Identification of a Subtype of Hepatocellular Carcinoma with Poor Prognosis Based on Expression of Genes within the Glucose Metabolic Pathway


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
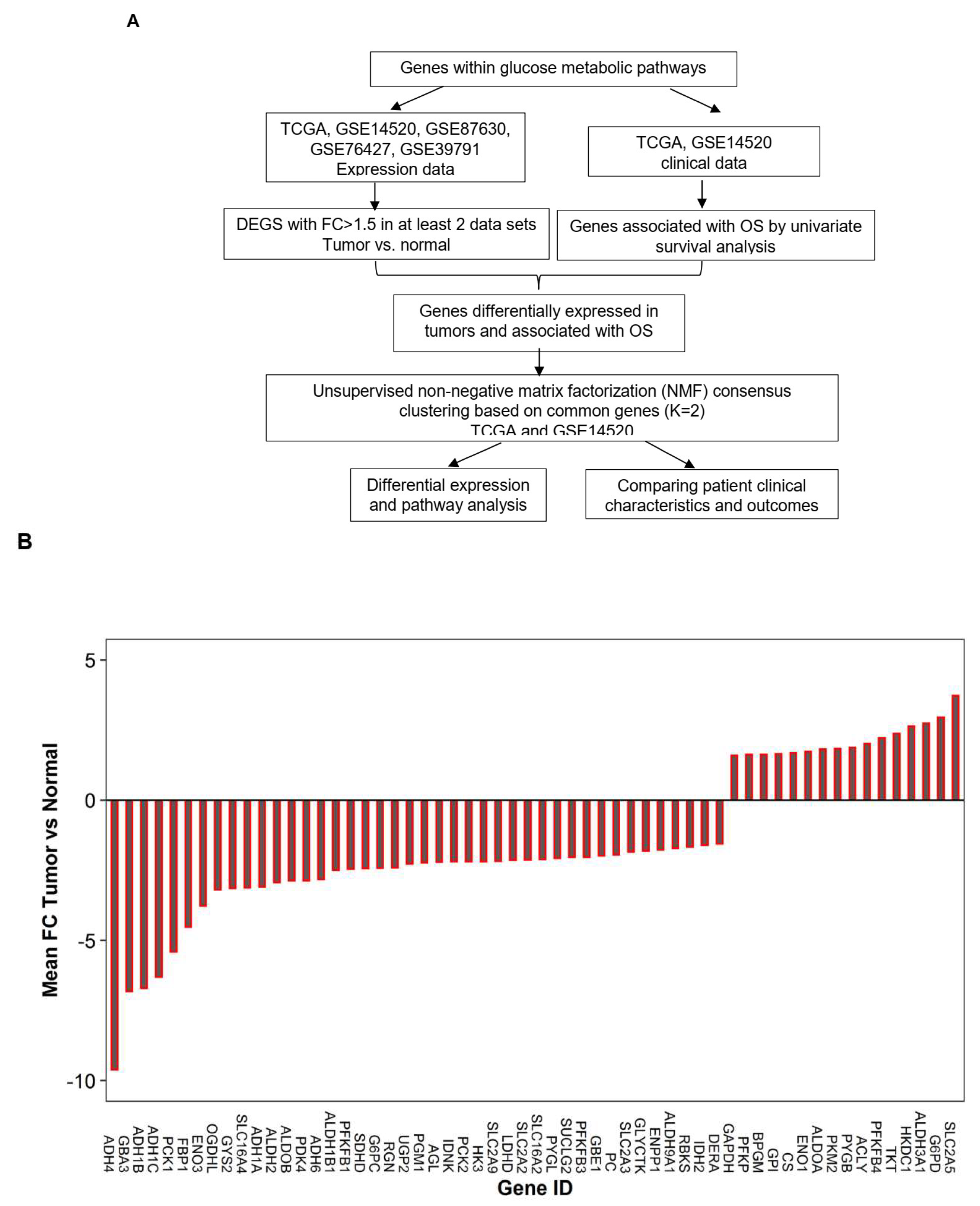
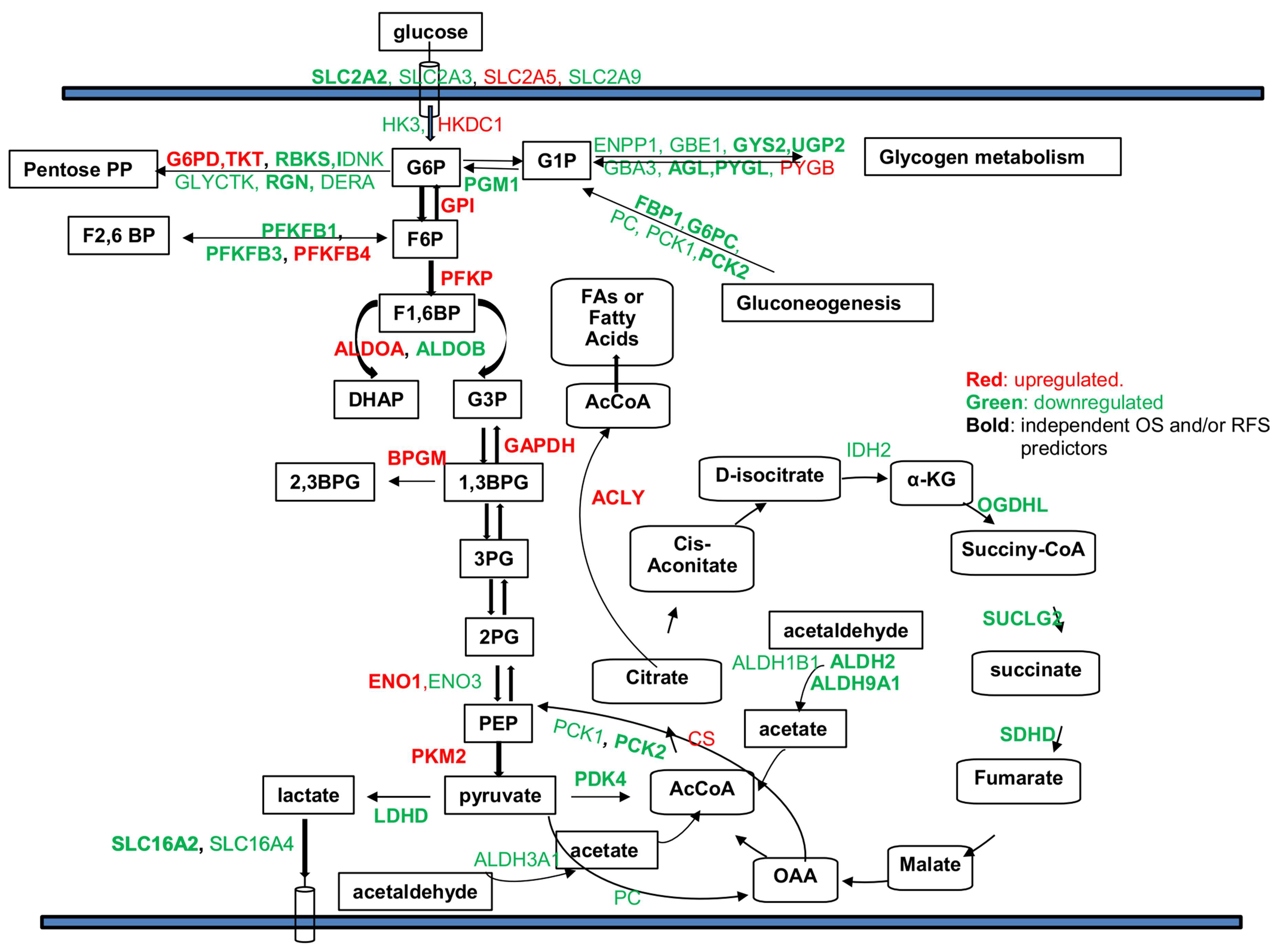
2.1. Genes within Glucose Metabolic Pathway Are Dysregulated in Human HCC Tumor Cells
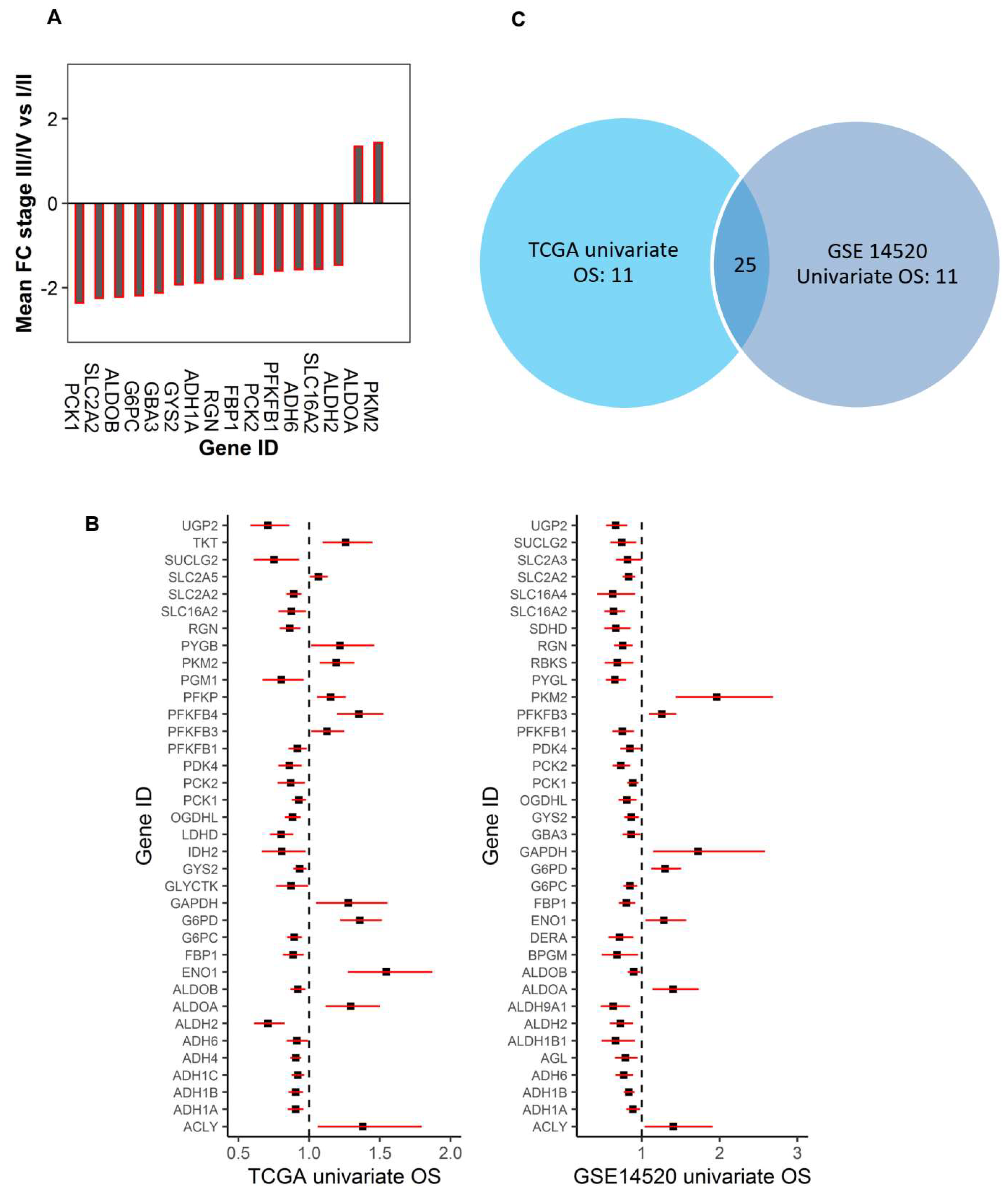
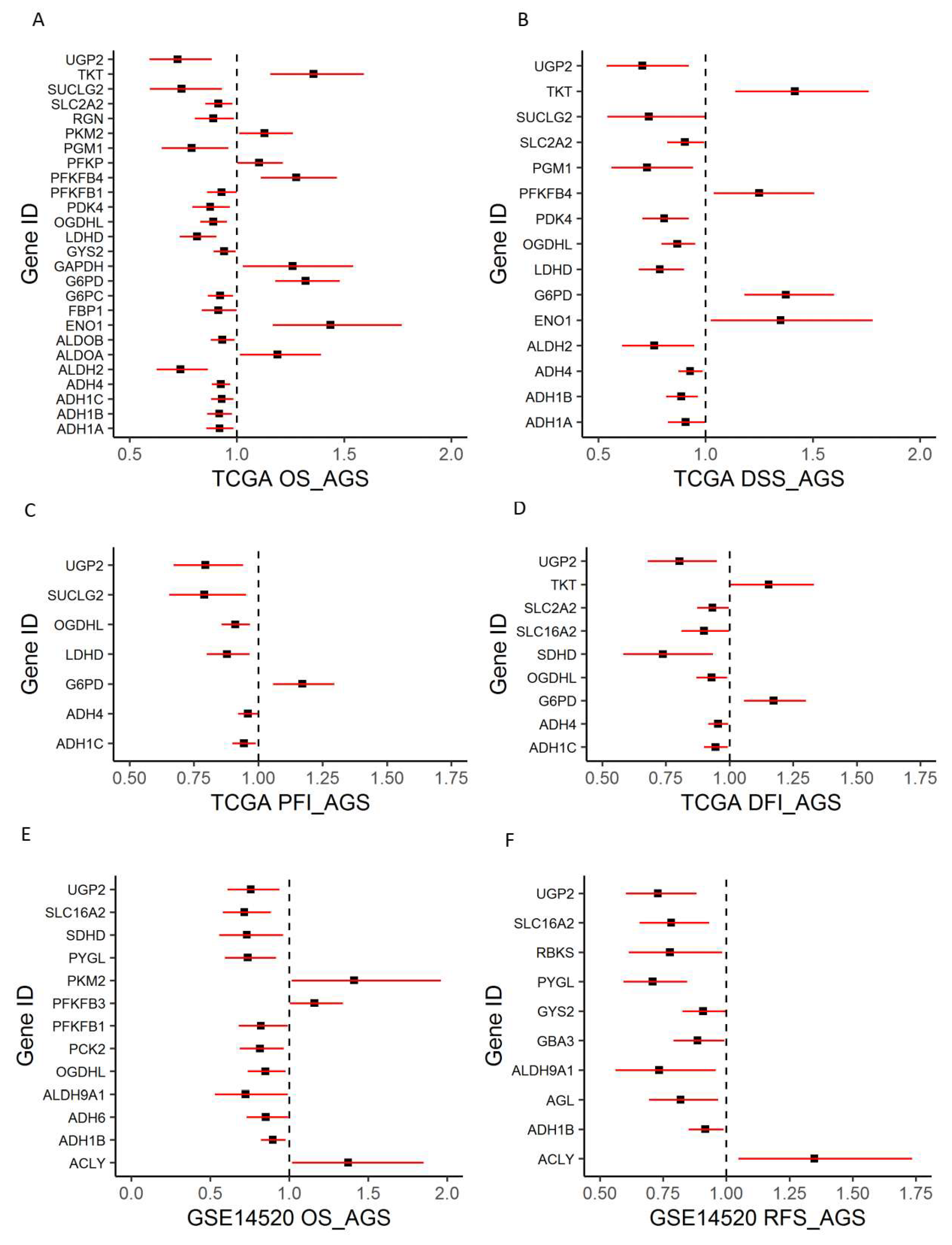
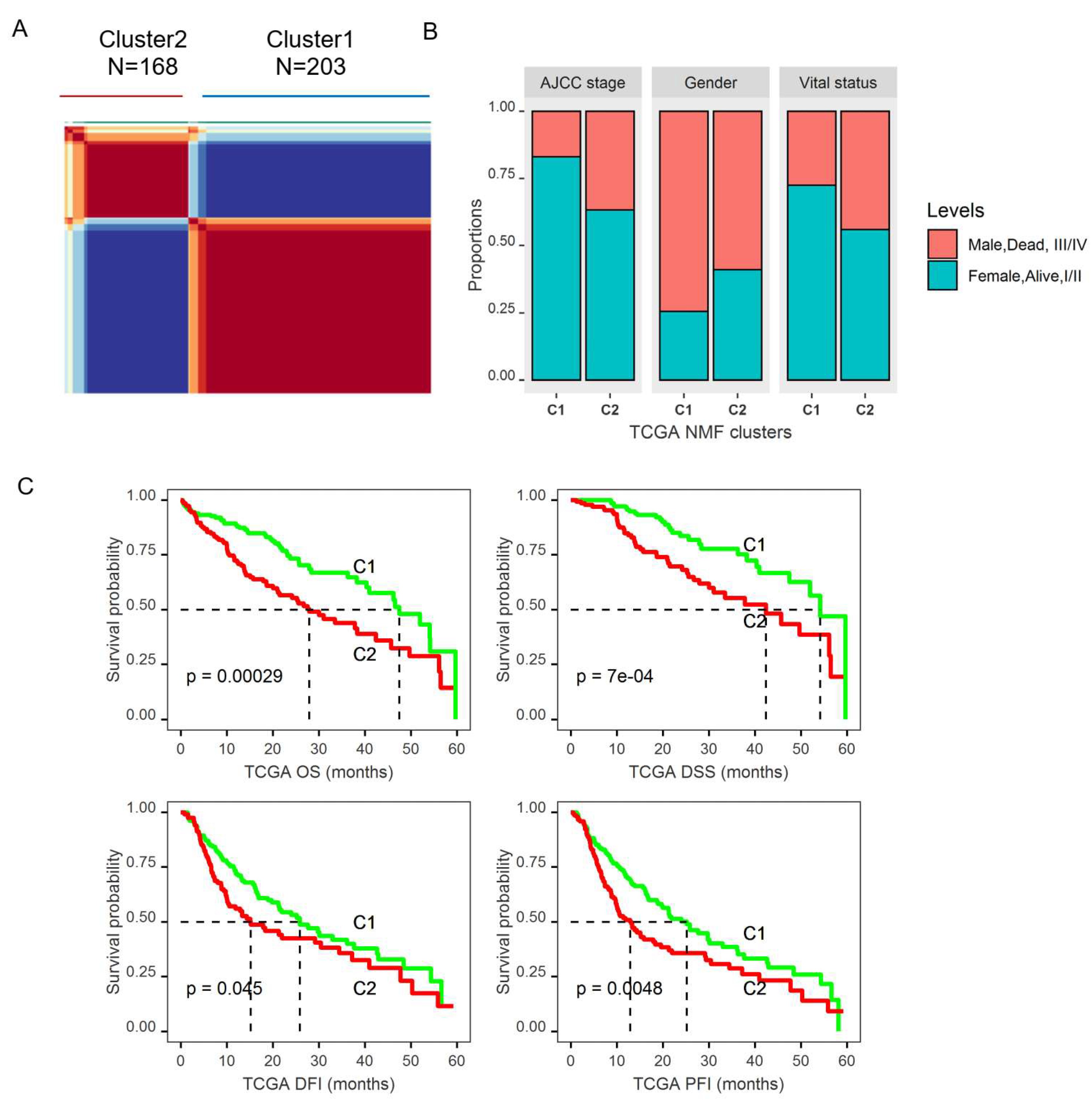
2.2. Dysregulated Genes within Glucose Metabolic Pathway Are Associated with HCC Patient Tumor Stage and Survival
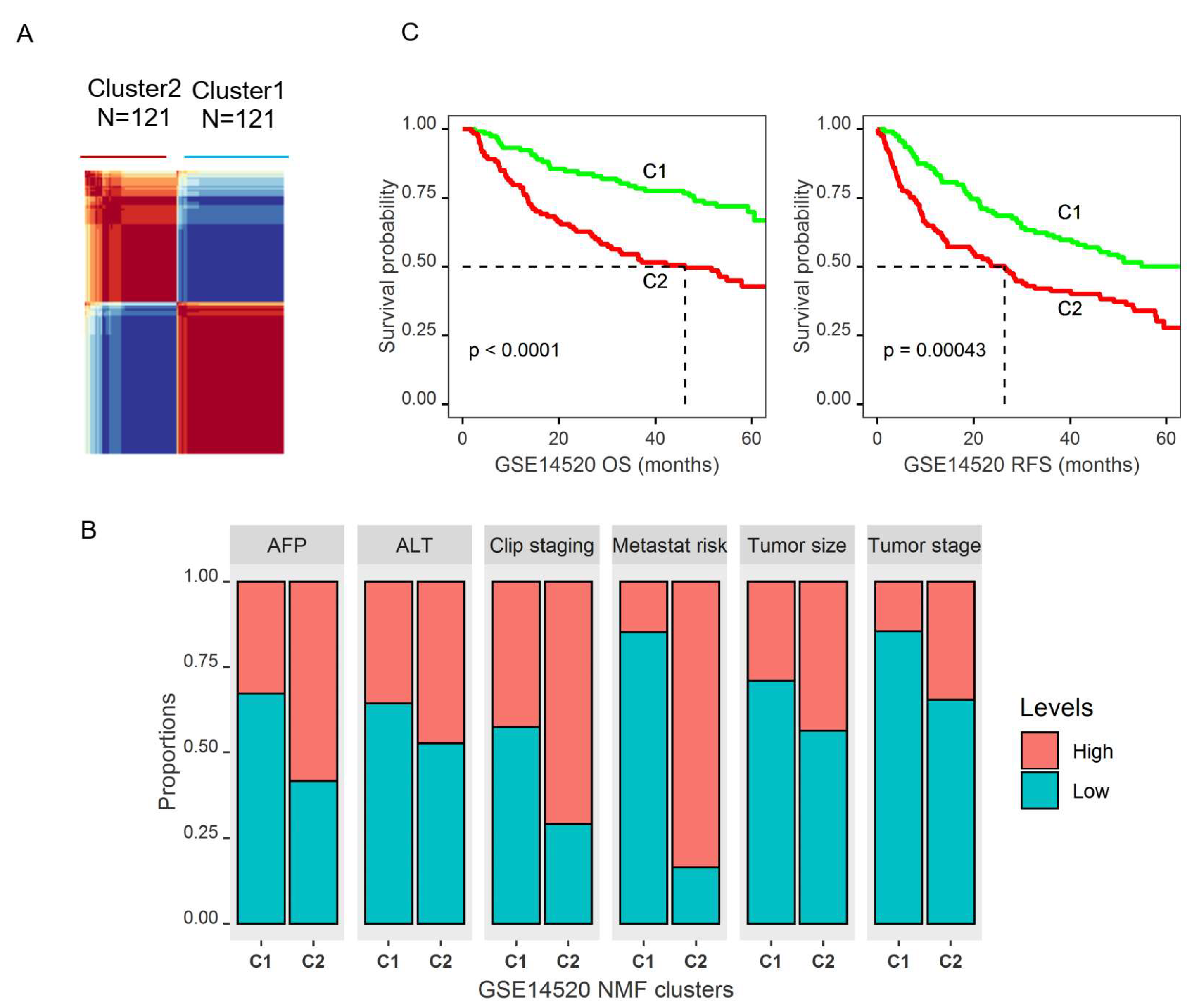
2.3. Molecular Subtypes of HCCs Based upon Genes Differentially Expressed in Tumors and Associated with Patient OS
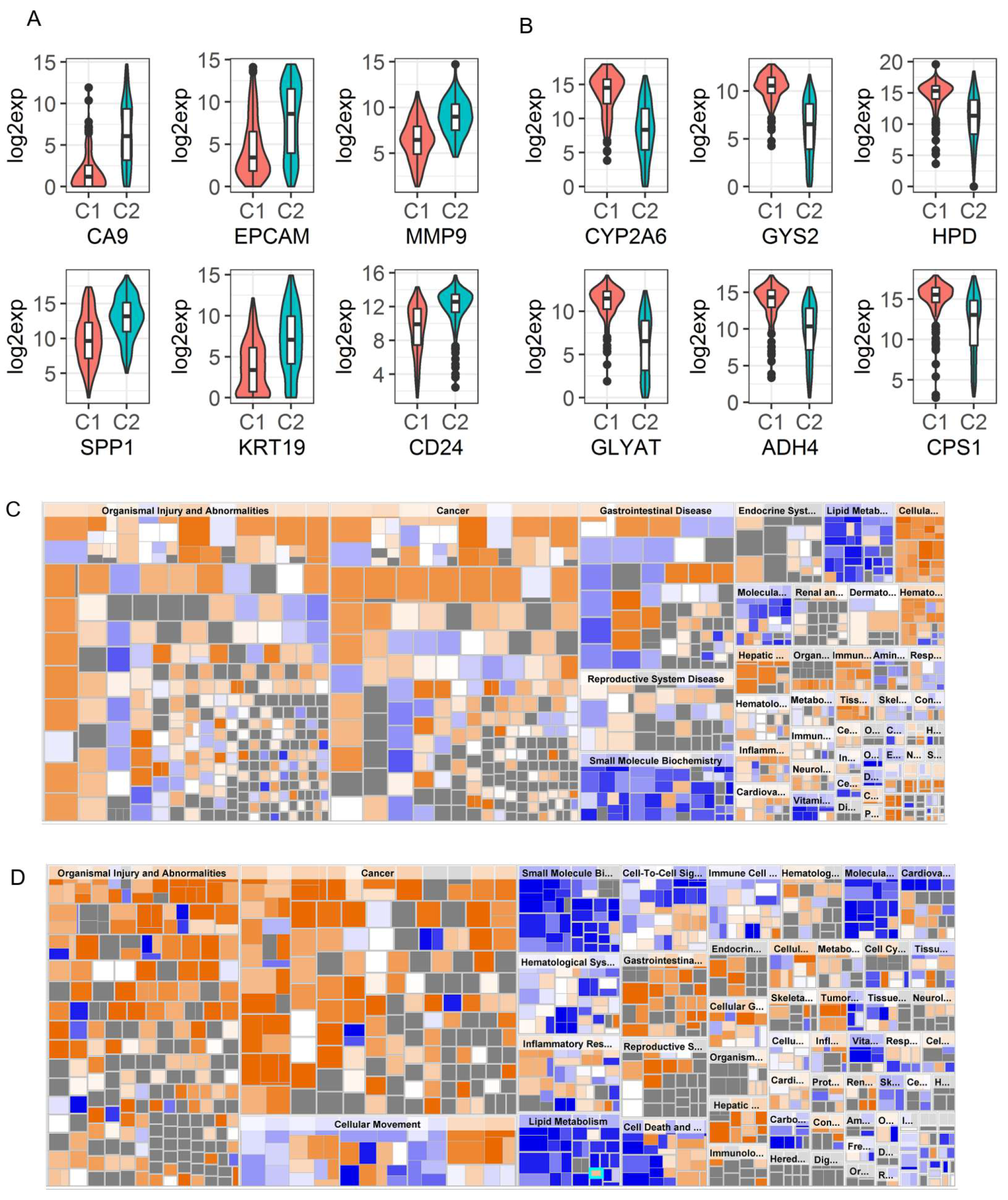
2.4. Identification of Genomic Key Features in the HCC Subtypes
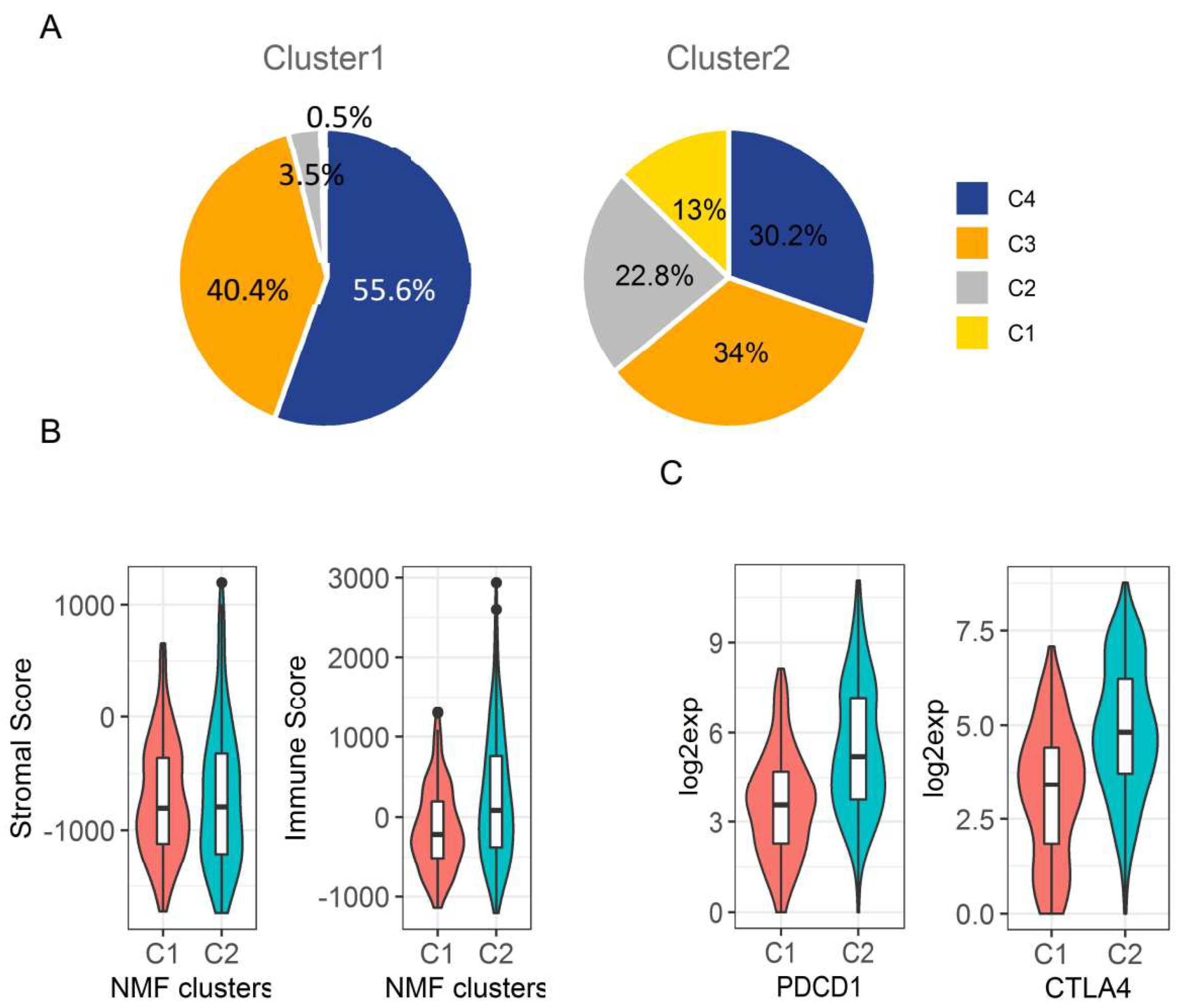
2.5. Immunologic Microenvironment in HCC Subtypes
3. Discussion
4. Materials and Methods
4.1. Gene List
4.2. Datasets and Gene Expression
4.3. Survival Analysis
4.4. Clustering Analysis
4.5. Comparison of NMF Clusters
4.6. Gene Set Enrichment Analysis between NMF Clusters
4.7. Tumour Microenvironment and Immune Subtype Analyses
4.8. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Altekruse, S.F.; McGlynn, K.A.; Reichman, M.E. Hepatocellular carcinoma incidence, mortality, and survival trends in the United States from 1975 to 2005. J. Clin. Oncol. 2009, 27, 1485–1491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calvisi, D.F.; Ladu, S.; Gorden, A.; Farina, M.; Lee, J.S.; Conner, E.A.; Schroeder, I.; Factor, V.M.; Thorgeirsson, S.S. Mechanistic and prognostic significance of aberrant methylation in the molecular pathogenesis of human hepatocellular carcinoma. J. Clin. Invest. 