Reduced Tumorigenicity of Mouse ES Cells and the Augmented Anti-Tumor Therapeutic Effects under Parg Deficiency

 ,
, 
Abstract
:1. Introduction
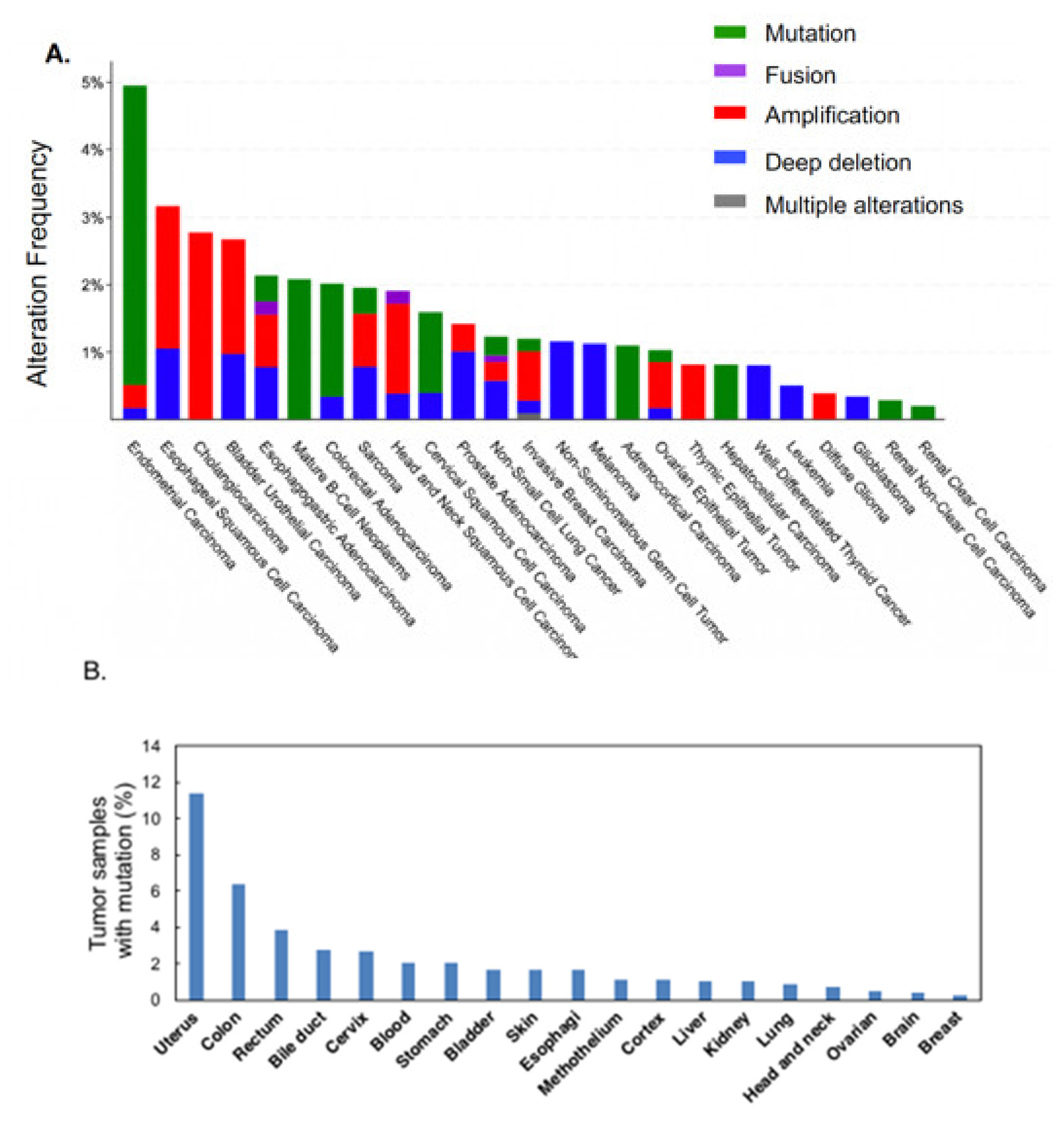
2. Results
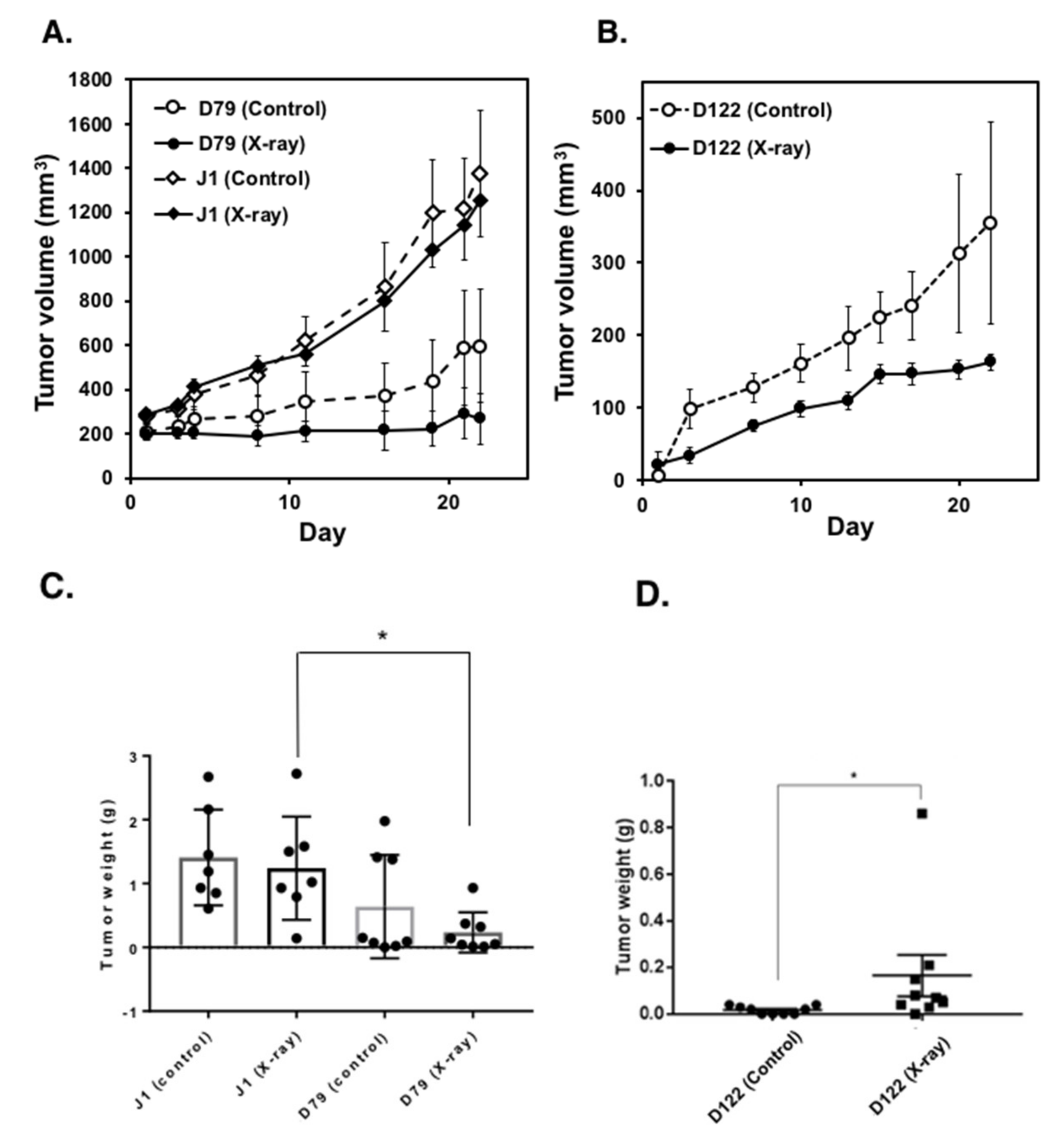
2.1. Parg−/− ES Cells Show Delayed Tumor Development
2.2. Characterization of Tumor Tissues
2.3. Time Course Analysis of Tumorigenesis
2.4. Augmented Anti-Tumor Therapeutic Effects under Parg Deficiency
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Tumorigenesis Analysis
4.3. Histological Analysis
4.4. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Schreiber, V.; Dantzer, F.; Ame, J.C.; de Murcia, G. Poly(ADP-ribose): Novel functions for an old molecule. Nat. Rev. Mol. Cell Biol. 2006, 7, 517–528. [Google Scholar] [CrossRef] [PubMed]
- Oka, S.; Kato, J.; Moss, J. Identification and characterization of a mammalian 39-kDa poly(ADP-ribose) glycohydrolase. J. Biol. Chem. 2006, 281, 705–713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ame, J.C.; Jacobson, E.L.; Jacobson, M.K. Molecular heterogeneity and regulation of poly(ADP-ribose) glycohydrolase. Mol. Cell Biochem. 1999, 193, 75–81. [Google Scholar] [CrossRef] [PubMed]
- Miwa, M.; Sugimura, T. Splitting of the ribose-ribose linkage of poly(adenosine diphosphate-robose) by a calf thymus extract. J. Biol. Chem. 1971, 246, 6362–6364. [Google Scholar]
- Wei, L.; Nakajima, S.; Hsieh, C.L.; Kanno, S.; Masutani, M.; Levine, A.S.; Yasui, A.; Lan, L. Damage response of XRCC1 at sites of DNA single strand breaks is regulated by phosphorylation and ubiquitylation after degradation of poly(ADP-ribose). J. Cell Sci. 2013, 126, 4414–4423. [Google Scholar] [CrossRef] [Green Version]
- Nakadate, Y.; Kodera, Y.; Kitamura, Y.; Tachibana, T.; Tamura, T.; Koizumi, F. Silencing of poly(ADP-ribose) glycohydrolase sensitizes lung cancer cells to radiation through the abrogation of DNA damage checkpoint. Biochem. Biophys. Res. Commun. 2013, 441, 793–798. [Google Scholar] [CrossRef]
- Shirai, H.; Fujimori, H.; Gunji, A.; Maeda, D.; Hirai, T.; Poetsch, A.R.; Harada, H.; Yoshida, T.; Sasai, K.; Okayasu, R.; et al. Parg deficiency confers radio-sensitization through enhanced cell death in mouse ES cells exposed to various forms of ionizing radiation. Biochem. Biophys. Res. Commun. 2013, 435, 100–106. [Google Scholar] [CrossRef]
