of human ubiquitome identifies E3 ubiquitin ligase DCST1 as a novel negative regulator of Type-I interferon signaling. Sci. Rep. 2016, 6, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Ismail, I.H.; McDonald, D.; Strickfaden, H.; Xu, Z.; Hendzel, M.J. A small molecule inhibitor of polycomb repressive complex 1 inhibits ubiquitin signaling at DNA double-strand breaks. J. Biol. Chem. 2013, 288, 26944–26954. [Google Scholar] [CrossRef] [Green Version]
- Cao, Y.; Wang, C.; Zhang, X.; Xing, G.; Lu, K.; Gu, Y.; He, F.; Zhang, L. Selective small molecule compounds increase BMP-2 responsiveness by inhibiting Smurf1-mediated Smad1/5 degradation. Sci. Rep. 2014, 4, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Zheng, H.; Qian, J.; Varghese, B.; Baker, D.P.; Fuchs, S. Ligand-Stimulated Downregulation of the Alpha Interferon Receptor: Role of Protein Kinase D2. Mol. Cell. Biol. 2011, 31, 710–720. [Google Scholar] [CrossRef] [Green Version]
- Azoitei, N.; Cobbaut, M.; Becher, A.; Van Lint, J.; Seufferlein, T. Protein kinase D2: A versatile player in cancer biology. Oncogene 2018, 37, 1263–1278. [Google Scholar] [CrossRef]
- Wei, N.; Chu, E.; Wipf, P.; Schmitz, J.C. Protein kinase D as a potential chemotherapeutic target for colorectal cancer. Mol. Cancer Ther. 2014, 13, 1130–1141. [Google Scholar] [CrossRef] [Green Version]
- Tandon, M.; Salamoun, J.M.; Carder, E.J.; Farber, E.; Xu, S.; Deng, F.; Tang, H.; Wipf, P.; Wang, Q.J. SD-208, a novel protein kinase D inhibitor, blocks prostate cancer cell proliferation and tumor Growth in Vivo by inducing G2/M cell cycle arrest. PLoS ONE 2015, 10, 1–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meissner, T.B.; Li, A.; Biswas, A.; Lee, K.H.; Liu, Y.J.; Bayir, E.; Iliopoulos, D.; Van Den Elsen, P.J.; Kobayashi, K.S. NLR family member NLRC5 is a transcriptional regulator of MHC class I genes. Proc. Natl. Acad. Sci. USA 2010, 107, 13794–13799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Staehli, F.; Ludigs, K.; Heinz, L.X.; Seguín-Estévez, Q.; Ferrero, I.; Braun, M.; Schroder, K.; Rebsamen, M.; Tardivel, A.; Mattmann, C.; et al. NLRC5 Deficiency Selectively Impairs MHC Class I- Dependent Lymphocyte Killing by Cytotoxic T Cells. J. Immunol. 2012, 188, 3820–3828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuenzel, S.; Till, A.; Winkler, M.; Häsler, R.; Lipinski, S.; Jung, S.; Grötzinger, J.; Fickenscher, H.; Schreiber, S.; Rosenstiel, P. The Nucleotide-Binding Oligomerization Domain-Like Receptor NLRC5 Is Involved in IFN-Dependent Antiviral Immune Responses. J. Immunol. 2010, 184, 1990–2000. [Google Scholar] [CrossRef]
- Kobayashi, K.S.; Van Den Elsen, P.J. NLRC5: A key regulator of MHC class I-dependent immune responses. Nat. Rev. Immunol. 2012, 12, 813–820. [Google Scholar] [CrossRef]
- Ozcan, M.; Janikovits, J.; von Knebel Doeberitz, M.; Kloor, M. Complex pattern of immune evasion in MSI colorectal cancer. Oncoimmunology 2018, 7, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Yoshihama, S.; Roszik, J.; Downs, I.; Meissner, T.B.; Vijayan, S.; Chapuy, B.; Sidiq, T.; Shipp, M.A.; Lizee, G.A.; Kobayashi, K.S. NLRC5/MHC class I transactivator is a target for immune evasion in cancer. Proc. Natl. Acad. Sci. USA 2016, 113, 5999–6004. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez, G.M.; Bobbala, D.; Serrano, D.; Mayhue, M.; Champagne, A.; Saucier, C.; Steimle, V.; Kufer, T.A.; Menendez, A.; Ramanathan, S.; et al. NLRC5 elicits antitumor immunity by enhancing processing and presentation of tumor antigens to CD8+ T lymphocytes. Oncoimmunology 2016, 5, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Tang, F.; Xu, Y.; Zhao, B. NLRC5: New cancer buster? Mol. Biol. Rep. 2020, 47, 2265–2277. [Google Scholar] [CrossRef] [PubMed]