2007, 117, 2713–2722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dekervel, J.; van Pelt, J.; Verslype, C. Advanced unresectable hepatocellular carcinoma: New biologics as fresh ammunition or clues to disease understanding? Curr. Opin. Oncol. 2013, 25, 409–416. [Google Scholar] [CrossRef] [PubMed]
- Llovet, J.M.; Villanueva, A.; Lachenmayer, A.; Finn, R.S. Advances in targeted therapies for hepatocellular carcinoma in the genomic era. Nat. Rev. Clin. Oncol. 2015, 12, 408–424. [Google Scholar] [CrossRef] [PubMed]
- Allemani, C.; Weir, H.K.; Carreira, H.; Harewood, R.; Spika, D.; Wang, X.S.; Bannon, F.; Ahn, J.V.; Johnson, C.J.; Bonaventure, A.; et al. Global surveillance of cancer survival 1995-2009: Analysis of individual data for 25,676,887 patients from 279 population-based registries in 67 countries (CONCORD-2). Lancet 2015, 385, 977–1010. [Google Scholar] [CrossRef] [Green Version]
- Nwosu, Z.C.; Megger, D.A.; Hammad, S.; Sitek, B.; Roessler, S.; Ebert, M.P.; Meyer, C.; Dooley, S. Identification of the Consistently Altered Metabolic Targets in Human Hepatocellular Carcinoma. Cell Mol. Gastroenterol. Hepatol. 2017, 4, 303–323. [Google Scholar] [CrossRef] [Green Version]
- Marbaniang, C.; Kma, L. Dysregulation of Glucose Metabolism by Oncogenes and Tumor Suppressors in Cancer Cells. Asian Pac. J. Cancer Prev. 2018, 19, 2377–2390. [Google Scholar] [CrossRef]
- Jones, R.G.; Thompson, C.B. Tumor suppressors and cell metabolism: A recipe for cancer growth. Genes Dev. 2009, 23, 537–548. [Google Scholar] [CrossRef] [Green Version]
