DCLK1 Monoclonal Antibody-Based CAR-T Cells as a Novel Treatment Strategy against Human Colorectal Cancers

 , ,
, ,
Abstract
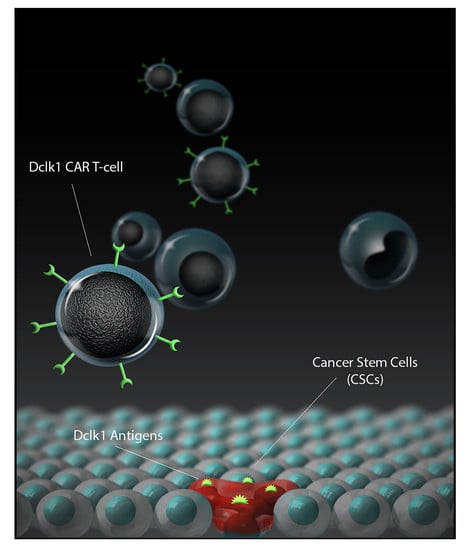
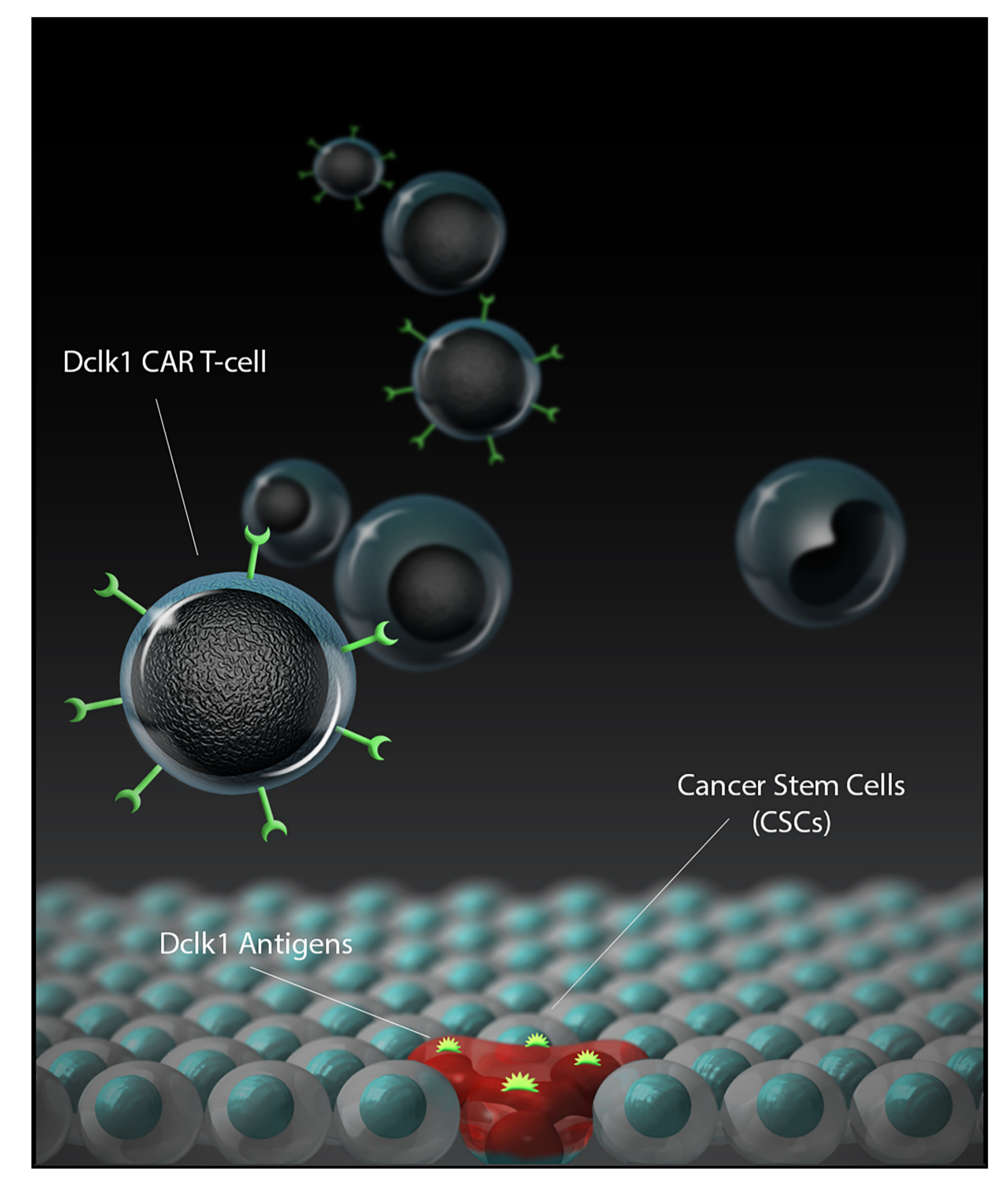
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. DCLK1 mAb CBT-15 Has High Binding Affinity to DCLK1
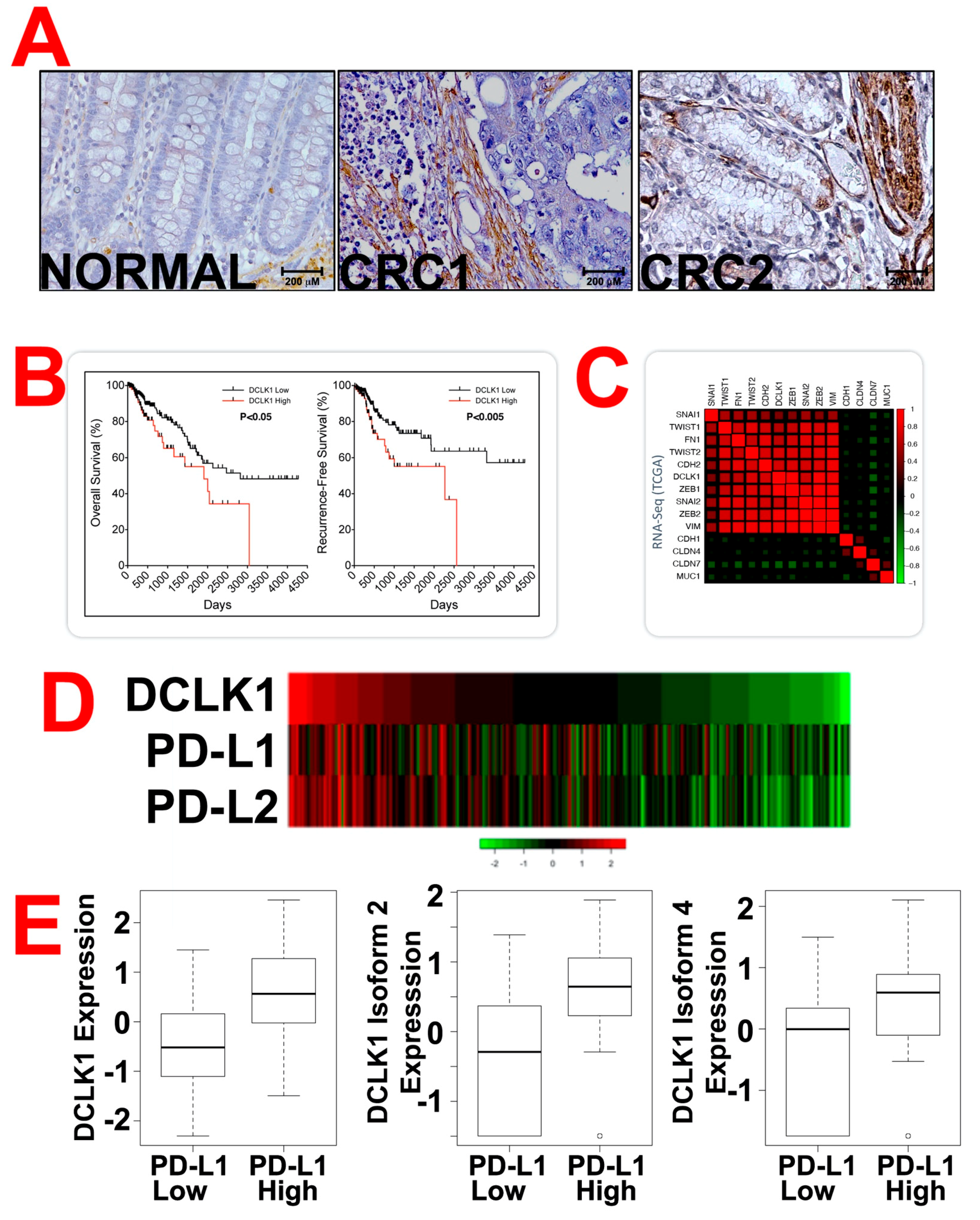