- Zampieri, M.; Passananti, C.; Calabrese, R.; Perilli, M.; Corbi, N.; De Cave, F.; Guastafierro, T.; Bacalini, M.G.; Reale, A.; Amicosante, G.; et al. Parp1 Localizes within the Dnmt1 Promoter and Protects Its Unmethylated State by Its Enzymatic Activity. PLoS ONE 2009, 4, e4717. [Google Scholar] [CrossRef]
- Koh, D.W.; Dawson, V.L.; Dawson, T.M. The road to survival goes through PARG. Cell Cycle. 2005, 4, 397–399. [Google Scholar] [CrossRef]
- Chen, L.; Gunji, A.; Uemura, A.; Fujihara, H.; Nakamoto, K.; Onodera, T.; Sasaki, Y.; Imamichi, S.; Isumi, M.; Nozaki, T.; et al. Development of renal failure in PargParp-1 null and Timm23 hypomorphic mice. Biochem. Pharmacol. 2019, 167, 116–124. [Google Scholar] [CrossRef]
- Slade, D.; Dunstan, M.S.; Barkauskaite, E.; Weston, R.; Lafite, P.; Dixon, N.; Ahel, M.; Leys, D.; Ahel, I. The structure and catalytic mechanism of a poly(ADP-ribose) glycohydrolase. Nature 2011, 477, 616–620. [Google Scholar] [CrossRef] [PubMed]
- Tucker, J.A.; Bennett, N.; Brassington, C.; Durant, S.T.; Hassall, G.; Holdgate, G.; McAlister, M.; Nissink, J.W.; Truman, C.; Watson, M. Structures of the human poly (ADP-ribose) glycohydrolase catalytic domain confirm catalytic mechanism and explain inhibition by ADP-HPD derivatives. PLoS ONE 2012, 7, e50889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Gagne, J.P.; Poirier, G.G.; Xu, W. Crystallographic and biochemical analysis of the mouse poly(ADP-ribose) glycohydrolase. PLoS ONE 2014, 9, e86010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poitras, M.F.; Koh, D.W.; Yu, S.W.; Andrabi, S.A.; Mandir, A.S.; Poirier, G.G.; Dawson, V.L.; Dawson, T.M. Spatial and functional relationship between poly(ADP-ribose) polymerase-1 and poly(ADP-ribose) glycohydrolase in the brain. Neuroscience 2007, 148, 198–211. [Google Scholar] [CrossRef] [Green Version]
- Meyer-Ficca, M.L.; Meyer, R.G.; Coyle, D.L.; Jacobson, E.L.; Jacobson, M.K. Human poly(ADP-ribose) glycohydrolase is expressed in alternative splice variants yielding isoforms that localize to different cell compartments. Exp. Cell Res. 2004, 297, 521–532. [Google Scholar] [CrossRef]