- Lunt, S.Y.; Vander Heiden, M.G. Aerobic glycolysis: Meeting the metabolic requirements of cell proliferation. Annu. Rev. Cell Dev. Biol. 2011, 27, 441–464. [Google Scholar] [CrossRef] [Green Version]
- Gatenby, R.A.; Gillies, R.J. Why do cancers have high aerobic glycolysis? Nat. Rev. Cancer 2004, 4, 891–899. [Google Scholar] [CrossRef]
- Kroemer, G.; Pouyssegur, J. Tumor cell metabolism: Cancer’s Achilles’ heel. Cancer Cell 2008, 13, 472–482. [Google Scholar] [CrossRef] [PubMed]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cancer Genome Atlas Research Network. Comprehensive and Integrative Genomic Characterization of Hepatocellular Carcinoma. Cell 2017, 169, 1327–1341.23. [Google Scholar] [CrossRef] [PubMed]
- Rousset, M.; Paris, H.; Chevalier, G.; Terrain, B.; Murat, J.C.; Zweibaum, A. Growth-related enzymatic control of glycogen metabolism in cultured human tumor cells. Cancer Res. 1984, 44, 154–160. [Google Scholar] [PubMed]
- Grasmann, G.; Smolle, E.; Olschewski, H.; Leithner, K. Gluconeogenesis in cancer cells - Repurposing of a starvation-induced metabolic pathway? Biochim. Biophys. Acta Rev. Cancer 2019, 1872, 24–36. [Google Scholar] [CrossRef]
- Li, Y.; Zhuang, H.; Zhang, X.; Li, Y.; Liu, Y.; Yi, X.; Qin, G.; Wei, W.; Chen, R. Multiomics Integration Reveals the Landscape of Prometastasis Metabolism in Hepatocellular Carcinoma. Mol. Cell Proteomics 2018, 17, 607–618. [Google Scholar] [CrossRef] [Green Version]