- Huynh, J.; Chand, A.; Gough, D.; Ernst, M. Therapeutically exploiting STAT3 activity in cancer—using tissue repair as a road map. Nat. Rev. Cancer 2019, 19, 82–96. [Google Scholar] [CrossRef]
- Blaskovich, M.A.; Sun, J.; Cantor, A.; Turkson, J.; Jove, R.; Sebti, S.M. Discovery of JSI-124 (cucurbitacin I), a selective Janus kinase/signal transducer and activator of transcription 3 signaling pathway inhibitor with potent antitumor activity against human and murine cancer cells in mice. Cancer Res. 2003, 63, 1270–1279. [Google Scholar]
- Nam, S.; Buettner, R.; Turkson, J.; Kim, D.; Cheng, J.Q.; Muehlbeyer, S.; Hippe, F.; Vatter, S.; Merz, K.H.; Eisenbrand, G.; et al. Indirubin derivatives inhibit Stat3 signaling and induce apoptosis in human cancer cells. Proc. Natl. Acad. Sci. USA 2005, 102, 5998–6003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kotha, A.; Sekharam, M.; Cilenti, L.; Siddiquee, K.; Khaled, A.; Zervos, A.S.; Carter, B.; Turkson, J.; Jove, R. Resveratrol inhibits Src and Stat3 signaling and induces the apoptosis of malignant cells containing activated Stat3 protein. Mol. Cancer Ther. 2006, 5, 621–629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schust, J.; Sperl, B.; Hollis, A.; Mayer, T.U.; Berg, T. Stattic: A Small-Molecule Inhibitor of STAT3 Activation and Dimerization. Chem. Biol. 2006, 13, 1235–1242. [Google Scholar] [CrossRef] [Green Version]
- Yang, F.; Van Meter, T.E.; Buettner, R.; Hedvat, M.; Liang, W.; Kowolik, C.M.; Mepani, N.; Mirosevich, J.; Nam, S.; Chen, M.Y.; et al. Sorafenib inhibits signal transducer and activator of transcription 3 signaling associated with growth arrest and apoptosis of medulloblastomas. Mol. Cancer Ther. 2008, 7, 3519–3526. [Google Scholar] [CrossRef] [Green Version]
- Xin, H.; Zhang, C.; Herrmann, A.; Du, Y.; Figlin, R.; Yu, H. Sunitinib inhibition of Stat3 induces renal cell carcinoma tumor cell apoptosis and reduces immunosuppressive cells. Cancer Res. 2009, 69, 2506–2513. [Google Scholar] [CrossRef] [Green Version]
- McFarland, B.C.; Gray, G.K.; Nozell, S.E.; Hong, S.W.; Benveniste, E.N. Activation of the NF-κB pathway by the STAT3 inhibitor JSI-124 in human glioblastoma cells. Mol. Cancer Res. 2013, 11, 494–505. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Y.; Ye, T.; Yu, X.; Lei, Q.; Yang, F.; Xia, Y.; Song, X.; Liu, L.; Deng, H.; Gao, T.; et al. Nifuroxazide exerts potent anti-tumor and anti-metastasis activity in melanoma. Sci. Rep. 2016, 6, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Yamazaki, T.; Pietrocola, F.; Zhou, H.; Zitvogel, L.; Ma, Y.; Kroemer, G. STAT3 inhibition enhances the therapeutic efficacy of immunogenic chemotherapy by stimulating type 1 interferon production by cancer cells. Cancer Res. 2015, 75, 3812–3822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, C.; Talukder, A.; Savage, N.M.; Singh, N.; Liu, K. JAK-STAT-mediated chronic inflammation impairs cytotoxic T lymphocyte activation to decrease anti-PD-1 immunotherapy efficacy in pancreatic cancer. Oncoimmunology 2017, 6, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Cohen, E.E.W.; Harrington, K.J.; Hong, D.S.; Mesia, R.; Brana, I.; Perez Segura, P.; Wise-Draper, T.; Scott, M.L.; Mitchell, P.D.; Mugundu, G.M.; et al. A phase Ib/II study (SCORES) of durvalumab (D) plus danvatirsen (DAN.; AZD9150) or AZD5069 (CX2i) in advanced solid malignancies and recurrent/metastatic head and neck squamous cell carcinoma (RM-HNSCC): Updated results. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2018, 29, viii372. [Google Scholar] [CrossRef]
