Immunotherapy in Pediatric Solid Tumors—A Systematic Review


Abstract
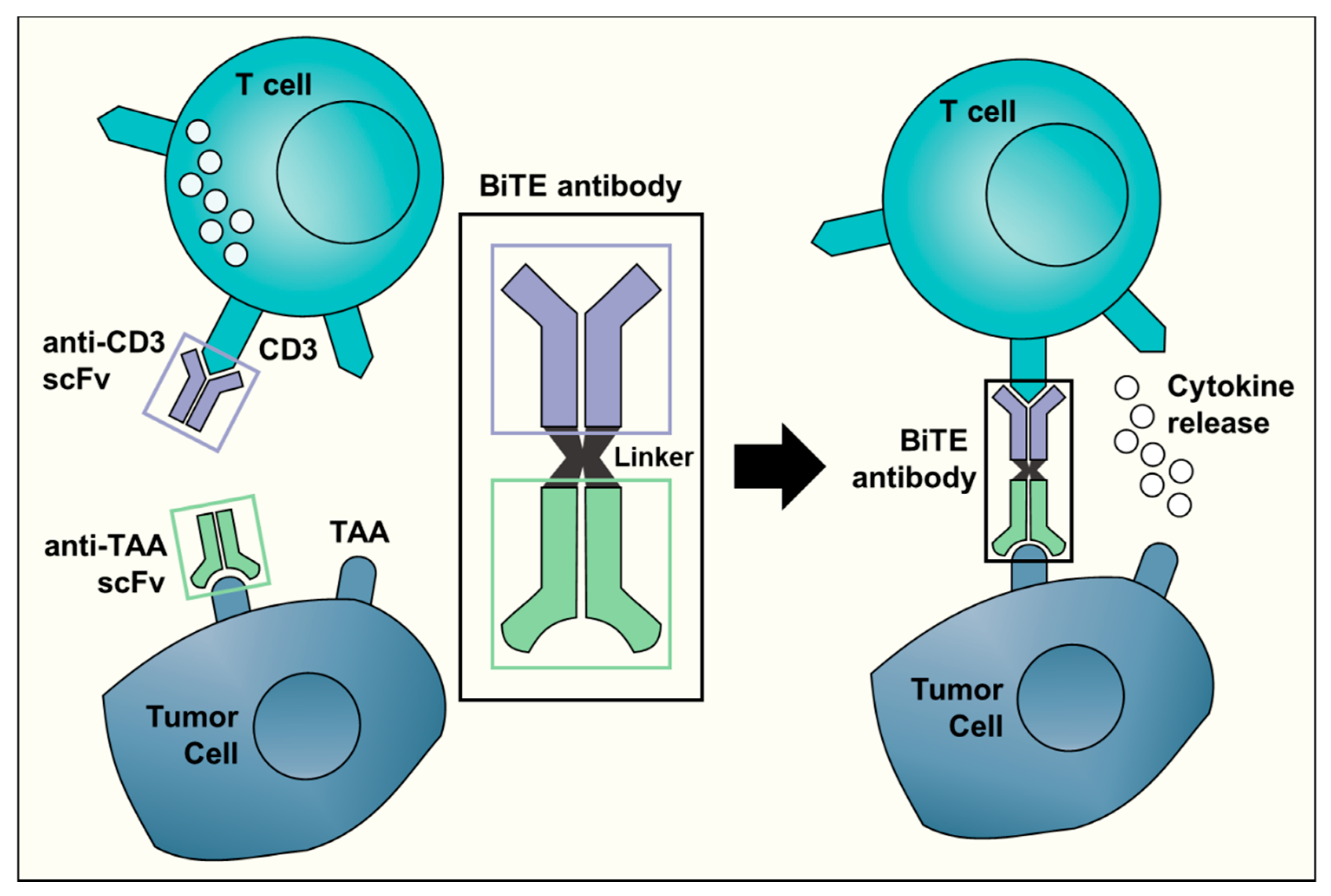
:1. Introduction
2. Direct Utilization of the Immune System
2.1. Oncolytic Virus-Based Therapy
2.2. Antigen-Targeting Therapy
2.3. Immune Checkpoint Inhibitors
3. Modulation of the Immune System
3.1. Tumor Microenvironment: Cancer-Associated Fibroblasts, Tumor-Associated Macrophages, and Myeloid-Derived Suppressor Cells
3.2. Cytokines and Growth Factors
3.3. Chimeric Antigen Receptor T Cell Therapy
3.4. Natural Killer Cell-Based Immunotherapy
3.5. Cancer Vaccines
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Kelly, E.; Russell, S.J. History of oncolytic viruses: Genesis to genetic engineering. Mol. Ther. 2007, 15, 651–659. [Google Scholar] [CrossRef] [PubMed]
- Sze, D.Y.; Reid, T.R.; Rose, S.C. Oncolytic virotherapy. J. Vasc. Interv. Radiol. 2013, 24, 1115–1122. [Google Scholar] [CrossRef] [PubMed]
- Coffey, M.C.; Strong, J.E.; Forsyth, P.A.; Lee, P.W. Reovirus therapy of tumors with activated Ras pathway. Science 1998, 282, 1332–1334. [Google Scholar] [CrossRef] [PubMed]
- Sanchala, D.S.; Bhatt, L.K.; Prabhavalkar, K.S. Oncolytic Herpes Simplex Viral Therapy: A Stride toward Selective Targeting of Cancer Cells. Front. Pharmacol. 2017, 8, 270. [Google Scholar] [CrossRef] [PubMed]
- Wedekind, M.F.; Denton, N.L.; Chen, C.Y.; Cripe, T.P. Pediatric Cancer Immunotherapy: Opportunities and Challenges. Paediatr Drugs 2018, 20, 395–408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, J.; Markert, J.M.; Leavenworth, J.W. Modulation of the Intratumoral Immune Landscape by Oncolytic Herpes Simplex Virus Virotherapy. Front. Oncol. 2017, 7, 136. [Google Scholar] [CrossRef] [Green Version]
- Pol, J.G.; Levesque, S.; Workenhe, S.T.; Gujar, S.; Le Boeuf, F.; Clements, D.R.; Fahrner, J.E.; Fend, L.; Bell, J.C.; Mossman, K.L.; et al. Trial Watch: Oncolytic viro-immunotherapy of hematologic and solid tumors. Oncoimmunology 2018, 7, e1503032. [Google Scholar] [CrossRef] [Green Version]
- Waters, A.M.; Friedman, G.K.; Ring, E.K.; Beierle, E.A. Oncolytic virotherapy for pediatric malignancies: Future prospects. Oncolytic Virother. 2016, 5, 73–80. [Google Scholar] [CrossRef] [Green Version]
- Tong, Y.; Qian, W. Targeting cancer stem cells with oncolytic virus. Stem. Cell Investig. 2014, 1, 20. [Google Scholar] [CrossRef]
- Foreman, P.M.; Friedman, G.K.; Cassady, K.A.; Markert, J.M. Oncolytic Virotherapy for the Treatment of Malignant Glioma. Neurotherapeutics 2017, 14, 333–344. [Google Scholar] [CrossRef] [Green Version]
- Bridle, B.W.; Stephenson, K.B.; Boudreau, J.E.; Koshy, S.; Kazdhan, N.; Pullenayegum, E.; Brunelliere, J.; Bramson, J.L.; Lichty, B.D.; Wan, Y. Potentiating cancer immunotherapy using an oncolytic virus. Mol. Ther. 2010, 18, 1430–1439. [Google Scholar] [CrossRef] [PubMed]
- Cantoni, C.; Grauwet, K.; Pietra, G.; Parodi, M.; Mingari, M.C.; Maria, A.D.; Favoreel, H.; Vitale, M. Role of NK cells in immunotherapy and virotherapy of solid tumors. Immunotherapy 2015, 7, 861–882. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Yin, J.; Li, T.; Huang, S.; Yan, H.; Leavenworth, J.; Wang, X. NK cell-based cancer immunotherapy: From basic biology to clinical application. Sci. China Life Sci. 2015, 58, 1233–1245. [Google Scholar] [CrossRef]
- Bommareddy, P.K.; Aspromonte, S.; Zloza, A.; Rabkin, S.D.; Kaufman, H.L. MEK inhibition enhances oncolytic virus immunotherapy through increased tumor cell killing and T cell activation. Sci. Transl. Med. 2018, 10. [Google Scholar] [CrossRef] [Green Version]
- Denton, N.L.; Chen, C.Y.; Hutzen, B.; Currier, M.A.; Scott, T.; Nartker, B.; Leddon, J.L.; Wang, P.Y.; Srinivas, R.; Cassady, K.A.; et al. Myelolytic Treatments Enhance Oncolytic Herpes Virotherapy in Models of Ewing Sarcoma by Modulating the Immune Microenvironment. Mol. Ther. Oncolytics 2018, 11, 62–74. [Google Scholar] [CrossRef] [Green Version]
- Rehman, H.; Silk, A.W.; Kane, M.P.; Kaufman, H.L. Into the clinic: Talimogene laherparepvec (T-VEC), a first-in-class intratumoral oncolytic viral therapy. J. Immunother. Cancer 2016, 4, 53. [Google Scholar] [CrossRef] [Green Version]
- Vedi, A.; Ziegler, D.S. Antibody therapy for pediatric leukemia. Front. Oncol. 2014, 4, 82. [Google Scholar] [CrossRef] [Green Version]
- Yu, A.L.; Gilman, A.L.; Ozkaynak, M.F.; London, W.B.; Kreissman, S.G.; Chen, H.X.; Smith, M.; Anderson, B.; Villablanca, J.G.; Matthay, K.K.; et al. Anti-GD2 antibody with GM-CSF, interleukin-2, and isotretinoin for neuroblastoma. N. Engl. J. Med. 2010, 363, 1324–1334. [Google Scholar] [CrossRef] [Green Version]
- Heiner, J.P.; Miraldi, F.; Kallick, S.; Makley, J.; Neely, J.; Smith-Mensah, W.H.; Cheung, N.K. Localization of GD2-specific monoclonal antibody 3F8 in human osteosarcoma. Cancer Res. 1987, 47, 5377–5381. [Google Scholar]