- Woo, H.G.; Choi, J.H.; Yoon, S.; Jee, B.A.; Cho, E.J.; Lee, J.H.; Yu, S.J.; Yoon, J.H.; Yi, N.J.; Lee, K.W.; et al. Integrative analysis of genomic and epigenomic regulation of the transcriptome in liver cancer. Nat. Commun. 2017, 8, e839. [Google Scholar] [CrossRef] [Green Version]
- Finkelmeier, F.; Canli, O.; Peiffer, K.H.; Walter, D.; Tal, A.; Koch, C.; Pession, U.; Vermehren, J.; Trojan, J.; Zeuzem, S.; et al. Circulating hypoxia marker carbonic anhydrase IX (CA9) in patients with hepatocellular carcinoma and patients with cirrhosis. PLoS ONE 2018, 13, e0200855. [Google Scholar] [CrossRef]
- Gao, X.; Sheng, Y.; Yang, J.; Wang, C.; Zhang, R.; Zhu, Y.; Zhang, Z.; Zhang, K.; Yan, S.; Sun, H.; et al. Osteopontin alters DNA methylation through up-regulating DNMT1 and sensitizes CD133+/CD44+ cancer stem cells to 5 azacytidine in hepatocellular carcinoma. J. Exp. Clin. Cancer Res. 2018, 37, e179. [Google Scholar] [CrossRef]
- Wang, G.; Liu, Y.; Huang, J.; Liu, J.; Wang, J.; Yang, J. PPARalpha Suppresses PD-L1-Mediated Immune Escape by Down-regulating SPP1 in Human Hepatocellular Carcinoma. Cancer Res. Treat 2019. [Google Scholar] [CrossRef]
- Nishida, N.; Kudo, M. Immunological Microenvironment of Hepatocellular Carcinoma and Its Clinical Implication. Oncology 2017, 92, 40–49. [Google Scholar] [CrossRef] [PubMed]
- Tamborero, D.; Rubio-Perez, C.; Muinos, F.; Sabarinathan, R.; Piulats, J.M.; Muntasell, A.; Dienstmann, R.; Lopez-Bigas, N.; Gonzalez-Perez, A. A Pan-cancer Landscape of Interactions between Solid Tumors and Infiltrating Immune Cell Populations. Clin. Cancer Res. 2018, 24, 3717–3728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thorsson, V.; Gibbs, D.L.; Brown, S.D.; Wolf, D.; Bortone, D.S.; Ou