- D’amico, S.; Shi, J.; Martin, B.L.; Crawford, H.C.; Petrenko, O.; Reich, N.C. STAT3 is a master regulator of epithelial identity and KRAS-driven tumorigenesis. Genes Dev. 2018, 32, 1175–1187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barber, G.N. STING: Infection, inflammation and cancer. Nat. Rev. Immunol. 2015, 15, 760–770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abe, T.; Barber, G.N. Cytosolic-DNA-Mediated, STING-Dependent Proinflammatory Gene Induction Necessitates Canonical NF- B Activation through TBK1. J. Virol. 2014, 88, 5328–5341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Challa, S.V.; Zhou, S.; Sheri, A.; Padmanabhan, S.; Meher, G.; Gimi, R.; Schmidt, D.; Cleary, D.; Afdhal, N.; Iyer, R. Preclinical studies of SB 11285, a novel STING agonist for immuno-oncology. J. Clin. Oncol. 2017, 35, e14616. [Google Scholar] [CrossRef]
- Wang-Bishop, L.; Wehbe, M.; Shae, D.; James, J.; Hacker, B.C.; Garland, K.; Chistov, P.P.; Rafat, M.; Balko, J.M.; Wilson, J.T. Potent STING activation stimulates immunogenic cell death to enhance antitumor immunity in neuroblastoma. J. Immunother. Cancer 2020, 8, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Ager, C.R.; Reilley, M.J.; Nicholas, C.; Bartkowiak, T.; Jaiswal, A.R.; Curran, M.A. Intratumoral STING activation with T-cell checkpoint modulation generates systemic antitumor immunity. Cancer Immunol. Res. 2017, 5, 676–684. [Google Scholar] [CrossRef] [Green Version]
- Ghaffari, A.; Peterson, N.; Khalaj, K.; Vitkin, N.; Robinson, A.; Francis, J.A.; Koti, M. Sting agonist therapy in combination with pd-1 immune checkpoint blockade enhances response to carboplatin chemotherapy in high-grade serous ovarian cancer. Br. J. Cancer 2018, 119, 440–449. [Google Scholar] [CrossRef]
- Meric-Bernstam, F.; Sandhu, S.K.; Hamid, O.; Spreafico, A.; Kasper, S.; Dummer, R.; Shimizu, T.; Steeghs, N.; Lewis, N.; Talluto, C.C.; et al. Phase Ib study of MIW815 (ADU-S100) in combination with spartalizumab (PDR001) in patients (pts) with advanced/metastatic solid tumors or lymphomas. J. Clin. Oncol. 2019, 37. [Google Scholar] [CrossRef]
- Seliger, B.; Harders, C.; Lohmann, S.; Momburg, F.; Urlinger, S.; Tampé, R.; Huber, C. Down-regulation of the MHC class I antigen-processing machinery after oncogenic transformation of murine fibroblasts. Eur. J. Immunol. 1998, 28, 122–133. [Google Scholar] [CrossRef]
- Herrmann, F.; Lehr, H.A.; Drexler, I.; Sutter, G.; Hengstler, J.; Wollscheid, U.; Seliger, B. HER-2/neu-Mediated Regulation of Components of the MHC Class I Antigen-Processing Pathway. Cancer Res. 2004, 64, 215–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bradley, S.D.; Chen, Z.; Melendez, B.; Talukder, A.; Khalili, J.S.; Rodriguez-Cruz, T.; Liu, S.; Whittington, M.; Deng, W.; Li, F.; et al. BRAFV600E co-opts a conserved MHC class I internalization pathway to diminish antigen presentation and CD8+ T-cell recognition of melanoma. Cancer Immunol. Res. 2015, 3, 602–609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brea, E.J.; Oh, C.Y.; Manchado, E.; Budhu, S.; Gejman, R.S.; Mo, G.; Mondello, P.; Han, J.E.; Jarvis, C.A.; Ulmert, D.; et al. Kinase regulation of human MHC class i molecule expression on cancer cells. Cancer Immunol. Res. 2016, 4, 936–947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Versteeg, R.; Noordermeer, I.A.; Krüse-Wolters, M.; Ruiter, D.J.; Schrier, P.I. c-myc down-regulates class I HLA expression in human melanomas. EMBO J. 1988, 7, 1023–1029. [Google Scholar] [CrossRef]