- Carpenter, E.L.; Haglund, E.A.; Mace, E.M.; Deng, D.; Martinez, D.; Wood, A.C.; Chow, A.K.; Weiser, D.A.; Belcastro, L.T.; Winter, C.; et al. Antibody targeting of anaplastic lymphoma kinase induces cytotoxicity of human neuroblastoma. Oncogene 2012, 31, 4859–4867. [Google Scholar] [CrossRef]
- Nguyen, R.; Moustaki, A.; Norrie, J.L.; Brown, S.; Akers, W.J.; Shirinifard, A.; Dyer, M.A. Interleukin-15 Enhances Anti-GD2 Antibody-Mediated Cytotoxicity in an Orthotopic PDX Model of Neuroblastoma. Clin. Cancer Res. 2019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ploessl, C.; Pan, A.; Maples, K.T.; Lowe, D.K. Dinutuximab: An Anti-GD2 Monoclonal Antibody for High-Risk Neuroblastoma. Ann. Pharmacother. 2016, 50, 416–422. [Google Scholar] [CrossRef] [PubMed]
- Kushner, B.H.; Cheung, I.Y.; Modak, S.; Basu, E.M.; Roberts, S.S.; Cheung, N.K. Humanized 3F8 Anti-GD2 Monoclonal Antibody Dosing With Granulocyte-Macrophage Colony-Stimulating Factor in Patients With Resistant Neuroblastoma: A Phase 1 Clinical Trial. JAMA Oncol. 2018, 4, 1729–1735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barry, W.E.; Jackson, J.R.; Asuelime, G.E.; Wu, H.W.; Sun, J.; Wan, Z.; Malvar, J.; Sheard, M.A.; Wang, L.; Seeger, R.C.; et al. Activated Natural Killer Cells in Combination with Anti-GD2 Antibody Dinutuximab Improve Survival of Mice after Surgical Resection of Primary Neuroblastoma. Clin. Cancer Res. 2019, 25, 325–333. [Google Scholar] [CrossRef] [Green Version]
- Baeuerle, P.A.; Reinhardt, C. Bispecific T-cell engaging antibodies for cancer therapy. Cancer Res. 2009, 69, 4941–4944. [Google Scholar] [CrossRef] [Green Version]
- Hoffman, L.M.; Gore, L. Blinatumomab, a Bi-Specific Anti-CD19/CD3 BiTE((R)) Antibody for the Treatment of Acute Lymphoblastic Leukemia: Perspectives and Current Pediatric Applications. Front. Oncol. 2014, 4, 63. [Google Scholar] [CrossRef] [Green Version]
- von Stackelberg, A.; Locatelli, F.; Zugmaier, G.; Handgretinger, R.; Trippett, T.M.; Rizzari, C.; Bader, P.; O’Brien, M.M.; Brethon, B.; Bhojwani, D.; et al. Phase I/Phase II Study of Blinatumomab in Pediatric Patients With Relapsed/Refractory Acute Lymphoblastic Leukemia. J. Clin. Oncol. 2016, 34, 4381–4389. [Google Scholar] [CrossRef] [Green Version]
- Elitzur, S.; Arad-Cohen, N.; Barzilai-Birenboim, S.; Ben-Harush, M.; Bielorai, B.; Elhasid, R.; Feuerstein, T.; Gilad, G.; Gural, A.; Kharit, M.; et al. Blinatumomab as a bridge to further therapy in cases of overwhelming toxicity in pediatric B-cell precursor acute lymphoblastic leukemia: Report from the Israeli Study Group of Childhood Leukemia. Pediatr. Blood Cancer 2019, 66, e27898. [Google Scholar] [CrossRef]
- Leach, D.R.; Krummel, M.F.; Allison, J.P. Enhancement of antitumor immunity by CTLA-4 blockade. Science 1996, 271, 1734–1736. [Google Scholar] [CrossRef] [Green Version]
- Walker, L.S. Treg and CTLA-4: Two intertwining pathways to immune tolerance. J. Autoimmun. 2013, 45, 49–57. [Google Scholar] [CrossRef] [Green Version]
- Contardi, E.; Palmisano, G.L.; Tazzari, P.L.; Martelli, A.M.; Fala, F.; Fabbi, M.; Kato, T.; Lucarelli, E.; Donati, D.; Polito, L.; et al. CTLA-4 is constitutively expressed on tumor cells and can trigger apoptosis upon ligand interaction. Int. J. Cancer 2005, 117, 538–550. [Google Scholar] [CrossRef] [PubMed]
- Hingorani, P.; Maas, M.L.; Gustafson, M.P.; Dickman, P.; Adams, R.H.; Watanabe, M.; Eshun, F.; Williams, J.; Seidel, M.J.; Dietz, A.B. Increased CTLA-4(+) T cells and an increased ratio of monocytes with loss of class II (CD14(+) HLA-DR(lo/neg)) found in aggressive pediatric sarcoma patients. J. Immunother. Cancer 2015, 3, 35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merchant, M.S.; Wright, M.; Baird, K.; Wexler, L.H.; Rodriguez-Galindo, C.; Bernstein, D.; Delbrook, C.; Lodish, M.; Bishop, R.; Wolchok, J.D.; et al. Phase I Clinical Trial of Ipilimumab in Pediatric Patients with Advanced Solid Tumors. Clin. Cancer Res. 2016, 22, 1364–1370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brahmer, J.R.; Tykodi, S.S.; Chow, L.Q.; Hwu, W.J.; Topalian, S.L.; Hwu, P.; Drake, C.G.; Camacho, L.H.; Kauh, J.; Odunsi, K.; et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N. Engl. J. Med. 2012, 366, 2455–2465. [Google Scholar] [CrossRef] [Green Version]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef] [Green Version]
- Majzner, R.G.; Simon, J.S.; Grosso, J.F.; Martinez, D.; Pawel, B.R.; Santi, M.; Merchant, M.S.; Geoerger, B.; Hezam, I.; Marty, V.; et al. Assessment of programmed death-ligand 1 expression and tumor-associated immune cells in pediatric cancer tissues. Cancer 2017, 123, 3807–3815. [Google Scholar] [CrossRef] [Green Version]