Yang, T.H.; Porta-Pardo, E.; Gao, G.F.; Plaisier, C.L.; Eddy, J.A.; et al. The Immune Landscape of Cancer. Immunity 2018, 48, 812–830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshihara, K.; Shahmoradgoli, M.; Martinez, E.; Vegesna, R.; Kim, H.; Torres-Garcia, W.; Trevino, V.; Shen, H.; Laird, P.W.; Levine, D.A.; et al. Inferring tumour purity and stromal and immune cell admixture from expression data. Nat. Commun. 2013, 4, e2612. [Google Scholar] [CrossRef]
- Binnewies, M.; Roberts, E.W.; Kersten, K.; Chan, V.; Fearon, D.F.; Merad, M.; Coussens, L.M.; Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Hedrick, C.C.; et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat. Med. 2018, 24, 541–550. [Google Scholar] [CrossRef]
- Pavlova, N.N.; Thompson, C.B. The Emerging Hallmarks of Cancer Metabolism. Cell Metab. 2016, 23, 27–47. [Google Scholar] [CrossRef] [Green Version]
- Hay, N. Reprogramming glucose metabolism in cancer: Can it be exploited for cancer therapy? Nat. Rev. Cancer 2016, 16, 635–649. [Google Scholar] [CrossRef] [Green Version]
- Ancey, P.B.; Contat, C.; Meylan, E. Glucose transporters in cancer - from tumor cells to the tumor microenvironment. FEBS J. 2018, 285, 2926–2943. [Google Scholar] [CrossRef]
- Kim, Y.H.; Jeong, D.C.; Pak, K.; Han, M.E.; Kim, J.Y.; Liangwen, L.; Kim, H.J.; Kim, T.W.; Kim, T.H.; Hyun, D.W.; et al. SLC2A2 (GLUT2) as a novel prognostic factor for hepatocellular carcinoma. Oncotarget 2017, 8, 68381–68392. [Google Scholar] [CrossRef] [Green Version]
- Patra, K.C.; Hay, N. Hexokinase 2 as oncotarget. Oncotarget 2013, 4, 1862–1863. [Google Scholar] [CrossRef]