- Yang, W.; Li, Y.; Gao, R.; Xiu, Z.; Sun, T. MHC class I dysfunction of glioma stem cells escapes from CTL-mediated immune response via activation of Wnt/β-catenin signaling pathway. Oncogene 2020, 39, 1098–1111. [Google Scholar] [CrossRef]
- Lulli, D.; Carbone, M.L.; Pastore, S. The MEK inhibitors trametinib and cobimetinib induce a type I interferon response in human keratinocytes. Int. J. Mol. Sci. 2017, 18, 2227. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, S.; Hayashi, H.; Haratani, K.; Shimizu, S.; Tanizaki, J.; Sakai, K.; Kawakami, H.; Yonesaka, K.; Tsurutani, J.; Togashi, Y.; et al. Mutational activation of the epidermal growth factor receptor down-regulates major histocompatibility complex class I expression via the extracellular signal-regulated kinase in non–small cell lung cancer. Cancer Sci. 2019, 110, 52–60. [Google Scholar] [CrossRef] [Green Version]
- Sapkota, B.; Hill, C.E.; Pollack, B.P. Vemurafenib enhances MHC induction in BRAFV600E homozygous melanoma cells. Oncoimmunology 2013, 2, e22890. [Google Scholar] [CrossRef] [Green Version]
- Mimura, K.; Ando, T.; Poschke, I.; Mougiakakos, D.; Johansson, C.C.; Ichikawa, J.; Okita, R.; Nishimura, M.I.; Handke, D.; Krug, N.; et al. T cell recognition of HLA-A2 restricted tumor antigens is impaired by the oncogene HER2. Int. J. Cancer 2011, 128, 390–401. [Google Scholar] [CrossRef] [PubMed]
- Maruyama, T.; Mimura, K.; Sato, E.; Watanabe, M.; Mizukami, Y.; Kawaguchi, Y.; Ando, T.; Kinouchi, H.; Fujii, H.; Kono, K. Inverse correlation of HER2 with MHC class i expression on oesophageal squamous cell carcinoma. Br. J. Cancer 2010, 103, 552–559. [Google Scholar] [CrossRef] [Green Version]
- Hastings, K.; Yu, H.A.; Wei, W.; Sanchez-Vega, F.; Deveaux, M.; Choi, J.; Rizvi, H.; Lisberg, A.; Truini, A.; Lydon, C.A.; et al. EGFR mutation subtypes and response to immune checkpoint blockade treatment in non-small-cell lung cancer. Ann. Oncol. 2019, 30, 1311–1320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zage, P.E.; Sirisaengtaksin, N.; Liu, Y.; Gireud, M.; Brown, B.S.; Palla, S.; Richards, K.N.; Hughes, D.P.M.; Bean, A.J. UBE4B levels are correlated with clinical outcomes in neuroblastoma patients and with altered neuroblastoma cell proliferation and sensitivity to epidermal growth factor receptor inhibitors. Cancer 2013, 119, 915–923. [Google Scholar] [CrossRef] [PubMed]
- Leibowitz, M.S.; Srivastava, R.M.; Filho, P.A.A.; Egloff, A.M.; Wang, L.; Seethala, R.R.; Ferrone, S.; Ferris, R.L. SHP2 is overexpressed and inhibits pSTAT1-mediated APM component expression, T cell attracting chemokine secretion, and CTL recognition in head and neck cancer cells. Clin. Cancer Res. 2013, 19, 798–808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agazie, Y.M.; Hayman, M.J. Molecular Mechanism for a Role of SHP2 in Epidermal Growth Factor Receptor Signaling. Mol. Cell. Biol. 2003, 23, 7875–7886. [Google Scholar] [CrossRef] [Green Version]
- Grandis, J.R.; Drenning, S.D.; Zeng, Q.; Watkins, S.C.; Melhem, M.F.; Endo, S.; Johnson, D.E.; Huang, L.; He, Y.; Kim, J.D. Constitutive activation of stat3 signaling abrogates apoptosis in squamous cell carcinogenesis in vivo. Proc. Natl. Acad. Sci. USA 2000, 97, 4227–4232. [Google Scholar] [CrossRef] [Green Version]