- Geoerger, B.; Kang, H.J.; Yalon-Oren, M.; Marshall, L.V.; Vezina, C.; Pappo, A.S.; Laetsch, T.W.; Petrilli, A.S.; Ebinger, M.; Toporski, J.; et al. KEYNOTE-051: An update on the phase 2 results of pembrolizumab (pembro) in pediatric patients (pts) with advanced melanoma or a PD-L1–positive advanced, relapsed or refractory solid tumor or lymphoma. J. Clin. Oncol. 2018, 36, 10525. [Google Scholar] [CrossRef]
- Zheng, W.; Xiao, H.; Liu, H.; Zhou, Y. Expression of programmed death 1 is correlated with progression of osteosarcoma. APMIS 2015, 123, 102–107. [Google Scholar] [CrossRef]
- Robert, C.; Schachter, J.; Long, G.V.; Arance, A.; Grob, J.J.; Mortier, L.; Daud, A.; Carlino, M.S.; McNeil, C.; Lotem, M.; et al. Pembrolizumab versus Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2015, 372, 2521–2532. [Google Scholar] [CrossRef]
- Topalian, S.L.; Hodi, F.S.; Brahmer, J.R.; Gettinger, S.N.; Smith, D.C.; McDermott, D.F.; Powderly, J.D.; Carvajal, R.D.; Sosman, J.A.; Atkins, M.B.; et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N. Engl. J. Med. 2012, 366, 2443–2454. [Google Scholar] [CrossRef]
- Blumenthal, D.T.; Yalon, M.; Vainer, G.W.; Lossos, A.; Yust, S.; Tzach, L.; Cagnano, E.; Limon, D.; Bokstein, F. Pembrolizumab: First experience with recurrent primary central nervous system (CNS) tumors. J. Neurooncol. 2016, 129, 453–460. [Google Scholar] [CrossRef]
- Gorsi, H.S.; Malicki, D.M.; Barsan, V.; Tumblin, M.; Yeh-Nayre, L.; Milburn, M.; Elster, J.D.; Crawford, J.R. Nivolumab in the Treatment of Recurrent or Refractory Pediatric Brain Tumors: A Single Institutional Experience. J. Pediatr. Hematol. Oncol. 2019, 41, e235–e241. [Google Scholar] [CrossRef] [PubMed]
- Bouffet, E.; Larouche, V.; Campbell, B.B.; Merico, D.; de Borja, R.; Aronson, M.; Durno, C.; Krueger, J.; Cabric, V.; Ramaswamy, V.; et al. Immune Checkpoint Inhibition for Hypermutant Glioblastoma Multiforme Resulting From Germline Biallelic Mismatch Repair Deficiency. J. Clin. Oncol. 2016, 34, 2206–2211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, X.; McDowell, M.M.; Newman, W.C.; Mason, G.E.; Greene, S.; Tamber, M.S. Severe cerebral edema following nivolumab treatment for pediatric glioblastoma: Case report. J. Neurosurg. Pediatr. 2017, 19, 249–253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Twyman-Saint Victor, C.; Rech, A.J.; Maity, A.; Rengan, R.; Pauken, K.E.; Stelekati, E.; Benci, J.L.; Xu, B.; Dada, H.; Odorizzi, P.M.; et al. Radiation and dual checkpoint blockade activate non-redundant immune mechanisms in cancer. Nature 2015, 520, 373–377. [Google Scholar] [CrossRef] [Green Version]
- Lussier, D.M.; Johnson, J.L.; Hingorani, P.; Blattman, J.N. Combination immunotherapy with alpha-CTLA-4 and alpha-PD-L1 antibody blockade prevents immune escape and leads to complete control of metastatic osteosarcoma. J. Immunother. Cancer 2015, 3, 21. [Google Scholar] [CrossRef]
- Sounni, N.E.; Noel, A. Targeting the tumor microenvironment for cancer therapy. Clin. Chem. 2013, 59, 85–93. [Google Scholar] [CrossRef] [Green Version]
- Zeine, R.; Salwen, H.R.; Peddinti, R.; Tian, Y.; Guerrero, L.; Yang, Q.; Chlenski, A.; Cohn, S.L. Presence of cancer-associated fibroblasts inversely correlates with Schwannian stroma in neuroblastoma tumors. Mod. Pathol. 2009, 22, 950–958. [Google Scholar] [CrossRef] [Green Version]
- Kock, A.; Larsson, K.; Bergqvist, F.; Eissler, N.; Elfman, L.H.M.; Raouf, J.; Korotkova, M.; Johnsen, J.I.; Jakobsson, P.J.; Kogner, P. Inhibition of Microsomal Prostaglandin E Synthase-1 in Cancer-Associated Fibroblasts Suppresses Neuroblastoma Tumor Growth. EBioMedicine 2018, 32, 84–92. [Google Scholar] [CrossRef]
- Kraman, M.; Bambrough, P.J.; Arnold, J.N.; Roberts, E.W.; Magiera, L.; Jones, J.O.; Gopinathan, A.; Tuveson, D.A.; Fearon, D.T. Suppression of antitumor immunity by stromal cells expressing fibroblast activation protein-alpha. Science 2010, 330, 827–830. [Google Scholar] [CrossRef] [Green Version]
- Asgharzadeh, S.; Salo, J.A.; Ji, L.; Oberthuer, A.; Fischer, M.; Berthold, F.; Hadjidaniel, M.; Liu, C.W.; Metelitsa, L.S.; Pique-Regi, R.; et al. Clinical significance of tumor-associated inflammatory cells in metastatic neuroblastoma. J. Clin. Oncol. 2012, 30, 3525–3532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, L.; Asgharzadeh, S.; Salo, J.; Engell, K.; Wu, H.W.; Sposto, R.; Ara, T.; Silverman, A.M.; DeClerck, Y.A.; Seeger, R.C.; et al. Valpha24-invariant NKT cells mediate antitumor activity via killing of tumor-associated macrophages. J. Clin. Investig. 2009, 119, 1524–1536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maximov, V.; Chen, Z.; Wei, Y.; Robinson, M.H.; Herting, C.J.; Shanmugam, N.S.; Rudneva, V.A.; Goldsmith, K.C.; MacDonald, T.J.; Northcott, P.A.; et al. Tumour-associated macrophages exhibit anti-tumoural properties in Sonic Hedgehog medulloblastoma. Nat. Commun. 2019, 10, 2410. [Google Scholar] [CrossRef] [PubMed]