- Burns, D.M.; Ying, W.; Kauppinen, T.M.; Zhu, K.; Swanson, R.A. Selective Down-Regulation of Nuclear Poly(ADP-Ribose) Glycohydrolase. PLoS ONE 2009, 4, e4896. [Google Scholar] [CrossRef] [Green Version]
- Whatcott, C.J.; Meyer-Ficca, M.L.; Meyer, R.G.; Jacobson, M.K. A specific isoform of poly(ADP-ribose) glycohydrolase is targeted to the mitochondrial matrix by a N-terminal mitochondrial targeting sequence. Exp. Cell Res. 2009, 315, 3477–3485. [Google Scholar] [CrossRef] [Green Version]
- Cortes, U.; Tong, W.M.; Coyle, D.L.; Meyer-Ficca, M.L.; Meyer, R.G.; Petrilli, V.; Herceg, Z.; Jacobson, E.L.; Jacobson, M.K.; Wang, Z.Q. Depletion of the 110-kilodalton isoform of poly(ADP-ribose) glycohydrolase increases sensitivity to genotoxic and endotoxic stress in mice. Mol. Cell Biol. 2004, 24, 7163–7178. [Google Scholar] [CrossRef] [Green Version]
- Koh, D.W.; Lawler, A.M.; Poitras, M.F.; Sasaki, M.; Wattler, S.; Nehls, M.C.; Stoger, T.; Poirier, G.G.; Dawson, V.L.; Dawson, T.M. Failure to degrade poly(ADP-ribose) causes increased sensitivity to cytotoxicity and early embryonic lethality. Proc. Natl. Acad. Sci. USA 2004, 101, 17699–17704. [Google Scholar] [CrossRef] [Green Version]
- Yu, S.W.; Wang, H.; Poitras, M.F.; Coombs, C.; Bowers, W.J.; Federoff, H.J.; Poirier, G.G.; Dawson, T.M.; Dawson, V.L. Mediation of poly(ADP-ribose) polymerase-1-dependent cell death by apoptosis-inducing factor. Science 2002, 297, 259–263. [Google Scholar] [CrossRef]
- Andrabi, S.A.; Kim, N.S.; Yu, S.W.; Wang, H.; Koh, D.W.; Sasaki, M.; Klaus, J.A.; Otsuka, T.; Zhang, Z.; Koehler, R.C.; et al. Poly(ADP-ribose) (PAR) polymer is a death signal. Proc. Natl. Acad. Sci. USA 2006, 103, 18308–18313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, W.; Fu, Y.; Deng, Y.; Zeng, Z.; Gu, P.; Liu, H.; Liu, J.; Xu, X.; Wu, D.; Luo, X.; et al. Regulation of Wnt Singaling Pathway by Poly (ADP-Ribose) Glycohydrolase (PARG) Silencing Suppresses Lung Cancer in Mice Induced by Benzo(a)pyrene Inhalation Exposure. Front. Pharmacol. 2019, 10, 338. [Google Scholar] [CrossRef] [PubMed]