- Patra, K.C.; Wang, Q.; Bhaskar, P.T.; Miller, L.; Wang, Z.; Wheaton, W.; Chandel, N.; Laakso, M.; Muller, W.J.; Allen, E.L.; et al. Hexokinase 2 is required for tumor initiation and maintenance and its systemic deletion is therapeutic in mouse models of cancer. Cancer Cell 2013, 24, 213–228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, M.W.; Priyadarshini, M.; Cordoba-Chacon, J.; Becker, T.C.; Layden, B.T. Hepatic hexokinase domain containing 1 (HKDC1) improves whole body glucose tolerance and insulin sensitivity in pregnant mice. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 678–687. [Google Scholar] [CrossRef] [PubMed]
- Pusec, C.M.; De Jesus, A.; Khan, M.W.; Terry, A.R.; Ludvik, A.E.; Xu, K.; Giancola, N.; Pervaiz, H.; Daviau Smith, E.; Ding, X.; et al. Hepatic HKDC1 Expression Contributes to Liver Metabolism. Endocrinology 2019, 160, 313–330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.; Huang, S.; Wang, H.; Wu, J.; Chen, D.; Peng, B.; Zhou, Q. High expression of hexokinase domain containing 1 is associated with poor prognosis and aggressive phenotype in hepatocarcinoma. Biochem. Biophys. Res. Commun. 2016, 474, 673–679. [Google Scholar] [CrossRef]
- Wang, J.; Zhang, P.; Zhong, J.; Tan, M.; Ge, J.; Tao, L.; Li, Y.; Zhu, Y.; Wu, L.; Qiu, J.; et al. The platelet isoform of phosphofructokinase contributes to metabolic reprogramming and maintains cell proliferation in clear cell renal cell carcinoma. Oncotarget 2016, 7, 27142–27157. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.H.; Liu, R.; Li, J.; Zhang, C.; Wang, Y.; Cai, Q.; Qian, X.; Xia, Y.; Zheng, Y.; Piao, Y.; et al. Stabilization of phosphofructokinase 1 platelet isoform by AKT promotes tumorigenesis. Nat. Commun. 2017, 8, e949. [Google Scholar] [CrossRef] [Green Version]