2.2. DCLK1 Is Upregulated in Human CRC and CBT-15 Specifically Recognizes DCLK1 in CRC
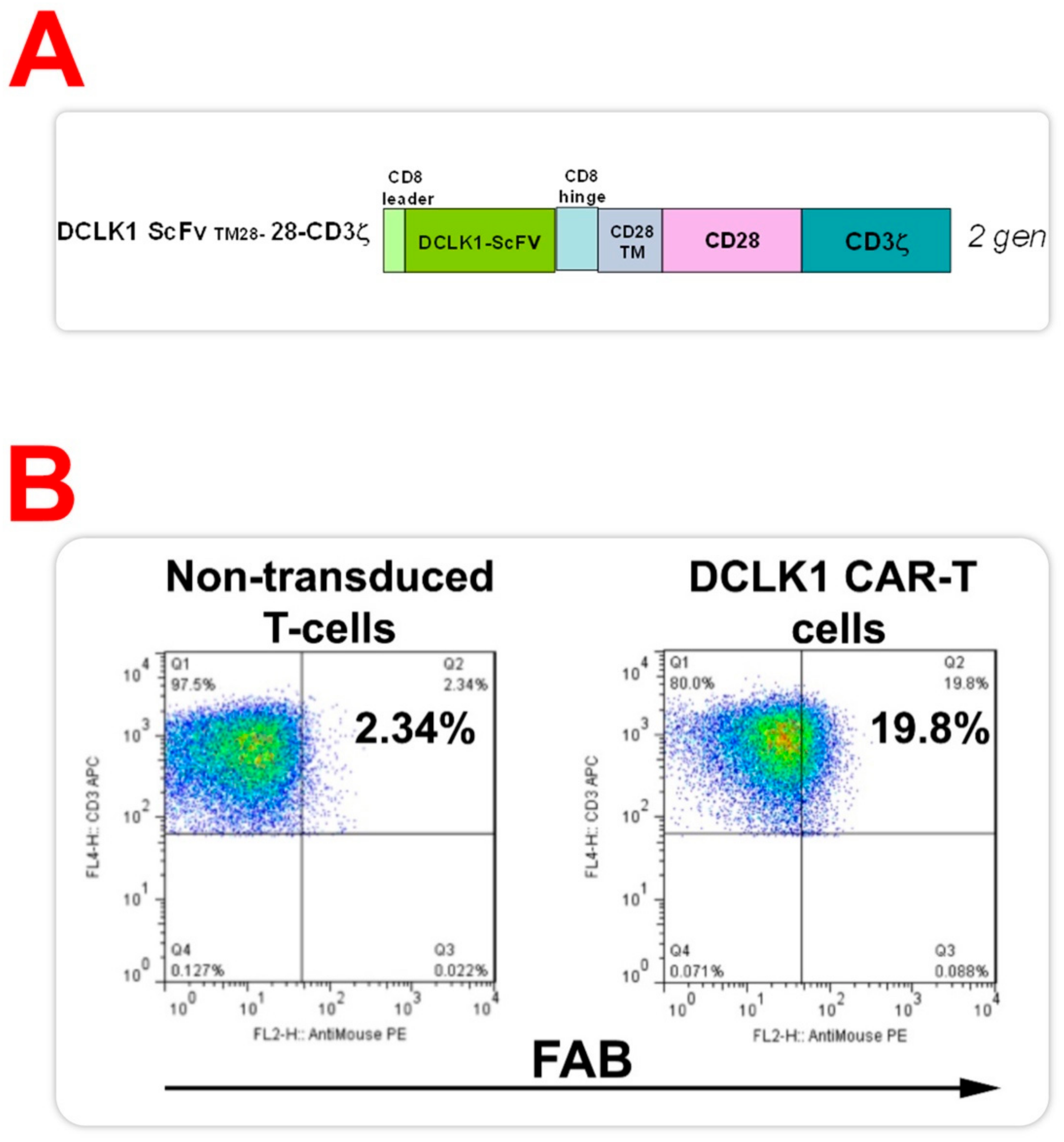
2.3. CBT-511, CAR-T Cells Generated with DCLK1 CBT-15 Antibody ScFv, Recognizes DCLK1 Protein
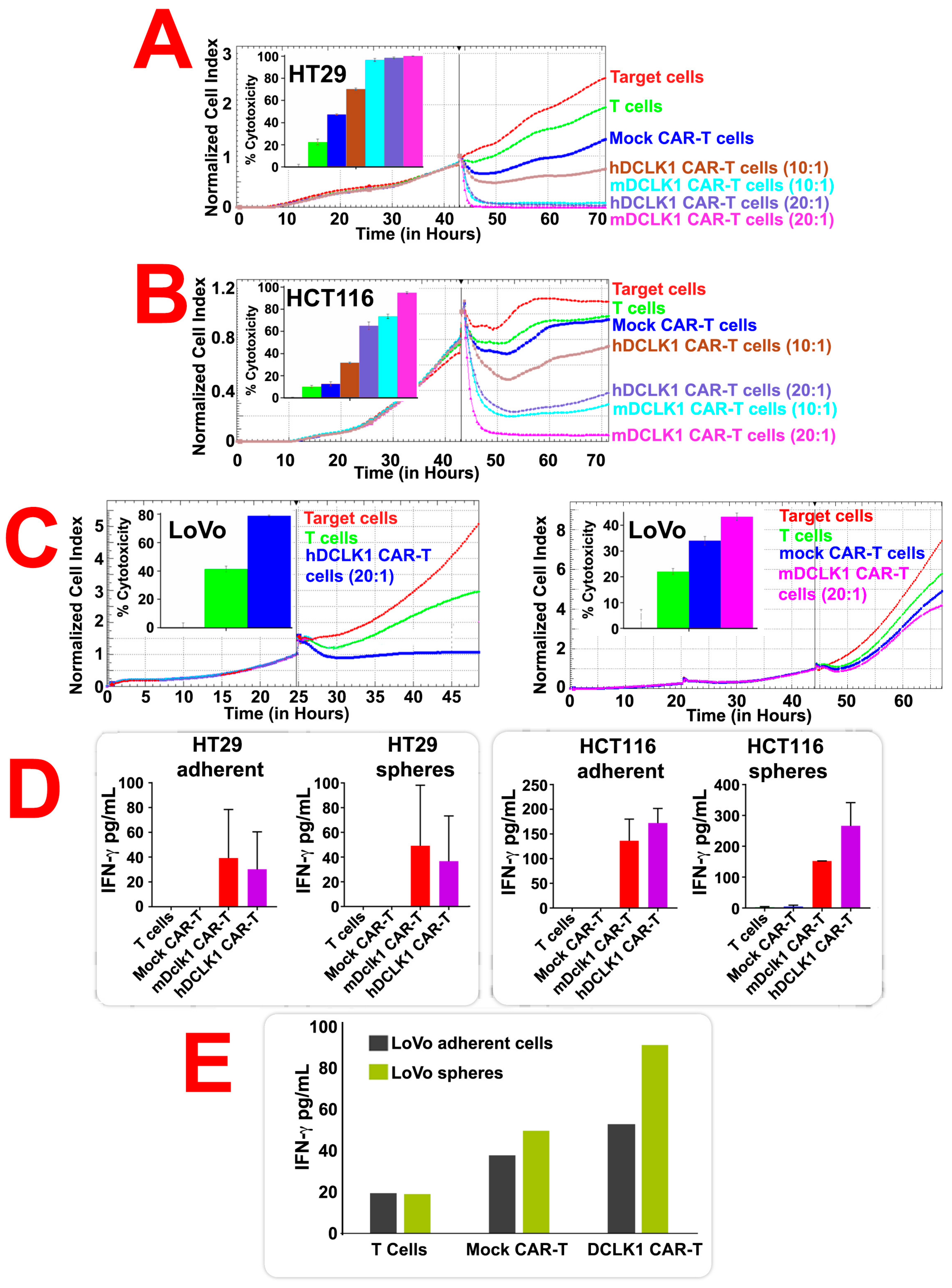
2.4. CBT-511 Effectively Kills CRC Cells and Induces the Secretion of IFN-γ
2.5. CBT-511 Blocks Subcutaneous LoVo CRC Cells-Derived Xenograft Tumor Growth In Vivo
3. Discussion
4. Materials and Methods
4.1. Cells, Primary Tissues
4.2. Monoclonal Antibody
4.3. Generation of CAR-Encoding Lentivirus
4.4. Generation and Expansion of CAR-T Cells
4.5. Flow Cytometry
4.6. Cytokine Induction Assay
4.7. Real-Time Cytotoxicity Assay (RTCA)
4.8. Mouse Xenograft Study
4.9. Statistical Analysis
5. Conclusions
6. Patents
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- American-Cancer-Society. What is the Survival Rates for Colorectal Cancer by Stage? Available online: http://www.cancer.org/cancer/colonandrectumcancer/detailedguide/colorectal-cancer-survival-rates (accessed on 23 June 2019).
