ALK Inhibitors-Induced M Phase Delay Contributes to the Suppression of Cell Proliferation


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
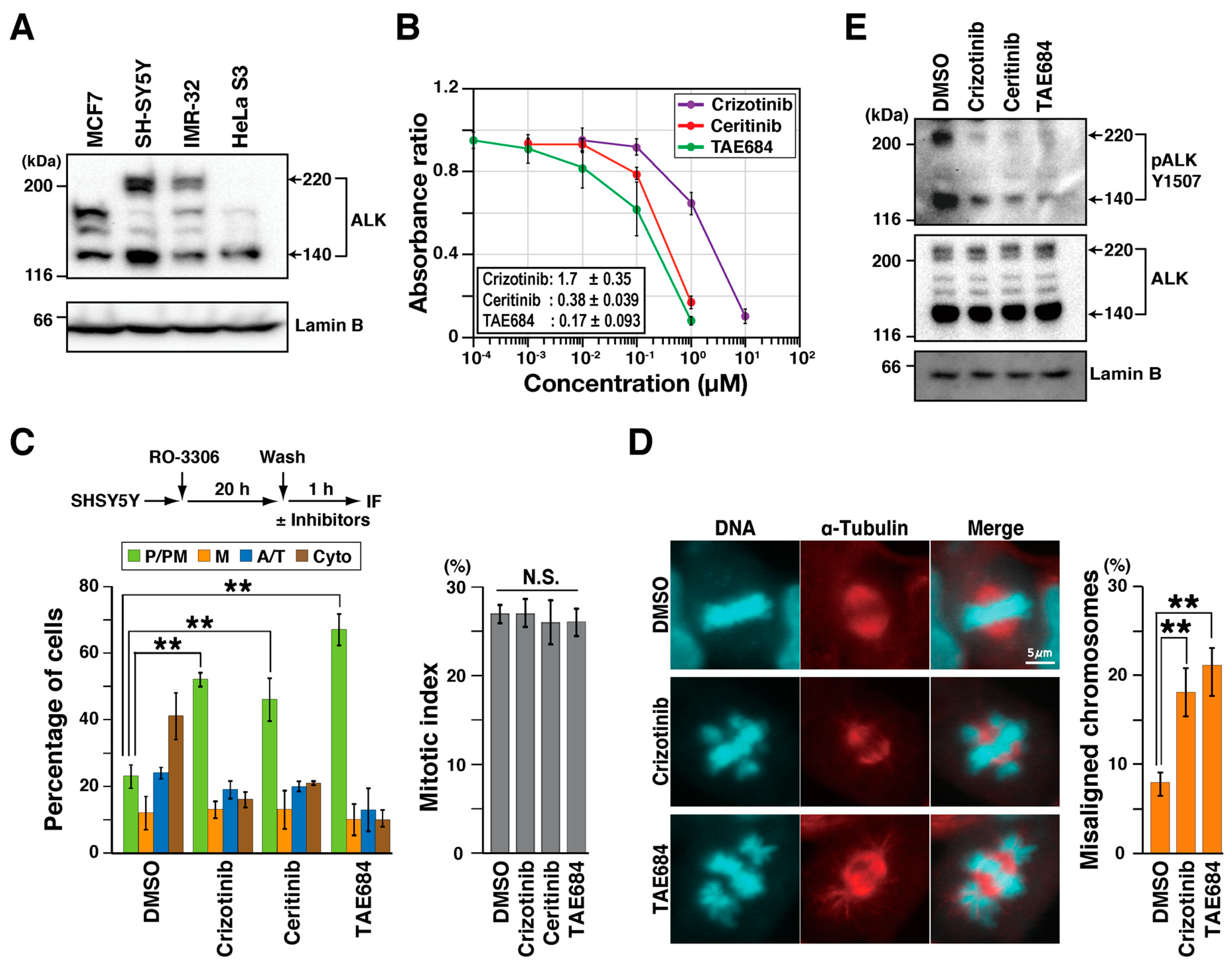
2.1. ALK Inhibitors Delay M Phase Progression
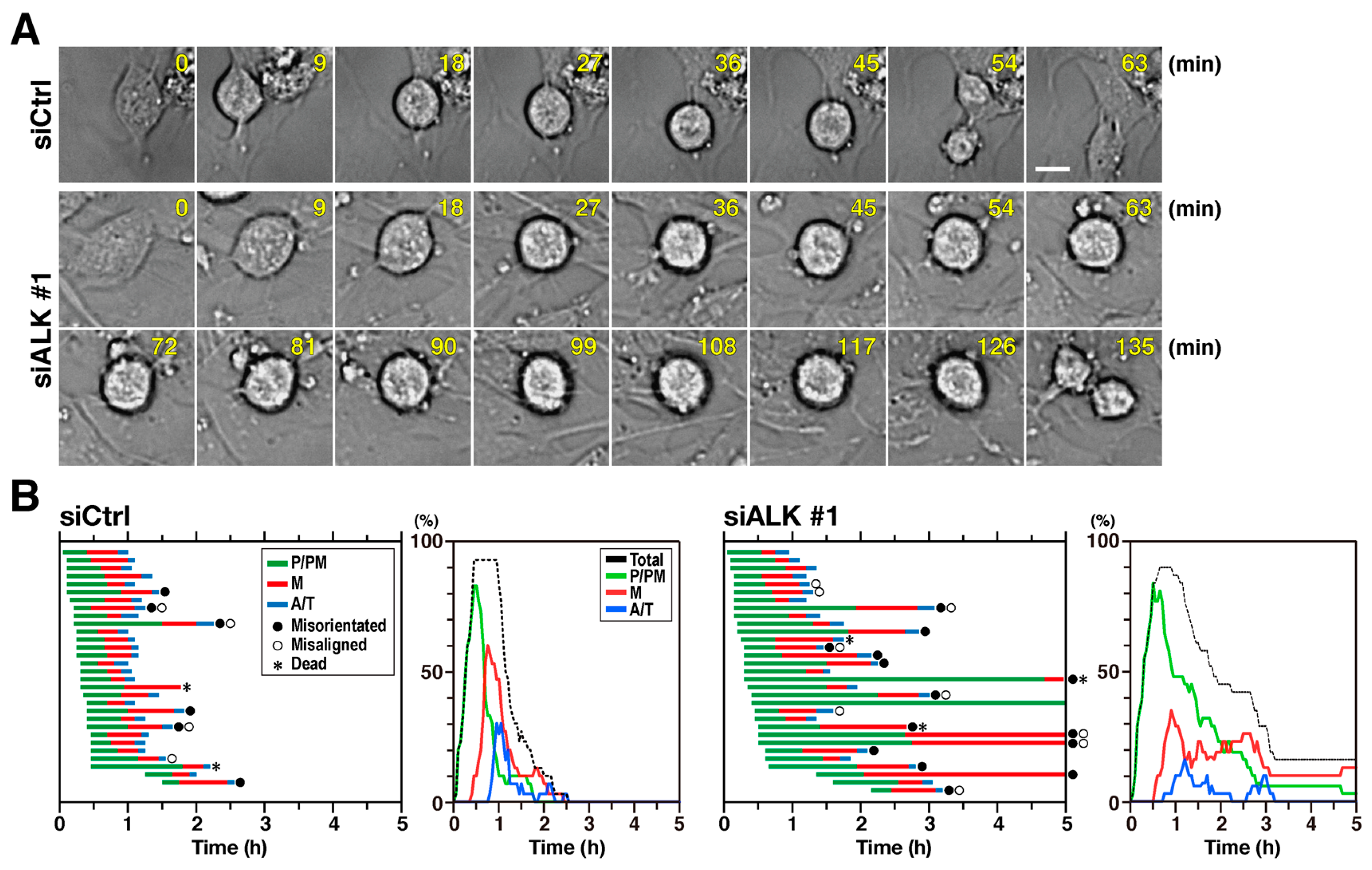
2.2. Knockdown of ALK Causes M Phase Delay
2.3. Depletion of ALK Prolonged both Prophase/Prometaphase and Metaphase
2.4. SAC Is Activated Indirectly in ALK-Inhibited Cells
2.5. Inhibition of EML4-ALK Fusion Delays M Phase Progression
3. Discussion
4. Materials and Methods
4.1. Cells
4.2. Reagents
4.3. Antibodies
4.4. Transfection
4.5. Cell Cycle Synchronization
4.6. Western Blotting
4.7. Immunofluorescence Microscopy
4.8. Time-Lapse Imaging
4.9. Proliferation Assay
4.10. Plasmid
4.11. Establishment of Stable Cell Lines by Lentiviral Transduction
4.12. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Iwahara, T.; Fujimoto, J.; Wen, D.; Cupples, R.; Bucay, N.; Arakawa, T.; Mori, S.; Ratzkin, B.; Yamamoto, T. Molecular characterization of ALK, a receptor tyrosine kinase expressed specifically in the nervous system. Oncogene 1997, 14, 439–449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weiss, J.B.; Xue, C.; Benice, T.; Xue, L.; Morris, S.W.; Raber, J. Anaplastic lymphoma kinase and leukocyte tyrosine kinase: Functions and genetic interactions in learning, memory and adult neurogenesis. Pharmacol. Biochem. Behav. 2012, 100, 566–574. [Google Scholar] [CrossRef] [PubMed]
- Bilsland, J.G.; Wheeldon, A.; Mead, A.; Znamenskiy, P.; Almond, S.; Waters, K.A.; Thakur, M.; Beaumont, V.; Bonnert, T.P.; Heavens, R.; et al. Behavioral and neurochemical alterations in mice deficient in anaplastic lymphoma kinase suggest therapeutic potential for psychiatric indications. Neuropsychopharmacology 2008, 33, 685–700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stoica, G.E.; Kuo, A.; Powers, C.; Bowden, E.T.; Sale, E.B.; Riegel, A.T.; Wellstein, A. Midkine binds to anaplastic lymphoma kinase (ALK) and acts as a growth factor for different cell types. J. Biol. Chem. 2002, 277, 35990–35998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perez-Pinera, P.; Zhang, W.; Chang, Y.; Vega, J.A.; Deuel, T.F. Anaplastic lymphoma kinase is activated through the pleiotrophin/receptor protein-tyrosine phosphatase β/ζ signaling pathway: An alternative