- Buddingh, E.P.; Kuijjer, M.L.; Duim, R.A.; Burger, H.; Agelopoulos, K.; Myklebost, O.; Serra, M.; Mertens, F.; Hogendoorn, P.C.; Lankester, A.C.; et al. Tumor-infiltrating macrophages are associated with metastasis suppression in high-grade osteosarcoma: A rationale for treatment with macrophage activating agents. Clin. Cancer Res. 2011, 17, 2110–2119. [Google Scholar] [CrossRef] [Green Version]
- Mori, K.; Ando, K.; Heymann, D. Liposomal muramyl tripeptide phosphatidyl ethanolamine: A safe and effective agent against osteosarcoma pulmonary metastases. Expert. Rev. Anticancer. Ther. 2008, 8, 151–159. [Google Scholar] [CrossRef]
- Kager, L.; Potschger, U.; Bielack, S. Review of mifamurtide in the treatment of patients with osteosarcoma. Ther. Clin. Risk. Manag. 2010, 6, 279–286. [Google Scholar] [CrossRef] [Green Version]
- Meyers, P.A.; Schwartz, C.L.; Krailo, M.D.; Healey, J.H.; Bernstein, M.L.; Betcher, D.; Ferguson, W.S.; Gebhardt, M.C.; Goorin, A.M.; Harris, M.; et al. Osteosarcoma: The addition of muramyl tripeptide to chemotherapy improves overall survival--a report from the Children’s Oncology Group. J. Clin. Oncol. 2008, 26, 633–638. [Google Scholar] [CrossRef]
- Chou, A.J.; Kleinerman, E.S.; Krailo, M.D.; Chen, Z.; Betcher, D.L.; Healey, J.H.; Conrad, E.U., 3rd; Nieder, M.L.; Weiner, M.A.; Wells, R.J.; et al. Addition of muramyl tripeptide to chemotherapy for patients with newly diagnosed metastatic osteosarcoma: A report from the Children’s Oncology Group. Cancer 2009, 115, 5339–5348. [Google Scholar] [CrossRef]
- Gabrilovich, D.I.; Nagaraj, S. Myeloid-derived suppressor cells as regulators of the immune system. Nat. Rev. Immunol. 2009, 9, 162–174. [Google Scholar] [CrossRef]
- Santilli, G.; Piotrowska, I.; Cantilena, S.; Chayka, O.; D’Alicarnasso, M.; Morgenstern, D.A.; Himoudi, N.; Pearson, K.; Anderson, J.; Thrasher, A.J.; et al. Polyphenon [corrected] E enhances the antitumor immune response in neuroblastoma by inactivating myeloid suppressor cells. Clin Cancer Res. 2013, 19, 1116–1125. [Google Scholar] [CrossRef] [Green Version]
- Liao, W.; Lin, J.X.; Leonard, W.J. Interleukin-2 at the crossroads of effector responses, tolerance, and immunotherapy. Immunity 2013, 38, 13–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Konjevic, G.; Mirjacic Martinovic, K.; Vuletic, A.; Babovic, N. In-vitro IL-2 or IFN-alpha-induced NKG2D and CD161 NK cell receptor expression indicates novel aspects of NK cell activation in metastatic melanoma patients. Melanoma Res. 2010, 20, 459–467. [Google Scholar] [CrossRef] [PubMed]
- Roper, M.; Smith, M.A.; Sondel, P.M.; Gillespie, A.; Reaman, G.H.; Hammond, G.D.; Levitt, D.; Rosolen, A.; Colamonici, O.R.; Neckers, L.M.; et al. A phase I study of interleukin-2 in children with cancer. Am. J. Pediatr. Hematol. Oncol. 1992, 14, 305–311. [Google Scholar] [CrossRef] [PubMed]
- Bauer, M.; Reaman, G.H.; Hank, J.A.; Cairo, M.S.; Anderson, P.; Blazar, B.R.; Frierdich, S.; Sondel, P.M. A phase II trial of human recombinant interleukin-2 administered as a 4-day continuous infusion for children with refractory neuroblastoma, non-Hodgkin’s lymphoma, sarcoma, renal cell carcinoma, and malignant melanoma. A childrens cancer group study. Cancer 1995, 75, 2959–2965. [Google Scholar] [CrossRef]
- Kalwak, K.; Ussowicz, M.; Gorczynska, E.; Turkiewicz, D.; Toporski, J.; Dobaczewski, G.; Latos-Grazynska, E.; Ryczan, R.; Noworolska-Sauren, D.; Chybicka, A. Immunologic effects of intermediate-dose IL-2 i.v. after autologous hematopoietic cell transplantation in pediatric solid tumors. J. Interferon Cytokine Res. 2003, 23, 173–181. [Google Scholar] [CrossRef]
- Hakansson, A.; Gustafsson, B.; Krysander, L.; Hakansson, L. Tumour-infiltrating lymphocytes in metastatic malignant melanoma and response to interferon alpha treatment. Br. J. Cancer 1996, 74, 670–676. [Google Scholar] [CrossRef] [Green Version]
- Navid, F.; Furman, W.L.; Fleming, M.; Rao, B.N.; Kovach, S.; Billups, C.A.; Cain, A.M.; Amonette, R.; Jenkins, J.J.; Pappo, A.S. The feasibility of adjuvant interferon alpha-2b in children with high-risk melanoma. Cancer 2005, 103, 780–787. [Google Scholar] [CrossRef]
- Warren, K.; Bent, R.; Wolters, P.L.; Prager, A.; Hanson, R.; Packer, R.; Shih, J.; Camphausen, K. A phase 2 study of pegylated interferon alpha-2b (PEG-Intron((R))) in children with diffuse intrinsic pontine glioma. Cancer 2012, 118, 3607–3613. [Google Scholar] [CrossRef]
- Jakacki, R.I.; Dombi, E.; Steinberg, S.M.; Goldman, S.; Kieran, M.W.; Ullrich, N.J.; Pollack, I.F.; Goodwin, A.; Manley, P.E.; Fangusaro, J.; et al. Phase II trial of pegylated interferon alfa-2b in young patients with neurofibromatosis type 1 and unresectable plexiform neurofibromas. Neuro Oncol. 2017, 19, 289–297. [Google Scholar] [CrossRef] [Green Version]