- Garrido, G.; Rabasa, A.; Garrido, C.; Chao, L.; Garrido, F.; García-Lora, Á.M.; Sánchez-Ramírez, B. Upregulation of HLA Class I Expression on Tumor Cells by the Anti-EGFR Antibody Nimotuzumab. Front. Pharmacol. 2017, 8, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Srivastava, R.M.; Trivedi, S.; Concha-Benavente, F.; Hyun-Bae, J.; Wang, L.; Seethala, R.R.; Branstetter IV, B.F.; Ferrone, S.; Ferris, R.L. Stat1-induced HLA class i upregulation enhances immunogenicity and clinical response to anti-EGFR mab cetuximab therapy in HNC patients. Cancer Immunol. Res. 2015, 3, 936–945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pollack, B.P.; Sapkota, B.; Cartee, T.V. Epidermal growth factor receptor inhibition augments the expression of MHC class I and II genes. Clin. Cancer Res. 2011, 17, 4400–4413. [Google Scholar] [CrossRef] [Green Version]
- Freeman, A.J.; Vervoort, S.J.; Ramsbottom, K.M.; Kelly, M.J.; Michie, J.; Pijpers, L.; Johnstone, R.W.; Kearney, C.J.; Oliaro, J. Natural Killer Cells Suppress T Cell-Associated Tumor Immune Evasion. Cell Rep. 2019, 28, 2784–2794.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, S.; Yin, T.; Li, D.; Gao, X.; Wan, Y.; Ma, X.; Ye, T.; Guo, F.; Sun, J.; Lin, Z.; et al. Enhanced interaction between natural killer cells and lung cancer cells: Involvement in gefitinib-mediated immunoregulation. J. Transl. Med. 2013, 11, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van’t Veer, L.J.; Beijersbergen, R.L.; Bernards, R. N-myc suppresses major histocompatibility complex class I gene expression through down-regulation of the p50 subunit of NF-kappa B. EMBO J. 1993, 12, 195–200. [Google Scholar] [CrossRef] [Green Version]
- Forloni, M.; Albini, S.; Limongi, M.Z.; Cifaldi, L.; Boldrini, R.; Nicotra, M.R.; Giannini, G.; Natali, P.G.; Giacomini, P.; Fruci, D. NF-κB, and not MYCN, regulates MHC class I and endoplasmic reticulum aminopeptidases in human neuroblastoma cells. Cancer Res. 2010, 70, 916–924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hodge, J.W.; Garnett, C.T.; Farsaci, B.; Palena, C.; Tsang, K.Y.; Ferrone, S.; Gameiro, S.R. Chemotherapy-induced immunogenic modulation of tumor cells enhances killing by cytotoxic T lymphocytes and is distinct from immunogenic cell death. Int. J. Cancer 2013, 133, 624–636. [Google Scholar] [CrossRef] [PubMed]
- Khallouf, H.; Märten, A.; Serba, S.; Teichgräber, V.; Büchler, M.W.; Jäger, D.; Schmidt, J. 5-fluorouracil and interferon-α immunochemotherapy enhances immunogenicity of murine pancreatic cancer through upregulation of NKG2D ligands and MHC class i. J. Immunother. 2012, 35, 245–253. [Google Scholar] [CrossRef]
- Wan, S.; Pestka, S.; Jubin, R.G.; Lyu, Y.L.; Tsai, Y.C.; Liu, L.F. Chemotherapeutics and radiation stimulate MHC class i expression through elevated interferon-beta signaling in breast cancer cells. PLoS ONE 2012, 7, e32542. [Google Scholar] [CrossRef]
- Iwai, T.; Sugimoto, M.; Wakita, D.; Yorozu, K.; Kurasawa, M.; Yamamoto, K. Topoisomerase I inhibitor, irinotecan, depletes regulatory T cells and up-regulates MHC class I and PD-L1 expression, resulting in a supra-additive antitumor effect when combined with anti- PD-L1 antibodies. Oncotarget 2018, 9, 31411–31421. [Google Scholar] [CrossRef]
- Alagkiozidis, I.; Facciabene, A.; Tsiatas, M.; Carpenito, C.; Benencia, F.; Adams, S.; Jonak, Z.; June, C.H.; Powell, D.J.; Coukos, G. Time-dependent cytotoxic drugs selectively cooperate with IL-18 for cancer chemo-immunotherapy. J. Transl. Med. 2011, 9, 77. [Google Scholar] [CrossRef] [Green Version]