mechanism of receptor tyrosine kinase activation. J. Biol. Chem. 2007, 282, 28683–28690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guan, J.; Umapathy, G.; Yamazaki, Y.; Wolfstetter, G.; Mendoza, P.; Pfeifer, K.; Mohammed, A.; Hugosson, F.; Zhang, H.; Hsu, A.W.; et al. FAM150A and FAM150B are activating ligands for anaplastic lymphoma kinase. Elife 2015, 4, 1–16. [Google Scholar] [CrossRef]
- Huang, H. Anaplastic lymphoma kinase (Alk) receptor tyrosine kinase: A catalytic receptor with many faces. Int. J. Mol. Sci. 2018, 19, 3448. [Google Scholar] [CrossRef] [Green Version]
- George, R.E.; Sanda, T.; Hanna, M.; Fröhling, S.; Luther, W.; Zhang, J.; Ahn, Y.; Zhou, W.; London, W.B.; McGrady, P.; et al. Activating mutations in ALK provide a therapeutic target in neuroblastoma. Nature 2008, 455, 975–978. [Google Scholar] [CrossRef]
- Chen, Y.; Takita, J.; Choi, Y.L.; Kato, M.; Ohira, M.; Sanada, M.; Wang, L.; Soda, M.; Kikuchi, A.; Igarashi, T.; et al. Oncogenic mutations of ALK kinase in neuroblastoma. Nature 2008, 455, 971–974. [Google Scholar] [CrossRef]
- Morris, S.W.; Kirstein, M.N.; Valentine, M.B.; Dittmer, K.G.; Shapiro, D.N.; Saltman, D.L.; Look, A.T. Fusion of a kinase gene, ALK, to a nucleolar protein gene, NPM, in non-Hodgkin’s lymphoma. Science 1994, 263, 1281–1284. [Google Scholar] [CrossRef]
- Holla, V.R.; Elamin, Y.Y.; Bailey, A.M.; Johnson, A.M.; Litzenburger, B.C.; Khotskaya, Y.B.; Sanchez, N.S.; Zeng, J.; Shufean, M.A.; Shaw, K.R.; et al. ALK: A tyrosine kinase target for cancer therapy. Mol. Case Stud. 2017, 3, a001115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slupianek, A.; Nieborowska-Skorska, M.; Hoser, G.; Morrione, A.; Majewski, M.; Xue, L.; Morris, S.W.; Wasik, M.A.; Skorski, T. Role of phosphatidylinositol 3-kinase-Akt pathway in nucleophosmin/anaplastic lymphoma kinase-mediated lymphomagenesis. Cancer Res. 2001, 61, 2194–2199. [Google Scholar] [PubMed]
- Schönherr, C.; Yang, H.-L.; Vigny, M.; Palmer, R.H.; Hallberg, B. Anaplastic lymphoma kinase activates the small GTPase Rap1 via the Rap1-specific GEF C3G in both neuroblastoma and PC12 cells. Oncogene 2010, 29, 2817–2830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zamo, A.; Chiarle, R.; Piva, R.; Howes, J.; Fan, Y.; Chilosi, M.; Levy, D.E.; Inghirami, G. Anaplastic lymphoma kinase (ALK) activates Stat3 and protects hematopoietic cells from cell death. Oncogene 2002, 21, 1038–1047. [Google Scholar] [CrossRef] [Green Version]