- Jordan, C.T.; Guzman, M.L.; Noble, M. Cancer stem cells. N. Engl. J. Med. 2006, 355, 1253–1261. [Google Scholar] [CrossRef] [PubMed]
- Clarke, M.F.; Dick, J.E.; Dirks, P.B.; Eaves, C.J.; Jamieson, C.H.; Jones, D.L.; Visvader, J.; Weissman, I.L.; Wahl, G.M. Cancer stem cells-perspectives on current status and future directions: AACR Workshop on cancer stem cells. Cancer Res. 2006, 66, 9339–9344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beatty, G.L.; Gladney, W.L. Immune escape mechanisms as a guide for cancer immunotherapy. Clin. Cancer Res. 2015, 21, 687–692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Markman, J.L.; Shiao, S.L. Impact of the immune system and immunotherapy in colorectal cancer. J. Gastrointest. Oncol. 2015, 6, 208–223. [Google Scholar] [CrossRef]
- Menon, S.; Shin, S.; Dy, G. Advances in cancer immunotherapy in solid tumors. Cancers 2016, 8, 106. [Google Scholar] [CrossRef] [Green Version]
- Yan, X.; Zhang, S.; Deng, Y.; Wang, P.; Hou, Q.; Xu, H. Prognostic factors for checkpoint inhibitor based immunotherapy: An update with new evidences. Front. Pharmacol. 2018, 9, 1050. [Google Scholar] [CrossRef] [Green Version]
- Kouidhi, S.; Ben Ayed, F.; Benammar Elgaaied, A. Targeting tumor metabolism: A new challenge to improve immunotherapy. Front. Immunol. 2018, 9, 353. [Google Scholar] [CrossRef] [Green Version]
- Noble, F.; Mellows, T.; McCormick Matthews, L.H.; Bateman, A.C.; Harris, S.; Underwood, T.J.; Byrne, J.P.; Bailey, I.S.; Sharland, D.M.; Kelly, J.J.; et al. Tumour infiltrating lymphocytes correlate with improved survival in patients with oesophageal adenocarcinoma. Cancer Immunol. Immunother. 2016, 65, 651–662. [Google Scholar] [CrossRef]
- Fan, F.; Samuel, S.; Evans, K.W.; Lu, J.; Xia, L.; Zhou, Y.; Sceusi, E.; Tozzi, F.; Ye, X.C.; Mani, S.A.; et al. Overexpression of snail induces epithelial-mesenchymal transition and a cancer stem cell-like phenotype in human colorectal cancer cells. Cancer Med. 2012, 1, 5–16. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, Y.; Lu, J.; Zhang, P.; Wang, Y.; Xu, Y.; Wang, Z.; Mao, J.H.; Wei, G. Rapamycin inhibits FBXW7 loss-induced epithelial-mesenchymal transition and cancer stem cell-like characteristics in colorectal cancer cells. Biochem. Biophys. Res. Commun. 2013, 434, 352–356. [Google Scholar] [CrossRef] [Green Version]
- Brabletz, S.; Bajdak, K.; Meidhof, S.; Burk, U.; Niedermann, G.; Firat, E.; Wellner, U.; Dimmler, A.; Faller, G.; Schubert, J.; et al. The ZEB1/miR-200 feedback loop controls Notch signalling in cancer cells. EMBO J. 2011, 30, 770–782. [Google Scholar] [CrossRef] [PubMed]
- Krantz, S.B.; Shields, M.A.; Dangi-Garimella, S.; Munshi, H.G.; Bentrem, D.J. Contribution of epithelial-to-mesenchymal transition and cancer stem cells to pancreatic cancer progression. J. Surg. Res. 2012, 173, 105–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Li, Y.; Kong, D.; Banerjee, S.; Ahmad, A.; Azmi, A.S.; Ali, S.; Abbruzzese, J.L.; Gallick, G.E.; Sarkar, F.H. Acquisition of epithelial-mesenchymal transition phenotype of gemcitabine-resistant pancreatic cancer cells is linked with activation of the notch signaling pathway. Cancer Res. 2009, 69, 2400–2407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wellner, U.; Schubert, J.; Burk, U.C.; Schmalhofer, O.; Zhu, F.; Sonntag, A.; Waldvogel, B.; Vannier, C.; Darling, D.; zur Hausen, A.; et al. The EMT-activator ZEB1 promotes tumorigenicity by repressing stemness-inhibiting microRNAs. Nat. Cell Biol. 2009, 11, 1487–1495. [Google Scholar] [CrossRef]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef] [Green Version]
- Brabletz, T. EMT and MET in metastasis: Where are the cancer stem cells? Cancer Cell 2012, 22, 699–701. [Google Scholar] [CrossRef] [Green Version]