- Nozaki, T.; Masutani, M.; Watanabe, M.; Ochiya, T.; Hasegawa, F.; Nakagama, H.; Suzuki, H.; Sugimura, T. Syncytiotrophoblastic giant cells in teratocarcinoma-like tumors derived from Parp-disrupted mouse embryonic stem cells. Proc. Natl. Acad. Sci. USA 1999, 96, 13345–13350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, K.; Mitsui, K.; Yamanaka, S. Role of ERas in promoting tumour-like properties in mouse embryonic stem cells. Nature 2003, 423, 541–545. [Google Scholar] [CrossRef]
- Hilberg, F.; Wagner, E.F. Embryonic stem (ES) cells lacking functional c-jun: Consequences for growth and differentiation, AP-1 activity and tumorigenicity. Oncogene 1992, 7, 2371–2380. [Google Scholar]
- Zhang, X.; Morham, S.G.; Langenbach, R.; Baggs, R.B.; Young, D.A. Lack of cyclooxygenase-2 inhibits growth of teratocarcinomas in mice. Exp. Cell Res. 2000, 254, 232–240. [Google Scholar] [CrossRef]
- Gidekel, S.; Pizov, G.; Bergman, Y.; Pikarsky, E. Oct-3/4 is a dose-dependent oncogenic fate determinant. Cancer Cell 2003, 4, 361–370. [Google Scholar] [CrossRef] [Green Version]
- Bryant, H.E.; Petermann, E.; Schultz, N.; Jemth, A.S.; Loseva, O.; Issaeva, N.; Johansson, F.; Fernandez, S.; McGlynn, P.; Helleday, T. PARP is activated at stalled forks to mediate Mre11-dependent replication restart and recombination. EMBO J. 2009, 28, 2601–2615. [Google Scholar] [CrossRef] [Green Version]
- Shirai, H.; Poetsch, A.R.; Gunji, A.; Maeda, D.; Fujimori, H.; Fujihara, H.; Yoshida, T.; Ogino, H.; Masutani, M. PARG dysfunction enhances DNA double strand break formation in S-phase after alkylation DNA damage and augments different cell death pathways. Cell Death Dis. 2013, 4, e656. [Google Scholar] [CrossRef]
- Virág, L.; Szabó, C. The Therapeutic Potential of Poly(ADP-Ribose) Polymerase Inhibitors. Pharmacol. Rev. 2002, 54, 375–429. [Google Scholar] [CrossRef]
- Gonçalves, A.; Finetti, P.; Sabatier, R.; Gilabert, M.; Adelaide, J.; Borg, J.-P.; Chaffanet, M.; Viens, P.; Birnbaum, D.; Bertucci, F. Poly(ADP-ribose) polymerase-1 mRNA expression in human breast cancer: A meta-analysis. Breast Cancer Res. Treat. 2011, 127, 273–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nosho, K.; Yamamoto, H.; Mikami, M.; Taniguchi, H.; Takahashi, T.; Adachi, Y.; Imamura, A.; Imai, K.; Shinomura, Y. Overexpression of poly(ADP-ribose) polymerase-1 (PARP-1) in the early stage of colorectal carcinogenesis. Eur. J. Cancer 2006, 42, 2374–2381. [Google Scholar] [CrossRef] [PubMed]