- Hrustanovic, G.; Olivas, V.; Pazarentzos, E.; Tulpule, A.; Asthana, S.; Blakely, C.M.; Okimoto, R.A.; Lin, L.; Neel, D.S.; Sabnis, A.; et al. RAS-MAPK dependence underlies a rational polytherapy strategy in EML4-ALK-positive lung cancer. Nat. Med. 2015, 21, 1038–1047. [Google Scholar] [CrossRef]
- Motegi, A.; Fujimoto, J.; Kotani, M.; Sakuraba, H.; Yamamoto, T. ALK receptor tyrosine kinase promotes cell growth and neurite outgrowth. J. Cell Sci. 2004, 117, 3319–3329. [Google Scholar] [CrossRef] [Green Version]
- Bresler, S.C.; Wood, A.C.; Haglund, E.A.; Courtright, J.; Belcastro, L.T.; Plegaria, J.S.; Cole, K.; Toporovskaya, Y.; Zhao, H.; Carpenter, E.L.; et al. Differential inhibitor sensitivity of anaplastic lymphoma kinase variants found in neuroblastoma. Sci. Transl. Med. 2011, 3, 108ra114. [Google Scholar] [CrossRef] [Green Version]
- Childress, M.A.; Himmelberg, S.M.; Chen, H.; Deng, W.; Davies, M.A.; Lovly, C.M. ALK fusion partners impact response to ALK inhibition: Differential effects on sensitivity, cellular phenotypes, and biochemical properties. Mol. Cancer Res. 2018, 16, 1724–1736. [Google Scholar] [CrossRef] [Green Version]
- Doebele, R.C.; Pilling, A.B.; Aisner, D.L.; Kutateladze, T.G.; Le, A.T.; Weickhardt, A.J.; Kondo, K.L.; Linderman, D.J.; Heasley, L.E.; Franklin, W.A.; et al. Mechanisms of resistance to crizotinib in patients with ALK gene rearranged non-small cell lung cancer. Clin. Cancer Res. 2012, 18, 1472–1482. [Google Scholar] [CrossRef] [Green Version]
- Nigg, E.A. Mitotic kinases as regulators of cell division and its checkpoints. Nat. Rev. Mol. Cell Biol. 2001, 2, 21–32. [Google Scholar] [CrossRef]
- Petronczki, M.; Lénárt, P.; Peters, J.-M. Polo on the rise—From mitotic entry to cytokinesis with Plk. Dev. Cell 2008, 14, 646–659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruchaud, S.; Carmena, M.; Earnshaw, W.C. Chromosomal passengers: Conducting cell division. Nat. Rev. Mol. Cell Biol. 2007, 8, 798–812. [Google Scholar] [CrossRef] [PubMed]