- Kohli, S.S.; Kohli, V.S. Role of RANKL-RANK/osteoprotegerin molecular complex in bone remodeling and its immunopathologic implications. Indian J. Endocrinol Metab. 2011, 15, 175–181. [Google Scholar] [CrossRef]
- Roodman, G.D. Mechanisms of bone metastasis. N. Engl. J. Med. 2004, 350, 1655–1664. [Google Scholar] [CrossRef] [PubMed]
- Fizazi, K.; Lipton, A.; Mariette, X.; Body, J.J.; Rahim, Y.; Gralow, J.R.; Gao, G.; Wu, L.; Sohn, W.; Jun, S. Randomized phase II trial of denosumab in patients with bone metastases from prostate cancer, breast cancer, or other neoplasms after intravenous bisphosphonates. J. Clin. Oncol. 2009, 27, 1564–1571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.A.; Jung, J.S.; Kim, D.H.; Lim, J.S.; Kim, M.S.; Kong, C.B.; Song, W.S.; Cho, W.H.; Jeon, D.G.; Lee, S.Y.; et al. RANKL expression is related to treatment outcome of patients with localized, high-grade osteosarcoma. Pediatr. Blood Cancer 2011, 56, 738–743. [Google Scholar] [CrossRef]
- Nakano, K.; Abe, S.; Sohmura, Y. Recombinant human tumor necrosis factor--I. Cytotoxic activity in vitro. Int. J. Immunopharmacol. 1986, 8, 347–355. [Google Scholar] [CrossRef]
- Lejeune, F.J.; Lienard, D.; Matter, M.; Ruegg, C. Efficiency of recombinant human TNF in human cancer therapy. Cancer Immunol. 2006, 6, 6. [Google Scholar]
- Seibel, N.L.; Dinndorf, P.A.; Bauer, M.; Sondel, P.M.; Hammond, G.D.; Reaman, G.H. Phase I study of tumor necrosis factor-alpha and actinomycin D in pediatric patients with cancer: A Children’s Cancer Group study. J. Immunother. Emphasis Tumor Immunol. 1994, 16, 125–131. [Google Scholar] [CrossRef]
- Meany, H.J.; Seibel, N.L.; Sun, J.; Finklestein, J.Z.; Sato, J.; Kelleher, J.; Sondel, P.; Reaman, G. Phase 2 trial of recombinant tumor necrosis factor-alpha in combination with dactinomycin in children with recurrent Wilms tumor. J. Immunother. 2008, 31, 679–683. [Google Scholar] [CrossRef] [Green Version]
- Daniels, R.A.; Turley, H.; Kimberley, F.C.; Liu, X.S.; Mongkolsapaya, J.; Ch’En, P.; Xu, X.N.; Jin, B.Q.; Pezzella, F.; Screaton, G.R. Expression of TRAIL and TRAIL receptors in normal and malignant tissues. Cell Res. 2005, 15, 430–438. [Google Scholar] [CrossRef]
- Picarda, G.; Lamoureux, F.; Geffroy, L.; Delepine, P.; Montier, T.; Laud, K.; Tirode, F.; Delattre, O.; Heymann, D.; Redini, F. Preclinical evidence that use of TRAIL in Ewing’s sarcoma and osteosarcoma therapy inhibits tumor growth, prevents osteolysis, and increases animal survival. Clin. Cancer Res. 2010, 16, 2363–2374. [Google Scholar] [CrossRef] [Green Version]
- Petak, I.; Douglas, L.; Tillman, D.M.; Vernes, R.; Houghton, J.A. Pediatric rhabdomyosarcoma cell lines are resistant to Fas-induced apoptosis and highly sensitive to TRAIL-induced apoptosis. Clin. Cancer Res. 2000, 6, 4119–4127. [Google Scholar]
- Merchant, M.S.; Geller, J.I.; Baird, K.; Chou, A.J.; Galli, S.; Charles, A.; Amaoko, M.; Rhee, E.H.; Price, A.; Wexler, L.H.; et al. Phase I trial and pharmacokinetic study of lexatumumab in pediatric patients with solid tumors. J. Clin. Oncol. 2012, 30, 4141–4147. [Google Scholar] [CrossRef] [PubMed]
- Baek, J.H.; Sohn, S.K.; Kim, D.H.; Kim, J.G.; Yang, D.H.; Kim, Y.K.; Lee, J.J.; Kim, H.J. Pilot remission induction therapy with idarubicin, plus an intensified dose of ara-C and priming with granulocyte colony-stimulating factor for acute myeloid leukemia. Acta Haematol. 2007, 117, 109–114. [Google Scholar] [CrossRef] [PubMed]
- Anderson, P.M.; Markovic, S.N.; Sloan, J.A.; Clawson, M.L.; Wylam, M.; Arndt, C.A.; Smithson, W.A.; Burch, P.; Gornet, M.; Rahman, E. Aerosol granulocyte macrophage-colony stimulating factor: A low toxicity, lung-specific biological therapy in patients with lung metastases. Clin. Cancer Res. 1999, 5, 2316–2323. [Google Scholar] [PubMed]
- Merchant, M.S.; Yang, X.; Melchionda, F.; Romero, M.; Klein, R.; Thiele, C.J.; Tsokos, M.; Kontny, H.U.; Mackall, C.L. Interferon gamma enhances the effectiveness of tumor necrosis factor-related apoptosis-inducing ligand receptor agonists in a xenograft model of Ewing’s sarcoma. Cancer Res. 2004, 64, 8349–8356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cartellieri, M.; Bachmann, M.; Feldmann, A.; Bippes, C.; Stamova, S.; Wehner, R.; Temme, A.; Schmitz, M. Chimeric antigen receptor-engineered T cells for immunotherapy of cancer. J. Biomed. Biotechnol. 2010, 2010, 956304. [Google Scholar] [CrossRef] [PubMed]
- Hombach, A.; Wieczarkowiecz, A.; Marquardt, T.; Heuser, C.; Usai, L.; Pohl, C.; Seliger, B.; Abken, H. Tumor-specific T cell activation by recombinant immunoreceptors: CD3 zeta signaling and CD28 costimulation are simultaneously required for efficient IL-2 secretion and can be integrated into one combined CD28/CD3 zeta signaling receptor molecule. J. Immunol. 2001, 167, 6123–6131. [Google Scholar] [CrossRef] [Green Version]