- Choi, J.E.; Bae, J.S.; Kang, M.J.; Chung, M.J.; Jang, K.Y.; Park, H.S.; Moon, W.S. Expression of epithelial-mesenchymal transition and cancer stem cell markers in colorectal adenocarcinoma: Clinicopathological significance. Oncol. Rep. 2017, 38, 1695–1705. [Google Scholar] [CrossRef] [Green Version]
- Sureban, S.M.; Madhoun, M.F.; May, R.; Qu, D.; Ali, N.; Fazili, J.; Weygant, N.; Chandrakesan, P.; Ding, K.; Lightfoot, S.A.; et al. Plasma DCLK1 is a marker of hepatocellular carcinoma (HCC): Targeting DCLK1 prevents HCC tumor xenograft growth via a microRNA-dependent mechanism. Oncotarget 2015, 6, 37200–37215. [Google Scholar] [CrossRef] [Green Version]
- Sureban, S.M.; May, R.; Lightfoot, S.A.; Hoskins, A.B.; Lerner, M.; Brackett, D.J.; Postier, R.G.; Ramanujam, R.; Mohammed, A.; Rao, C.V.; et al. DCAMKL-1 regulates epithelial-mesenchymal transition in human pancreatic cells through a miR-200a-dependent mechanism. Cancer Res. 2011, 71, 2328–2338. [Google Scholar] [CrossRef] [Green Version]
- Sureban, S.M.; May, R.; Mondalek, F.G.; Qu, D.; Ponnurangam, S.; Pantazis, P.; Anant, S.; Ramanujam, R.P.; Houchen, C.W. Nanoparticle-based delivery of siDCAMKL-1 increases microRNA-144 and inhibits colorectal cancer tumor growth via a Notch-1 dependent mechanism. J. Nanobiotechnol. 2011, 9, 40. [Google Scholar] [CrossRef] [Green Version]
- Sureban, S.M.; May, R.; Qu, D.; Weygant, N.; Chandrakesan, P.; Ali, N.; Lightfoot, S.A.; Pantazis, P.; Rao, C.V.; Postier, R.G.; et al. DCLK1 regulates pluripotency and angiogenic factors via microRNA-dependent mechanisms in pancreatic cancer. PLoS ONE 2013, 8, e73940. [Google Scholar] [CrossRef] [PubMed]
- Sureban, S.M.; May, R.; Ramalingam, S.; Subramaniam, D.; Natarajan, G.; Anant, S.; Houchen, C.W. Selective blockade of DCAMKL-1 results in tumor growth arrest by a Let-7a MicroRNA-dependent mechanism. Gastroenterology 2009, 137, 649–659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sureban, S.M.; May, R.; Weygant, N.; Qu, D.; Chandrakesan, P.; Bannerman-Menson, E.; Ali, N.; Pantazis, P.; Westphalen, C.B.; Wang, T.C.; et al. XMD8-92 inhibits pancreatic tumor xenograft growth via a DCLK1-dependent mechanism. Cancer Lett. 2014, 351, 151–161. [Google Scholar] [CrossRef] [PubMed]
- Nakanishi, Y.; Seno, H.; Fukuoka, A.; Ueo, T.; Yamaga, Y.; Maruno, T.; Nakanishi, N.; Kanda, K.; Komekado, H.; Kawada, M.; et al. Dclk1 distinguishes between tumor and normal stem cells in the intestine. Nat. Genet. 2013, 45, 98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Westphalen, C.B.; Asfaha, S.; Hayakawa, Y.; Takemoto, Y.; Lukin, D.J.; Nuber, A.H.; Brandtner, A.; Setlik, W.; Remotti, H.; Muley, A.; et al. Long-lived intestinal tuft cells serve as colon cancer-initiating cells. J. Clin. Invest. 2014, 124, 1283–1295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weygant, N.; Ge, Y.; Qu, D.; Kaddis, J.S.; Berry, W.L.; May, R.; Chandrakesan, P.; Bannerman-Menson, E.; Vega, K.J.; Tomasek, J.J.; et al. Survival of patients with gastrointestinal cancers can be predicted by a surrogate microRNA signature for cancer stem-like cells marked by dclk1 kinase. Cancer Res. 2016, 76, 4090–4099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maus, M.V. Designing CAR T cells for glioblastoma. Oncoimmunology 2015, 4, e1048956. [Google Scholar] [CrossRef] [Green Version]
- Gross, G.; Waks, T.; Eshhar, Z. Expression of immunoglobulin-T-cell receptor chimeric molecules as functional receptors with antibody-type specificity. Proc. Natl. Acad. Sci. USA 1989, 86, 10024–10028. [Google Scholar] [CrossRef] [Green Version]
- Gross, G.; Eshhar, Z. Therapeutic potential of T cell chimeric antigen receptors (CARs) in cancer treatment: Counteracting off-tumor toxicities for safe CAR T cell therapy. Annu. Rev. Pharmacol. Toxicol. 2016, 56, 59–83. [Google Scholar] [CrossRef]