- Staibano, S.; Pepe, S.; Muzio, L.L.; Somma, P.; Mascolo, M.; Argenziano, G.; Scalvenzi, M.; Salvatore, G.; Fabbrocini, G.; Molea, G.; et al. Poly(adenosine diphosphate-ribose) polymerase 1 expression in malignant melanomas from photoexposed areas of the head and neck region. Hum. Pathol. 2005, 36, 724–731. [Google Scholar] [CrossRef] [PubMed]
- Sevigny, M.B.; Silva, J.M.; Lan, W.C.; Alano, C.C.; Swanson, R.A. Expression and activity of poly(ADP-ribose) glycohydrolase in cultured astrocytes, neurons, and C6 glioma cells. Brain Res. Mol. Brain Res. 2003, 117, 213–220. [Google Scholar] [CrossRef]
- Pillay, N.; Tighe, A.; Nelson, L.; Littler, S.; Coulson-Gilmer, C.; Bah, N.; Golder, A.; Bakker, B.; Spierings, D.C.J.; James, D.I.; et al. DNA Replication Vulnerabilities Render Ovarian Cancer Cells Sensitive to Poly(ADP-Ribose) Glycohydrolase Inhibitors. Cancer Cell 2019, 35, 519–533. [Google Scholar] [CrossRef] [Green Version]
- Fujihara, H.; Ogino, H.; Maeda, D.; Shirai, H.; Nozaki, T.; Kamada, N.; Jishage, K.; Tanuma, S.; Takato, T.; Ochiya, T.; et al. Poly(ADP-ribose) Glycohydrolase deficiency sensitizes mouse ES cells to DNA damaging agents. Curr. Cancer Drug Targets 2009, 9, 953–962. [Google Scholar] [CrossRef]
- Jain, A.; Agostini, L.C.; McCarthy, G.A.; Chand, S.N.; Ramirez, A.; Nevler, A.; Cozzitorto, J.; Schultz, C.W.; Lowder, C.Y.; Smith, K.M.; et al. Poly (ADP) Ribose Glycohydrolase Can Be Effectively Targeted in Pancreatic Cancer. Cancer Res. 2019, 79, 4491–4502. [Google Scholar] [CrossRef]
- Fauzee, N.J.; Li, Q.; Wang, Y.L.; Pan, J. Silencing Poly (ADP-Ribose) Glycohydrolase (PARG) Expression Inhibits Growth of Human Colon Cancer Cells In Vitro via PI3K/Akt/NFkappa-B Pathway. Pathol. Oncol. Res. 2011. [Google Scholar] [CrossRef]
- Sasaki, Y.; Fujimori, H.; Hozumi, M.; Onodera, T.; Nozaki, T.; Murakami, Y.; Ashizawa, K.; Inoue, K.; Koizumi, F.; Masutani, M. Dysfunction of Poly (ADP-Ribose) Glycohydrolase Induces a Synthetic Lethal Effect in Dual Specificity Phosphatase 22-Deficient Lung Cancer Cells. Cancer Res. 2019, 79, 3851–3861. [Google Scholar] [CrossRef]
- cBioPortal for Cancer Genomics. Available online: https://www.cbioportal.org (accessed on 30 December 2019).