- Pule, M.A.; Straathof, K.C.; Dotti, G.; Heslop, H.E.; Rooney, C.M.; Brenner, M.K. A chimeric T cell antigen receptor that augments cytokine release and supports clonal expansion of primary human T cells. Mol. Ther. 2005, 12, 933–941. [Google Scholar] [CrossRef]
- Zhong, X.S.; Matsushita, M.; Plotkin, J.; Riviere, I.; Sadelain, M. Chimeric antigen receptors combining 4-1BB and CD28 signaling domains augment PI3kinase/AKT/Bcl-XL activation and CD8+ T cell-mediated tumor eradication. Mol. Ther. 2010, 18, 413–420. [Google Scholar] [CrossRef]
- Huang, M.A.; Krishnadas, D.K.; Lucas, K.G. Cellular and Antibody Based Approaches for Pediatric Cancer Immunotherapy. J. Immunol. Res. 2015, 2015, 675269. [Google Scholar] [CrossRef] [Green Version]
- Louis, C.U.; Savoldo, B.; Dotti, G.; Pule, M.; Yvon, E.; Myers, G.D.; Rossig, C.; Russell, H.V.; Diouf, O.; Liu, E.; et al. Antitumor activity and long-term fate of chimeric antigen receptor-positive T cells in patients with neuroblastoma. Blood 2011, 118, 6050–6056. [Google Scholar] [CrossRef]
- Pule, M.A.; Savoldo, B.; Myers, G.D.; Rossig, C.; Russell, H.V.; Dotti, G.; Huls, M.H.; Liu, E.; Gee, A.P.; Mei, Z.; et al. Virus-specific T cells engineered to coexpress tumor-specific receptors: Persistence and antitumor activity in individuals with neuroblastoma. Nat. Med. 2008, 14, 1264–1270. [Google Scholar] [CrossRef] [PubMed]
- Heczey, A.; Louis, C.U. Advances in chimeric antigen receptor immunotherapy for neuroblastoma. Discov. Med. 2013, 16, 287–294. [Google Scholar] [PubMed]
- Walker, A.J.; Majzner, R.G.; Zhang, L.; Wanhainen, K.; Long, A.H.; Nguyen, S.M.; Lopomo, P.; Vigny, M.; Fry, T.J.; Orentas, R.J.; et al. Tumor Antigen and Receptor Densities Regulate Efficacy of a Chimeric Antigen Receptor Targeting Anaplastic Lymphoma Kinase. Mol. Ther. 2017, 25, 2189–2201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orentas, R.J.; Lee, D.W.; Mackall, C. Immunotherapy targets in pediatric cancer. Front. Oncol. 2012, 2, 3. [Google Scholar] [CrossRef] [Green Version]
- Ragab, S.M.; Samaka, R.M.; Shams, T.M. HER2/neu expression: A predictor for differentiation and survival in children with Wilms tumor. Pathol. Oncol. Res. 2010, 16, 61–67. [Google Scholar] [CrossRef]
- Ahmed, N.; Salsman, V.S.; Yvon, E.; Louis, C.U.; Perlaky, L.; Wels, W.S.; Dishop, M.K.; Kleinerman, E.E.; Pule, M.; Rooney, C.M.; et al. Immunotherapy for osteosarcoma: Genetic modification of T cells overcomes low levels of tumor antigen expression. Mol. Ther. 2009, 17, 1779–1787. [Google Scholar] [CrossRef]
- Hegde, M.; Moll, A.J.; Byrd, T.T.; Louis, C.U.; Ahmed, N. Cellular immunotherapy for pediatric solid tumors. Cytotherapy 2015, 17, 3–17. [Google Scholar] [CrossRef] [Green Version]
- Rainusso, N.; Brawley, V.S.; Ghazi, A.; Hicks, M.J.; Gottschalk, S.; Rosen, J.M.; Ahmed, N. Immunotherapy targeting HER2 with genetically modified T cells eliminates tumor-initiating cells in osteosarcoma. Cancer Gene Ther. 2012, 19, 212–217. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, N.; Brawley, V.S.; Hegde, M.; Robertson, C.; Ghazi, A.; Gerken, C.; Liu, E.; Dakhova, O.; Ashoori, A.; Corder, A.; et al. Human Epidermal Growth Factor Receptor 2 (HER2) -Specific Chimeric Antigen Receptor-Modified T Cells for the Immunotherapy of HER2-Positive Sarcoma. J. Clin. Oncol. 2015, 33, 1688–1696. [Google Scholar] [CrossRef]
- Kawakami, M.; Kawakami, K.; Takahashi, S.; Abe, M.; Puri, R.K. Analysis of interleukin-13 receptor alpha2 expression in human pediatric brain tumors. Cancer 2004, 101, 1036–1042. [Google Scholar] [CrossRef]
- Okada, H.; Low, K.L.; Kohanbash, G.; McDonald, H.A.; Hamilton, R.L.; Pollack, I.F. Expression of glioma-associated antigens in pediatric brain stem and non-brain stem gliomas. J. Neurooncol. 2008, 88, 245–250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, C.E.; Starr, R.; Aguilar, B.; Shami, A.F.; Martinez, C.; D’Apuzzo, M.; Barish, M.E.; Forman, S.J.; Jensen, M.C. Stem-like tumor-initiating cells isolated from IL13Ralpha2 expressing gliomas are targeted and killed by IL13-zetakine-redirected T Cells. Clin. Cancer Res. 2012, 18, 2199–2209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kahlon, K.S.; Brown, C.; Cooper, L.J.; Raubitschek, A.; Forman, S.J.; Jensen, M.C. Specific recognition and killing of glioblastoma multiforme by interleukin 13-zetakine redirected cytolytic T cells. Cancer Res. 2004, 64, 9160–9166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brudno, J.N.; Kochenderfer, J.N. Recent advances in CAR T-cell toxicity: Mechanisms, manifestations and management. Blood Rev. 2019, 34, 45–55. [Google Scholar] [CrossRef] [PubMed]
- Teachey, D.T.; Lacey, S.F.; Shaw, P.A.; Melenhorst, J.J.; Maude, S.L.; Frey, N.; Pequignot, E.; Gonzalez, V.E.; Chen, F.; Finklestein, J.; et al. Identification of Predictive Biomarkers for Cytokine Release Syndrome after Chimeric Antigen Receptor T-cell Therapy for Acute Lymphoblastic Leukemia. Cancer Discov. 2016, 6, 664–679. [Google Scholar] [CrossRef] [Green Version]