- Moog-Lutz, C.; Degoutin, J.; Gouzi, J.Y.; Frobert, Y.; Brunet-De Carvalho, N.; Bureau, J.; Créminon, C.; Vigny, M. Activation and inhibition of anaplastic lymphoma kinase receptor tyrosine kinase by monoclonal antibodies and absence of agonist activity of pleiotrophin. J. Biol. Chem. 2005, 280, 26039–26048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vassilev, L.T.; Tovar, C.; Chen, S.; Knezevic, D.; Zhao, X.; Sun, H.; Heimbrook, D.C.; Chen, L. Selective small-molecule inhibitor reveals critical mitotic functions of human CDK. Proc. Natl. Acad. Sci. USA 2006, 103, 10660–10665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyazaki, M.; Otomo, R.; Matsushima-Hibiya, Y.; Suzuki, H.; Nakajima, A.; Abe, N.; Tomiyama, A.; Ichimura, K.; Matsuda, K.; Watanabe, T.; et al. The p53 activator overcomes resistance to ALK inhibitors by regulating p53-target selectivity in ALK-driven neuroblastomas. Cell Death Discov. 2018, 4, 56. [Google Scholar] [CrossRef] [PubMed]
- Lara-Gonzalez, P.; Westhorpe, F.G.; Taylor, S.S. The spindle assembly checkpoint. Curr. Biol. 2012, 22, R966–R980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hewitt, L.; Tighe, A.; Santaguida, S.; White, A.M.; Jones, C.D.; Musacchio, A.; Green, S.; Taylor, S.S. Sustained Mps1 activity is required in mitosis to recruit O-Mad2 to the Mad1-C-Mad2 core complex. J. Cell Biol. 2010, 190, 25–34. [Google Scholar] [CrossRef] [Green Version]
- Musacchio, A. The molecular biology of spindle assembly checkpoint signaling dynamics. Curr. Biol. 2015, 25, R1002–R1018. [Google Scholar] [CrossRef] [Green Version]
- Soda, M.; Choi, Y.L.; Enomoto, M.; Takada, S.; Yamashita, Y.; Ishikawa, S.; Fujiwara, S.; Watanabe, H.; Kurashina, K.; Hatanaka, H.; et al. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature 2007, 448, 561–566. [Google Scholar] [CrossRef]
- Ducray, S.P.; Natarajan, K.; Garland, G.D.; Turner, S.D.; Egger, G. The transcriptional roles of ALK fusion proteins in tumorigenesis. Cancers 2019, 11, 1074. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Ye, X.; Liu, J.; Zha, J.; Pei, L. Evaluation of eml4-alk fusion proteins in non-small cell lung cancer using small molecule inhibitors. Neoplasia 2011, 13, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wan, W.; Albom, M.S.; Lu, L.; Quail, M.R.; Becknell, N.C.; Weinberg, L.R.; Reddy, D.R.; Holskin, B.P.; Angeles, T.S.; Underiner, T.L.; et al. Anaplastic lymphoma kinase activity is essential for the proliferation and survival of anaplastic large-cell lymphoma cells. Blood 2006, 107, 1617–1623. [Google Scholar] [CrossRef] [PubMed]