- De Mora-García, M.L.; Duenas-González, A.; Hernández-Montes, J.; De la Cruz-Hernández, E.; Pérez-Cárdenas, E.; Weiss-Steider, B.; Santiago-Osorio, E.; Ortíz-Navarrete, V.F.; Rosales, V.H.; Cantú, D.; et al. Up-regulation of HLA class-I antigen expression and antigen-specific CTL response in cervical cancer cells by the demethylating agent hydralazine and the histone deacetylase inhibitor valproic acid. J. Transl. Med. 2006, 4, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Khan, A.N.H.; Gregorie, C.J.; Tomasi, T.B. Histone deacetylase inhibitors induce TAP, LMP, Tapasin genes and MHC class I antigen presentation by melanoma cells. Cancer Immunol. Immunother. 2008, 57, 647–654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ritter, C.; Fan, K.; Paschen, A.; Reker Hardrup, S.; Ferrone, S.; Nghiem, P.; Ugurel, S.; Schrama, D.; Becker, J.C. Epigenetic priming restores the HLA class-I antigen processing machinery expression in Merkel cell carcinoma. Sci. Rep. 2017, 7, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Setiadi, A.F.; Omilusik, K.; David, M.D.; Seipp, R.P.; Hartikainen, J.; Gopaul, R.; Choi, K.B.; Jefferies, W.A. Epigenetic enhancement of antigen processing and presentation promotes immune recognition of tumors. Cancer Res. 2008, 68, 9601–9607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suraweera, A.; O’Byrne, K.J.; Richard, D.J. Combination therapy with histone deacetylase inhibitors (HDACi) for the treatment of cancer: Achieving the full therapeutic potential of HDACi. Front. Oncol. 2018, 8, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serrano, A.; Tanzarella, S.; Lionello, I.; Mendez, R.; Traversari, C.; Ruiz-Cabello, F.; Garrido, F. Expression of HLA class I antigens and restoration of antigen-specific CTL response in melanoma cells following 5-aza-2′-deoxycytidine treatment. Int. J. Cancer 2001, 94, 243–251. [Google Scholar] [CrossRef]
- Luo, N.; Nixon, M.J.; Gonzalez-Ericsson, P.I.; Sanchez, V.; Opalenik, S.R.; Li, H.; Zahnow, C.A.; Nickels, M.L.; Liu, F.; Tantawy, M.N.; et al. DNA methyltransferase inhibition upregulates MHC-I to potentiate cytotoxic T lymphocyte responses in breast cancer. Nat. Commun. 2018, 9, 248. [Google Scholar] [CrossRef]
- Fonsatti, E.; Nicolay, H.J.M.; Sigalotti, L.; Calabrò, L.; Pezzani, L.; Colizzi, F.; Altomonte, M.; Guidoboni, M.; Marincola, F.M.; Maio, M. Functional up-regulation of human leukocyte antigen class I antigens expression by 5-aza-2′-deoxycytidine in cutaneous melanoma: Immunotherapeutic implications. Clin. Cancer Res. 2007, 13, 3333–3338. [Google Scholar] [CrossRef] [Green Version]
- Santin, A.D.; Hermonat, P.L.; Ravaggi, A.; Chiriva-Internati, M.; Pecorelli, S.; Parham, G.P. Retinoic acid up-regulates the expression of major histocompatibility complex molecules and adhesion/costimulation molecules (specifically, intercellular adhesion molecule ICAM-1) in human cervical cancer. In Proceedings of the American Journal of Obstetrics and Gynecology; Mosby Inc.: St. Louis, MO, USA, 1998; Volume 179, pp. 1020–1025. [Google Scholar]
- Castriconi, R.; Dondero, A.; Cilli, M.; Ognio, E.; Pezzolo, A.; De Giovanni, B.; Gambini, C.; Pistoia, V.; Moretta, L.; Moretta, A.; et al. Human NK cell infusions prolong survival of metastatic human neuroblastoma-bearing NOD/scid mice. Cancer Immunol. Immunother. 2007, 56, 1733–1742. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Regulation | Pathway | Effector | Described Mechanism(s) | Potential Therapeutic Strategy | Clinical Status |