- Maude, S.L.; Barrett, D.; Teachey, D.T.; Grupp, S.A. Managing cytokine release syndrome associated with novel T cell-engaging therapies. Cancer J. 2014, 20, 119–122. [Google Scholar] [CrossRef]
- Straathof, K.C.; Pule, M.A.; Yotnda, P.; Dotti, G.; Vanin, E.F.; Brenner, M.K.; Heslop, H.E.; Spencer, D.M.; Rooney, C.M. An inducible caspase 9 safety switch for T-cell therapy. Blood 2005, 105, 4247–4254. [Google Scholar] [CrossRef]
- Di Stasi, A.; Tey, S.K.; Dotti, G.; Fujita, Y.; Kennedy-Nasser, A.; Martinez, C.; Straathof, K.; Liu, E.; Durett, A.G.; Grilley, B.; et al. Inducible apoptosis as a safety switch for adoptive cell therapy. N. Engl. J. Med. 2011, 365, 1673–1683. [Google Scholar] [CrossRef] [Green Version]
- Boyton, R.J.; Altmann, D.M. Natural killer cells, killer immunoglobulin-like receptors and human leucocyte antigen class I in disease. Clin. Exp. Immunol. 2007, 149, 1–8. [Google Scholar] [CrossRef]
- Leung, W.; Iyengar, R.; Turner, V.; Lang, P.; Bader, P.; Conn, P.; Niethammer, D.; Handgretinger, R. Determinants of antileukemia effects of allogeneic NK cells. J. Immunol. 2004, 172, 644–650. [Google Scholar] [CrossRef]
- Koscielniak, E.; Gross-Wieltsch, U.; Treuner, J.; Winkler, P.; Klingebiel, T.; Lang, P.; Bader, P.; Niethammer, D.; Handgretinger, R. Graft-versus-Ewing sarcoma effect and long-term remission induced by haploidentical stem-cell transplantation in a patient with relapse of metastatic disease. J. Clin. Oncol. 2005, 23, 242–244. [Google Scholar] [CrossRef] [PubMed]
- Lang, P.; Pfeiffer, M.; Muller, I.; Schumm, M.; Ebinger, M.; Koscielniak, E.; Feuchtinger, T.; Foll, J.; Martin, D.; Handgretinger, R. Haploidentical stem cell transplantation in patients with pediatric solid tumors: Preliminary results of a pilot study and analysis of graft versus tumor effects. Klin. Padiatr. 2006, 218, 321–326. [Google Scholar] [CrossRef] [PubMed]
- Kasow, K.A.; Handgretinger, R.; Krasin, M.J.; Pappo, A.S.; Leung, W. Possible allogeneic graft-versus-tumor effect in childhood melanoma. J. Pediatr. Hematol. Oncol. 2003, 25, 982–986. [Google Scholar] [CrossRef] [PubMed]
- Inaba, H.; Handgretinger, R.; Furman, W.; Hale, G.; Leung, W. Allogeneic graft-versus-hepatoblastoma effect. Pediatr. Blood Cancer 2006, 46, 501–505. [Google Scholar] [CrossRef]
- Perez-Martinez, A.; Leung, W.; Munoz, E.; Iyengar, R.; Ramirez, M.; Vicario, J.L.; Lassaletta, A.; Sevilla, J.; Gonzalez-Vicent, M.; Madero, L.; et al. KIR-HLA receptor-ligand mismatch associated with a graft-versus-tumor effect in haploidentical stem cell transplantation for pediatric metastatic solid tumors. Pediatr. Blood Cancer 2009, 53, 120–124. [Google Scholar] [CrossRef]
- Ibrahim, E.C.; Guerra, N.; Lacombe, M.J.; Angevin, E.; Chouaib, S.; Carosella, E.D.; Caignard, A.; Paul, P. Tumor-specific up-regulation of the nonclassical class I HLA-G antigen expression in renal carcinoma. Cancer Res. 2001, 61, 6838–6845. [Google Scholar]
- Wan, R.; Wang, Z.W.; Li, H.; Peng, X.D.; Liu, G.Y.; Ou, J.M.; Cheng, A.Q. Human Leukocyte Antigen-G Inhibits the Anti-Tumor Effect of Natural Killer Cells via Immunoglobulin-Like Transcript 2 in Gastric Cancer. Cell. Physiol. Biochem. 2017, 44, 1828–1841. [Google Scholar] [CrossRef]
- Kailayangiri, S.; Altvater, B.; Spurny, C.; Jamitzky, S.; Schelhaas, S.; Jacobs, A.H.; Wiek, C.; Roellecke, K.; Hanenberg, H.; Hartmann, W.; et al. Targeting Ewing sarcoma with activated and GD2-specific chimeric antigen receptor-engineered human NK cells induces upregulation of immune-inhibitory HLA-G. Oncoimmunology 2017, 6, e1250050. [Google Scholar] [CrossRef] [Green Version]
- Maki, G.; Hayes, G.M.; Naji, A.; Tyler, T.; Carosella, E.D.; Rouas-Freiss, N. Gregory SANK resistance of tumor cells from multiple myeloma chronic lymphocytic leukemia patients: Implication of, H.L.A.-G. Leukemia 2008, 22, 998–1006. [Google Scholar] [CrossRef] [Green Version]
- Hutzen, B.; Ghonime, M.; Lee, J.; Mardis, E.R.; Wang, R.; Lee, D.A.; Cairo, M.S.; Roberts, R.D.; Cripe, T.P.; Cassady, K.A. Immunotherapeutic Challenges for Pediatric Cancers. Mol. Ther. Oncolytics 2019, 15, 38–48. [Google Scholar] [CrossRef] [Green Version]
- Elster, J.D.; Krishnadas, D.K.; Lucas, K.G. Dendritic cell vaccines: A review of recent developments and their potential pediatric application. Hum. Vaccin. Immunother. 2016, 12, 2232–2239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jarnjak-Jankovic, S.; Hammerstad, H.; Saeboe-Larssen, S.; Kvalheim, G.; Gaudernack, G. A full scale comparative study of methods for generation of functional Dendritic cells for use as cancer vaccines. BMC Cancer 2007, 7, 119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fields, R.C.; Shimizu, K.; Mule, J.J. Murine dendritic cells pulsed with whole tumor lysates mediate potent antitumor immune responses in vitro and in vivo. Proc. Natl. Acad. Sci. USA 1998, 95, 9482–9487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meadors, J.L.; Cui, Y.; Chen, Q.R.; Song, Y.K.; Khan, J.; Merlino, G.; Tsokos, M.; Orentas, R.J.; Mackall, C.L. Murine rhabdomyosarcoma is immunogenic and responsive to T-cell-based immunotherapy. Pediatr. Blood Cancer 2011, 57, 921–929. [Google Scholar] [CrossRef]