- Guan, J.; Tucker, E.R.; Wan, H.; Chand, D.; Danielson, L.S.; Ruuth, K.; El Wakil, A.; Wite, B.; Jamin, Y.; Umapathy, G.; et al. The ALK inhibitor PF-06463922 is effective as a single agent in neuroblastoma driven by expression of ALK and MYCN. Dis. Model. Mech. 2016, 9, 941–952. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiba, R.; Akiya, M.; Hashimura, M.; Oguri, Y.; Inukai, M.; Hara, A.; Saegusa, M. ALK signaling cascade confers multiple advantages to glioblastoma cells through neovascularization and cell proliferation. PLoS ONE 2017, 12, e0183516. [Google Scholar] [CrossRef]
- Alam, M.W.; Borenäs, M.; Lind, D.E.; Cervantes-Madrid, D.; Umapathy, G.; Palmer, R.H.; Hallberg, B. Alectinib, an anaplastic lymphoma kinase inhibitor, abolishes ALK activity and growth in ALK-positive neuroblastoma cells. Front. Oncol. 2019, 9, 21–23. [Google Scholar] [CrossRef]
- Webb, T.R.; Slavish, J.; George, R.E.; Look, A.T.; Xue, L.; Jiang, Q.; Cui, X.; Rentrop, W.B.; Morris, S.W. Anaplastic lymphoma kinase: Role in cancer pathogenesis and small-molecule inhibitor development for therapy. Expert Rev. Anticancer Ther. 2009, 9, 331–356. [Google Scholar] [CrossRef] [Green Version]
- Tanizaki, J.; Okamoto, I.; Takezawa, K.; Sakai, K.; Azuma, K.; Kuwata, K.; Yamaguchi, H.; Hatashita, E.; Nishio, K.; Janne, P.A.; et al. Combined effect of ALK and MEK inhibitors in EML4-ALK-positive non-small-cell lung cancer cells. Br. J. Cancer 2012, 106, 763–767. [Google Scholar] [CrossRef] [Green Version]
- Yamada, T.; Takeuchi, S.; Nakade, J.; Kita, K.; Nakagawa, T.; Nanjo, S.; Nakamura, T.; Matsumoto, K.; Soda, M.; Mano, H.; et al. Paracrine receptor activation by microenvironment triggers bypass survival signals and ALK inhibitor resistance in EML4-ALK lung cancer cells. Clin. Cancer Res. 2012, 18, 3592–3602. [Google Scholar] [CrossRef] [Green Version]
- Kunda, P.; Baum, B. The actin cytoskeleton in spindle assembly and positioning. Trends Cell Biol. 2009, 19, 174–179. [Google Scholar] [CrossRef]
- Dogterom, M.; Koenderink, G.H. Actin-microtubule crosstalk in cell biology. Nat. Rev. Mol. Cell Biol. 2019, 20, 38–54. [Google Scholar] [CrossRef]