- Abken, H. Adoptive therapy with CAR redirected T cells: The challenges in targeting solid tumors. Immunotherapy 2015, 7, 535–544. [Google Scholar] [CrossRef]
- Lanitis, E.; Dangaj, D.; Irving, M.; Coukos, G. Mechanisms regulating T-cell infiltration and activity in solid tumors. Ann. Oncol. 2017, 28, xii18–xii32. [Google Scholar] [CrossRef] [PubMed]
- Maimela, N.R.; Liu, S.; Zhang, Y. Fates of CD8+ T cells in tumor microenvironment. Comput. Struct. Biotechnol. J. 2019, 17, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Martinez, M.; Moon, E.K. CAR T cells for solid tumors: New strategies for finding, infiltrating, and surviving in the tumor microenvironment. Front. Immunol. 2019, 10, 128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, L.; Liu, B. Critical factors in chimeric antigen receptor-modified T-cell (CAR-T) therapy for solid tumors. Onco Targets Ther. 2019, 12, 193–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miliotou, A.N.; Papadopoulou, L.C. CAR T-cell therapy: A new era in cancer immunotherapy. Curr. Pharm. Biotechnol. 2018, 19, 5–18. [Google Scholar] [CrossRef]
- Brown, C.E.; Mackall, C.L. CAR T cell therapy: Inroads to response and resistance. Nat. Rev. Immunol. 2019, 19, 73–74. [Google Scholar] [CrossRef]
- Cheadle, E.J.; Gornall, H.; Baldan, V.; Hanson, V.; Hawkins, R.E.; Gilham, D.E. CAR T cells: Driving the road from the laboratory to the clinic. Immunol. Rev. 2014, 257, 91–106. [Google Scholar] [CrossRef]
- Berahovich, R.; Zhou, H.; Xu, S.; Wei, Y.; Guan, J.; Guan, J.; Harto, H.; Fu, S.; Yang, K.; Zhu, S.; et al. CAR-T cells based on novel BCMA monoclonal antibody block multiple myeloma cell growth. Cancers 2018, 10, 323. [Google Scholar] [CrossRef] [Green Version]
- Golubovskaya, V.M.; Berahovich, R.; Xu, Q.; Zhou, H.; Xu, S.; Guan, J.; Harto, H.; Li, L.; Wu, L. GITR domain inside CAR co-stimulates activity of CAR-T cells against cancer. Front. Biosci. 2018, 23, 2245–2254. [Google Scholar] [CrossRef]
- Golubovskaya, V. CAR-T cell therapy: From the bench to the bedside. Cancers 2017, 9, 150. [Google Scholar] [CrossRef] [Green Version]
- Golubovskaya, V.; Berahovich, R.; Zhou, H.; Xu, S.; Harto, H.; Li, L.; Chao, C.C.; Mao, M.M.; Wu, L. CD47-CAR-T Cells Effectively Kill Target Cancer Cells and Block Pancreatic Tumor Growth. Cancers 2017, 9, 139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berahovich, R.; Xu, S.; Zhou, H.; Harto, H.; Xu, Q.; Garcia, A.; Liu, F.; Golubovskaya, V.M.; Wu, L. FLAG-tagged CD19-specific CAR-T cells eliminate CD19-bearing solid tumor cells in vitro and in vivo. Front. Biosci. 2017, 22, 1644–1654. [Google Scholar]
- Berahovich, R.; Liu, X.; Zhou, H.; Tsadik, E.; Xu, S.; Golubovskaya, V.; Wu, L. Hypoxia selectively impairs CAR-T Cells in vitro. Cancers 2019, 11, 602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Golubovskaya, V.; Wu, L. Different subsets of T cells, memory, effector functions, and CAR-T immunotherapy. Cancers 2016, 8, 36. [Google Scholar] [CrossRef] [Green Version]
- Xu, Q.; Harto, H.; Berahovich, R.; Xu, S.; Zhou, H.; Golubovskaya, V.; Wu, L. Generation of CAR-T cells for cancer immunotherapy. Methods Mol. Biol. 2019, 1884, 349–360. [Google Scholar] [CrossRef]
- Ge, Y.; Weygant, N.; Qu, D.; May, R.; Berry, W.L.; Yao, J.; Chandrakesan, P.; Zheng, W.; Zhao, L.; Zhao, K.L.; et al. Alternative splice variants of DCLK1 mark cancer stem cells, promote self-renewal and drug-resistance, and can be targeted to inhibit tumorigenesis in kidney cancer. Int. J. Cancer 2018, 143, 1162–1175. [Google Scholar] [CrossRef]
- Qu, D.; Weygant, N.; Yao, J.; Chandrakesan, P.; Berry, W.L.; May, R.; Pitts, K.; Husain, S.; Lightfoot, S.; Li, M.; et al. Overexpression of DCLK1-AL increases tumor cell invasion, drug resistance, and KRAS activation and can be targeted to inhibit tumorigenesis in pancreatic cancer. J. Oncol. 2019, 2019, 6402925. [Google Scholar] [CrossRef] [Green Version]