- Geiger, J.D.; Hutchinson, R.J.; Hohenkirk, L.F.; McKenna, E.A.; Yanik, G.A.; Levine, J.E.; Chang, A.E.; Braun, T.M.; Mule, J.J. Vaccination of pediatric solid tumor patients with tumor lysate-pulsed dendritic cells can expand specific T cells and mediate tumor regression. Cancer Res. 2001, 61, 8513–8519. [Google Scholar]
- Krishnadas, D.K.; Shapiro, T.; Lucas, K. Complete remission following decitabine/dendritic cell vaccine for relapsed neuroblastoma. Pediatrics 2013, 131, e336–e341. [Google Scholar] [CrossRef] [Green Version]
- Krishnadas, D.K.; Shusterman, S.; Bai, F.; Diller, L.; Sullivan, J.E.; Cheerva, A.C.; George, R.E.; Lucas, K.G. A phase I trial combining decitabine/dendritic cell vaccine targeting MAGE-A1, MAGE-A3 and NY-ESO-1 for children with relapsed or therapy-refractory neuroblastoma and sarcoma. Cancer Immunol. Immunother. 2015, 64, 1251–1260. [Google Scholar] [CrossRef]


{kind=link}
| Immunotherapy Approach | Disease | Target | Agent/Compound | NCT # | Phase of Study |
|---|---|---|---|---|---|
| Viral therapy | Cerebellar Brain Tumor | N/A | G207 (HSV) | 03911388 | Phase I (recruiting) |
| Viral therapy | Supratentorial Brain Tumor | N/A | G207 (HSV) +/− radiation | 02457845 | Phase I (recruiting) |
| Viral therapy | DIPG | N/A | DNX-2401 (adenovirus) | 03178032 | Phase I (recruiting) |
| Viral therapy | Glioma | N/A | Recombinant Polio/Rhinovirus | 03043391 | Phase I (recruiting) |
| Antigen-targeting and growth factor therapy | Neuroblastoma | GD2 | hu3F8 (mAB against GD2) and GM-CSF | 01757626 | Phase I/II (recruiting) |
| Immune checkpoint inhibitor | Solid tumors | CTLA-4 | Ipilimumab | 01445379 | Phase I (completed) |
| Immune checkpoint inhibitor | Solid tumors or lymphoma | PD-1 | Nivolumab with chemotherapy | 03585465 | Phase I/II (recruiting) |
| Immune checkpoint inhibitor | Hypermutated malignancies | PD-1 | Nivolumab | 02992964 | Phase I/II (recruiting) |
| Immune checkpoint inhibitor | Solid tumors | PD-1 | Nivolumab | 02901145 | Phase I/II (not yet recruiting) |
| Immune checkpoint inhibitor | Solid tumors or sarcoma | PD-1/CTLA-4 | Nivolumab +/− ipilimumab | 02304458 | Phase I/II (recruiting) |
| Cytokine therapy | DIPG | N/A | Pegylated IFN-α2b | 00041145 | Phase II (completed) |
| Cytokine therapy | Plexiform neurofibroma | N/A | Pegylated IFN-α2b | 00678951 | Phase II (completed) |
| Cytokine therapy | Osteosarcoma | N/A | Pegylated IFN-α2b | 00134030 | Phase III (active, not recruiting) |
| Cytokine targeted therapy | Osteosarcoma | RANKL | Denosumab (mAB against RANKL) | 02470091 | Phase II (active, not recruiting) |
| Cytokine targeted therapy | Solid tumors | TRAIL-R2 | Lexatumumab (mAB against TRAIL-R2) | 00428272 | Phase I (terminated) |
| Growth factor therapy | Osteosarcoma, Ewing sarcoma | N/A | Inhaled GM-CSF (Sargramostim) | 00673179 | Phase I (terminated) |
| CAR T cells | Neuroblastoma | GD2 | Anti-GD2 CAR T cells | 01822652 | Phase I (active, not recruiting) |
| CAR T cells | Sarcoma | HER2 | Anti-HER2 CAR T cells | 00902044 | Phase I (recruiting) |
| NK cells with cytokine therapy | Brain tumors, sarcoma, Wilms tumor, RMS | N/A | NK cells +/− rhIL-15 after lympho-depletion | 01875601 | Phase I (completed) |
| NK cells with antigen targeted therapy | Neuroblastoma | GD2 | hu14.18K322A (anti-GD2), NK cells | 01576692 | Phase I (completed) |
| NK cells with antigen targeted therapy | Neuroblastoma | GD2 | hu14.18K322A (anti-GD2), NK cells | 01857934 | Phase II (active, not recruiting) |
| NK cells | Solid tumors | N/A | NK cells | 01287104 | Phase I (completed) |
| NK cells | Ewing sarcoma, RMS | N/A | NK cells | 00640796 | Phase I (completed) |
| Cancer Vaccine | Neuroblastoma, sarcoma, RMS | Cancer testes antigen | Decitabine and DC vaccine + adjuvant | 01241162 | Phase I (Completed) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marayati, R.; Quinn, C.H.; Beierle, E.A. Immunotherapy in Pediatric Solid Tumors—A Systematic Review. Cancers 2019, 11, 2022. https://doi.org/10.3390/cancers11122022
Marayati R, Quinn CH, Beierle EA. Immunotherapy in Pediatric Solid Tumors—A Systematic Review. Cancers. 2019; 11(12):2022. https://doi.org/10.3390/cancers11122022
Chicago/Turabian StyleMarayati, Raoud, Colin H. Quinn, and Elizabeth A. Beierle. 2019. "Immunotherapy in Pediatric Solid Tumors—A Systematic Review" Cancers 11, no. 12: 2022. https://doi.org/10.3390/cancers11122022