- Tanaka, K. Dynamic regulation of kinetochore-microtubule interaction during mitosis. J. Biochem. 2012, 152, 415–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maiato, H.; Gomes, A.M.; Sousa, F.; Barisic, M. Mechanisms of chromosome congression during mitosis. Biology 2017, 6, 13. [Google Scholar] [CrossRef] [PubMed]
- Okada, K.; Araki, M.; Sakashita, T.; Ma, B.; Kanada, R.; Yanagitani, N.; Horiike, A.; Koike, S.; Oh-hara, T.; Watanabe, K.; et al. Prediction of ALK mutations mediating ALK-TKIs resistance and drug re-purposing to overcome the resistance. EBioMedicine 2019, 41, 105–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sasaki, T.; Koivunen, J.; Ogino, A.; Yanagita, M.; Nikiforow, S.; Zheng, W.; Lathan, C.; Marcoux, J.P.; Du, J.; Okuda, K.; et al. A novel ALK secondary mutation and EGFR signaling cause resistance to ALK kinase inhibitors. Cancer Res. 2011, 71, 6051–6060. [Google Scholar] [CrossRef] [Green Version]
- Lovly, C.M.; McDonald, N.T.; Chen, H.; Ortiz-Cuaran, S.; Heukamp, L.C.; Yan, Y.; Florin, A.; Ozretić, L.; Lim, D.; Wang, L.; et al. Rationale for co-targeting IGF-1R and ALK in ALK fusion-positive lung cancer. Nat. Med. 2014, 20, 1027–1034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katayama, R.; Shaw, A.T.; Khan, T.M.; Mino-Kenudson, M.; Solomon, B.J.; Halmos, B.; Jessop, N.A.; Wain, J.C.; Yeo, A.T.; Benes, C.; et al. Mechanisms of acquired crizotinib resistance in ALK-rearranged lung cancers. Sci. Transl. Med. 2012, 4, 120ra17. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, R.; Sasaki, T.; Minami, Y.; Hibino, Y.; Okumura, S.; Sado, M.; Miyokawa, N.; Hayashi, S.; Kitada, M.; Ohsaki, Y. Activation of Src signaling mediates acquired resistance to ALK inhibition in lung cancer. Int. J. Oncol. 2017, 51, 1533–1540. [Google Scholar] [CrossRef] [Green Version]
- Dardaei, L.; Wang, H.Q.; Singh, M.; Fordjour, P.; Shaw, K.X.; Yoda, S.; Kerr, G.; Yu, K.; Liang, J.; Cao, Y.; et al. SHP2 inhibition restores sensitivity in ALK-rearranged non-small-cell lung cancer resistant to ALK inhibitors. Nat. Med. 2018, 24, 512–517. [Google Scholar] [CrossRef]
- Kuniyasu, K.; Iemura, K.; Tanaka, K. Delayed chromosome alignment to the spindle equator increases the rate of chromosome missegregation in cancer cell lines. Biomolecules 2019, 9, 10. [Google Scholar] [CrossRef] [Green Version]