|---|---|---|---|---|---|
| Negative Regulators | NFkB pathway | N4BP1 | 1. Cezanne-1 stabilization 2. Preventing recruitment of NEMO to RIP1 | miRNA-28-5p [65,66,67,70,71] | Pre-clinical |
| Cezanne-1 | Deubiquitination and stabilization of TRAF3 | Aspecific DUB inhibitors (e.g., Ubal) [87] miR-1180 [69], miR-486 [68] Cyanopyrrolidine derivates (WO2017109488) | Pre-clinical | ||
| TNIP1 | 1. Preventing recruitment of NEMO to RIP1 2. Preventing degradation of the IkB p105 into p50 3. A20 stabilization | IL-17 [63] miR-1180 [69], miR-486 [68] | Pre-clinical | ||
| A20 | 1. Deubiquitination of NEMO 2. Deubiquitination of RIP-1 and TRAF6 3. Degradation of RIP-1 | Aspecific DUB inhibitors (e.g., Ubal) [87] miR-1180 [69], miR-486 [68] | Pre-clinical | ||
| TAX1BP1 | Itch recruitment to A20 | ||||
| Itch | Controlling interaction between A20 and RIP1/TRAF6 | Clomipramine and norclomipramine [80] 1,4-naphthoquinone 10E [81] | FDA-approved antidepressants Pre-clinical | ||
| CYLD | 1. Deubiquitination of NEMO 2. Deubiquitination of TRAF2 | miR-1288 [84], miR-186 [85], miR-362-5p [86] | Pre-clinical | ||
| Type I/II IFN pathway | DUX4 | 1. JAK 1/2 downregulation 2. STAT2 downregulation | p300 inhibitors [115] p38 inhibitors [117] | Pre-clinical Several Phase II Trials (Rheumatoid Arthritis, Asthma, LMNA-related cardiomyopathy) | |
| LNK (SH2B3) | Dephosphorylation of STAT1 | miR-29b [122], miR-30-5p [121], miR-98 [121], miR181a-5p [121] | |||
| PTPN1 (PTP1B) | 1. Dephosphorylation of TYK2 2. Dephosphorylation of JAK2 | MSI-1436C [135] | Phase I Trial, metastatic breast cancer | ||
| PTPN2 (TC-PTP) | 1. Dephosphorylation of JAK1 | miRNA-155 [136] | Pre-clinical | ||
| PTPN11 (SHP2) | 1. Dephosphorylation of JAK1 2. Dephosphorylation of STAT1 3. Dephosphorylation of STAT2 | Sodium stibogluconate [134] | Phase II Trial, Leishmaniasis, stage IV melanoma, advanced solid tumor | ||
| RNF2 | Polyubiquitination of STAT1, resulting in release from DNA | Trametinib [143] PRT4165 [148] | FDA-approved in several advanced-stage cancers Pre-clinical | ||
| Smurf1 | Degradation of STAT1 | A01 [149] | Pre-clinical | ||
| PIAS | Inhibiting STAT1 promotor recruitment | ||||
| Type I IFN pathway | DCST1 | Ubiquitination and degradation of STAT2 | |||
| PKD2 | Stimulating ubiquitination and endocytosis of IFNAR1 | CRT0066101 [152] SD-208 [153] | Pre-clinical | ||
| Pin1 | Ubiquitination and degradation of IRF3 | miR-200b [126], miR-200c [127], miR-296-5p [128] ATRA [129] KPT-6566 [130] Juglone [125] | Pre-clinical FDA-approved in cancer Pre-clinical Pre-clinical | ||
| NFkB and type I/II IFN pathway | STAT3 | 1. Negative feedback loop of pro-inflammatory type I signaling 2. Inhibition of IKK activation | STAT3 inhibitors: JSI-124 [169] Indirubin [164] Resveratrol [165] Nifuroxazide [170] Static [166] Sorafenib [167] Sunitinib [168] | Pre-clinical Phase 3/4 trials in acute promyelocytic leukemia, phase 2/3 in dermatitis and psoriasis Phase 2/3 in congestive heart failure, Friedreich Ataxia, Gulf War Illness, Lymphangioleiomyomatosis, and infertility FDA-approved antibiotic Pre-clinical Pre-clinical Pre-clinical | |
| EGFR receptor family (including HER2/neu) | 1. STAT1 inactivation 2. Stimulation of MAPK pathway 3. STAT3 activation 4. Fhit degradation | Tyrosine Kinase inhibitors: EGFR inhibitors (e.g., nimotuzumab [198], cetuximab [199], afatinib [185], erlotinib [185], gefitinib [202]). | FDA-approved in cancer | ||