- CanSAR database. Available online: https://cansarblack.icr.ac.uk/ (accessed on 29 December 2019).
- Masutani, M.; Nozaki, T.; Nishiyama, E.; Ochiya, T.; Wakabayashi, K.; Suzuki, H.; and Sugimura, T. Establishment of poly(ADP-ribose) polymerase-deficient mouse embryonic stem cell lines. Proc. Jpn. Acad. 1998, 74, 233–236. [Google Scholar] [CrossRef] [Green Version]
- Islam, R.; Koizumi, F.; Kodera, Y.; Inoue, K.; Okawara, T.; Masutani, M. Design and synthesis of phenolic hydrazide hydrazones as potent poly(ADP-ribose) glycohydrolase (PARG) inhibitors. Bioorg. Med. Chem. Lett. 2014, 24, 3802–3806. [Google Scholar] [CrossRef] [PubMed]
- Gogola, E.; Duarte, A.A.; de Ruiter, J.R.; Wiegant, W.W.; Schmid, J.A.; de Bruijn, R.; James, D.I.; Llobet, S.G.; Vis, D.J.; Annunziato, S.; et al. Selective Loss of PARG Restores PARylation and Counteracts PARP Inhibitor-Mediated Synthetic Lethality. Cancer Cell 2019, 35, 950–952. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Houl, J.H.; Ye, Z.; Brosey, C.A.; Balapiti-Modarage, L.P.F.; Namjoshi, S.; Bacolla, A.; Laverty, D.; Walker, B.L.; Pourfarjam, Y.; Warden, L.S.; et al. Selective small molecule PARG inhibitor causes replication fork stalling and cancer cell death. Nat. Commun. 2019, 10, 5654. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Tissue Type | Parg+/+(J1) | Parg−/− (D79) | Parg−/− (D122) |
|---|---|---|---|
| Embryonal carcinoma | + | + | + |
| Hemorrhage | − | − | − |
| Trophoblast giant cells | − | − | − |
| Ectodermal derivatives | |||
| Primitive neuroepithelium | + | + | + |
| Mature neural tissue | + | + | + |
| Keratinized epithelium | + | + | + |
| Mesodermal derivatives | |||
| Cartilage | + | + | + |
| Bone | + | + | + |
| Blood vessel | + | + | + |
| Lymphocyte and blood cell | + | + | + |
| Muscle | + | + | + |
| Endodermal derivatives | |||
| Ciliated epithelium | + | + | + |
| Gut epithelium | + | + | + |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sonoda, Y.; Sasaki, Y.; Gunji, A.; Shirai, H.; Araki, T.; Imamichi, S.; Onodera, T.; Rydén, A.-M.; Watanabe, M.; Itami, J.; et al. Reduced Tumorigenicity of Mouse ES Cells and the Augmented Anti-Tumor Therapeutic Effects under Parg Deficiency. Cancers 2020, 12, 1056. https://doi.org/10.3390/cancers12041056
Sonoda Y, Sasaki Y, Gunji A, Shirai H, Araki T, Imamichi S, Onodera T, Rydén A-M, Watanabe M, Itami J, et al. Reduced Tumorigenicity of Mouse ES Cells and the Augmented Anti-Tumor Therapeutic Effects under Parg Deficiency. Cancers. 2020; 12(4):1056. https://doi.org/10.3390/cancers12041056
Chicago/Turabian StyleSonoda, Yuki, Yuka Sasaki, Akemi Gunji, Hidenori Shirai, Tomonori Araki, Shoji Imamichi, Takae Onodera, Anna-Margareta Rydén, Masatoshi Watanabe, Jun Itami, and et al. 2020. "Reduced Tumorigenicity of Mouse ES Cells and the Augmented Anti-Tumor Therapeutic Effects under Parg Deficiency" Cancers 12, no. 4: 1056. https://doi.org/10.3390/cancers12041056