- Gascoigne, K.E.; Taylor, S.S. How do anti-mitotic drugs kill cancer cells? J. Cell Sci. 2009, 122, 2579–2585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, K.; Hirota, T. Chromosomal instability: A common feature and a therapeutic target of cancer. Biochim. Biophys. Acta Rev. Cancer 2016, 1866, 64–75. [Google Scholar] [CrossRef] [PubMed]
- Okumura, D.; Hagino, M.; Yamagishi, A.; Kaibori, Y.; Munira, S.; Saito, Y.; Nakayama, Y. Inhibitors of the VEGF receptor suppress HeLa S3 cell proliferation via misalignment of chromosomes and rotation of the mitotic spindle, causing a delay in M-phase progression. Int. J. Mol. Sci. 2018, 19, 4014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakayama, Y.; Saito, Y.; Soeda, S.; Iwamoto, E.; Ogawa, S.; Yamagishi, N.; Kuga, T.; Yamaguchi, N. Genistein induces cytokinesis failure through RhoA delocalization and anaphase chromosome bridging. J. Cell. Biochem. 2014, 115, 763–771. [Google Scholar] [CrossRef] [PubMed]
- Iwamoto, E.; Ueta, N.; Matsui, Y.; Kamijo, K.; Kuga, T.; Saito, Y.; Yamaguchi, N.; Nakayama, Y. ERK plays a role in chromosome alignment and participates in M-phase progression. J. Cell. Biochem. 2016, 117, 1340–1351. [Google Scholar] [CrossRef]
- Horiuchi, M.; Kuga, T.; Saito, Y.; Nagano, M.; Adachi, J.; Tomonaga, T.; Yamaguchi, N.; Nakayama, Y. The tyrosine kinase v-Src causes mitotic slippage by phosphorylating an inhibitory tyrosine residue of Cdk. J. Biol. Chem. 2018, 293, 15524–15537. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Boehm, J.S.; Yang, X.; Salehi-Ashtiani, K.; Hao, T.; Shen, Y.; Lubonja, R.; Thomas, S.R.; Alkan, O.; Bhimdi, T.; et al. A public genome-scale lentiviral expression library of human ORFs. Nat. Methods 2011, 8, 659–661. [Google Scholar] [CrossRef] [Green Version]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Munira, S.; Yuki, R.; Saito, Y.; Nakayama, Y. ALK Inhibitors-Induced M Phase Delay Contributes to the Suppression of Cell Proliferation. Cancers 2020, 12, 1054. https://doi.org/10.3390/cancers12041054
Munira S, Yuki R, Saito Y, Nakayama Y. ALK Inhibitors-Induced M Phase Delay Contributes to the Suppression of Cell Proliferation. Cancers. 2020; 12(4):1054. https://doi.org/10.3390/cancers12041054
Chicago/Turabian StyleMunira, Sirajam, Ryuzaburo Yuki, Youhei Saito, and Yuji Nakayama. 2020. "ALK Inhibitors-Induced M Phase Delay Contributes to the Suppression of Cell Proliferation" Cancers 12, no. 4: 1054. https://doi.org/10.3390/cancers12041054