| NMYC | Inhibition of p50 (although inconsistent) | ||||
| MAPK pathway | 1. Decreases IRF1 activity 2. Decreases STAT1 expression | Tyrosine Kinase inhibitors: MEK-inhibitors (e.g., trametinib [188,189], cobimetinib [188], selumetinib [2]) BRAF inhibitors (e.g., vemurafenib [184,190], dabrafenib [41]) | FDA-approved in cancer, selumetinib in phase 3 FDA-approved in cancer | ||
| NFkB pathway | NFkB-inducing receptor stimulation | 1. TNFR superfamily stimulation 2. PRR Receptor stimulation 3. IL-1 receptor stimulation | TNFα [40] | Phase I-III trials in cancer, all localized infusions | |
| IKK/IkBα | 1. Boosting IKK-activity 2. Degradation of IkBα | Betulinic acid [45] | Phase I trial, Anxiety | ||
| IKK | Boosting IKK-activity | Calcium/calcineurin + prostratin [51] | Pre-clinical | ||
| Nedd4 | 1. Degradation of N4BP1 2. Degradation of TRAF3 | ||||
| p50, RelA, and LMP-2/-7/-10 | 1. Enhanced expression of p50 2. Enhanced expression of RelA 3. Enhances expression of LMP-2/-7/-10 | Retinoids [41,218] | FDA-approved in several diseases, including cancers | ||
| Type I IFN pathway | IFN-inducing receptor stimulation | 1. PRR Receptors stimulation (e.g., RNF135, TRIM25, ISG15, NAB2) 2. IFNA Receptor stimulation | IFNα [100,101] IFNβ therapy [101] | FDA-approved in hepatitis B & high-risk melanoma Phase III trial in relapsing multiple sclerosis, Phase I in refractory solid tumors | |
| Type II IFN pathway | IFN-inducing receptor stimulation | IFNG Receptor stimulation | IFNγ-1b NK cell therapy [19,33,103,106,107,219] | FDA approved as localized injection in chronic granulomatous disease (CGD) and severe, malignant ostepetrosis (SMO), phase I-III in cancers Phase I/II Trial in several cancer types | |
| NFkB- and type I IFN pathway | STING | 1. Phosphorylation and activation of RelA 2. Phosphorylation and activation of IRF3 | STING agonists (e.g., SB 11285 [177]) | Phase I Trial in patients with advanced solid tumors | |
| NFkB- and type I IFN pathway | 1. Increasing APM expression (Calreticulin, TAP2, calnexin) 2. Enhancing type I IFN signaling 3. NFkB stabilization 4. IFNβ secretion | Docetaxel [205] 5-Fluorouracil [206] Topoisomerase inhibitors (e.g., topotecan [207,209], irinotecan [208], and etoposide [207] Microtubule stabilizers (e.g., paclitaxel [207,209] and vinblastine [207]) Cisplatin [207] | All FDA-approved in cancer | ||
| Epigenetic modification | NFkB and type I/II IFN pathway | 1. Histon acetylation to decrease genome accessibility 2. DNA methylation to decrease genome accessibility | HDAC inhibitors: (e.g., Romidepsin, Vorinostat [212] and Panobinostat) [1,211,212,213,214] DNMTis (e.g., 5-azacytidine [212], Decitabine [54], and Guadecitabine [216]) | All FDA-approved in cancer FDA approved, Guadecitabine phase II trials in cancer). |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cornel, A.M.; Mimpen, I.L.; Nierkens, S. MHC Class I Downregulation in Cancer: Underlying Mechanisms and Potential Targets for Cancer Immunotherapy. Cancers 2020, 12, 1760. https://doi.org/10.3390/cancers12071760
Cornel AM, Mimpen IL, Nierkens S. MHC Class I Downregulation in Cancer: Underlying Mechanisms and Potential Targets for Cancer Immunotherapy. Cancers. 2020; 12(7):1760. https://doi.org/10.3390/cancers12071760
Chicago/Turabian StyleCornel, Annelisa M., Iris L. Mimpen, and Stefan Nierkens. 2020. "MHC Class I Downregulation in Cancer: Underlying Mechanisms and Potential Targets for Cancer Immunotherapy" Cancers 12, no. 7: 1760. https://doi.org/10.3390/cancers12071760