- Sarkar, S.; Popov, V.L.; O’Connell, M.R.; Stevenson, H.L.; Lee, B.S.; Obeid, R.A.; Luthra, G.K.; Singh, P. A novel antibody against cancer stem cell biomarker, DCLK1-S, is potentially useful for assessing colon cancer risk after screening colonoscopy. Lab. Investig. 2017, 97, 1245–1261. [Google Scholar] [CrossRef]
- Mu, L.; Huang, K.; Hu, Y.; Yan, C.; Li, X.; Tao, D.; Gong, J.; Qin, J. Small-sized colorectal cancer cells harbor metastatic tumor-initiating cells. Oncotarget 2017, 8, 107907–107919. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, D.; Eide, P.W.; Eilertsen, I.A.; Danielsen, S.A.; Eknaes, M.; Hektoen, M.; Lind, G.E.; Lothe, R.A. Epigenetic and genetic features of 24 colon cancer cell lines. Oncogenesis 2013, 2, e71. [Google Scholar] [CrossRef]
- Ji, D.; Zhan, T.; Li, M.; Yao, Y.; Jia, J.; Yi, H.; Qiao, M.; Xia, J.; Zhang, Z.; Ding, H.; et al. Enhancement of sensitivity to chemo/radiation therapy by using miR-15b against DCLK1 in colorectal cancer. Stem Cell Rep. 2018, 11, 1506–1522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turano, M.; Costabile, V.; Cerasuolo, A.; Duraturo, F.; Liccardo, R.; Delrio, P.; Pace, U.; Rega, D.; Dodaro, C.A.; Milone, M.; et al. Characterisation of mesenchymal colon tumour-derived cells in tumourspheres as a model for colorectal cancer progression. Int. J. Oncol. 2018, 53, 2379–2396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bao, Y.; Lu, Y.; Wang, X.; Feng, W.; Sun, X.; Guo, H.; Tang, C.; Zhang, X.; Shi, Q.; Yu, H. Eukaryotic translation initiation factor 5A2 (eIF5A2) regulates chemoresistance in colorectal cancer through epithelial mesenchymal transition. Cancer Cell Int. 2015, 15, 109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larson, S.M.; Truscott, L.C.; Chiou, T.T.; Patel, A.; Kao, R.; Tu, A.; Tyagi, T.; Lu, X.; Elashoff, D.; De Oliveira, S.N. Pre-clinical development of gene modification of haematopoietic stem cells with chimeric antigen receptors for cancer immunotherapy. Hum. Vaccines Immunother. 2017, 13, 1094–1104. [Google Scholar] [CrossRef]
- Maliar, A.; Servais, C.; Waks, T.; Chmielewski, M.; Lavy, R.; Altevogt, P.; Abken, H.; Eshhar, Z. Redirected T cells that target pancreatic adenocarcinoma antigens eliminate tumors and metastases in mice. Gastroenterology 2012, 143, 1375–1384. [Google Scholar] [CrossRef]
- Weygant, N.; Qu, D.; May, R.; Chandrakesan, P.; Ge, Y.; Ryan, C.D.; An, G.; Schlosser, M.J.; Bannerman-Menson, E.; Houchen, C.W. Systemic delivery of CBT-15G DCLK1-targeted monoclonal antibody dramatically decreases tumorigenesis in a xenograft model of pancreatic cancer. Cancer Res. 2016, 76, Am2016–Am2577. [Google Scholar]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sureban, S.M.; Berahovich, R.; Zhou, H.; Xu, S.; Wu, L.; Ding, K.; May, R.; Qu, D.; Bannerman-Menson, E.; Golubovskaya, V.; et al. DCLK1 Monoclonal Antibody-Based CAR-T Cells as a Novel Treatment Strategy against Human Colorectal Cancers. Cancers 2020, 12, 54. https://doi.org/10.3390/cancers12010054
Sureban SM, Berahovich R, Zhou H, Xu S, Wu L, Ding K, May R, Qu D, Bannerman-Menson E, Golubovskaya V, et al. DCLK1 Monoclonal Antibody-Based CAR-T Cells as a Novel Treatment Strategy against Human Colorectal Cancers. Cancers. 2020; 12(1):54. https://doi.org/10.3390/cancers12010054
Chicago/Turabian StyleSureban, Sripathi M., Robert Berahovich, Hua Zhou, Shirley Xu, Lijun Wu, Kai Ding, Randal May, Dongfeng Qu, Edwin Bannerman-Menson, Vita Golubovskaya, and et al. 2020. "DCLK1 Monoclonal Antibody-Based CAR-T Cells as a Novel Treatment Strategy against Human Colorectal Cancers" Cancers 12, no. 1: 54. https://doi.org/10.3390/cancers12010054