- Bartrons, R.; Simon-Molas, H.; Rodriguez-Garcia, A.; Castano, E.; Navarro-Sabate, A.; Manzano, A.; Martinez-Outschoorn, U.E. Fructose 2,6-Bisphosphate in Cancer Cell Metabolism. Front Oncol. 2018, 8, e331. [Google Scholar] [CrossRef]
- Atsumi, T.; Chesney, J.; Metz, C.; Leng, L.; Donnelly, S.; Makita, Z.; Mitchell, R.; Bucala, R. High expression of inducible 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase (iPFK-2; PFKFB3) in human cancers. Cancer Res. 2002, 62, 5881–5887. [Google Scholar]
- Marotta, L.L.; Almendro, V.; Marusyk, A.; Shipitsin, M.; Schemme, J.; Walker, S.R.; Bloushtain-Qimron, N.; Kim, J.J.; Choudhury, S.A.; Maruyama, R.; et al. The JAK2/STAT3 signaling pathway is required for growth of CD44(+)CD24(-) stem cell-like breast cancer cells in human tumors. J. Clin Invest. 2011, 121, 2723–2735. [Google Scholar] [CrossRef]
- Gao, R.; Li, D.; Xun, J.; Zhou, W.; Li, J.; Wang, J.; Liu, C.; Li, X.; Shen, W.; Qiao, H.; et al. CD44ICD promotes breast cancer stemness via PFKFB4-mediated glucose metabolism. Theranostics 2018, 8, 6248–6262. [Google Scholar] [CrossRef]
- Taylor, C.; Mannion, D.; Miranda, F.; Karaminejadranjbar, M.; Herrero-Gonzalez, S.; Hellner, K.; Zheng, Y.; Bartholomeusz, G.; Bast, R.C., Jr.; Ahmed, A.A. Loss of PFKFB4 induces cell death in mitotically arrested ovarian cancer cells. Oncotarget 2017, 8, 17960–17980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.; Zeng, F.; Sun, Y.; Qiu, Q.; Zhang, J.; Huang, W.; Huang, J.; Huang, X.; Guo, L. Etk Interaction with PFKFB4 Modulates Chemoresistance of Small-cell Lung Cancer by Regulating Autophagy. Clin. Cancer Res. 2018, 24, 950–962. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Lu, C.; Fang, M.; Yan, W.; Chen, M.; Ji, Y.; He, S.; Liu, T.; Chen, T.; Xiao, J. HIF-1alpha activates hypoxia-induced PFKFB4 expression in human bladder cancer cells. Biochem. Biophys. Res. Commun. 2016, 476, 146–152. [Google Scholar] [CrossRef] [PubMed]
- Dayton, T.L.; Jacks, T.; Vander Heiden, M.G. PKM2, cancer metabolism, and the road ahead. EMBO Rep. 2016, 17, 1721–1730. [Google Scholar] [CrossRef] [Green Version]
- Chang, Y.C.; Chiou, J.; Yang, Y.F.; Su, C.Y.; Lin, Y.F.; Yang, C.N.; Lu, P.J.; Huang, M.S.; Yang, C.J.; Hsiao, M. Therapeutic Targeting of Aldolase A Interactions Inhibits Lung Cancer Metastasis and Prolongs Survival. Cancer Res. 2019, 79, 4754–4766. [Google Scholar] [CrossRef] [Green Version]
- Gizak, A.; Wisniewski, J.; Heron, P.; Mamczur, P.; Sygusch, J.; Rakus, D. Targeting a moonlighting function of aldolase induces apoptosis in cancer cells. Cell Death Dis. 2019, 10, e712. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Wang, F.; Gao, H.; Huang, S.; Cai, F.; Sun, J. ALDOLASE A regulates invasion of bladder cancer cells via E-cadherin-EGFR signaling. J. Cell Biochem. 2019, 120, 13694–13705. [Google Scholar] [CrossRef]
- Cancemi, P.; Buttacavoli, M.; Roz, E.; Feo, S. Expression of Alpha-Enolase (ENO1), Myc Promoter-Binding Protein-1 (MBP-1) and Matrix Metalloproteinases (MMP-2 and MMP-9) Reflect the Nature and Aggressiveness of Breast Tumors. Int. J. Mol. Sci. 2019, 20, 3952. [Google Scholar] [CrossRef] [Green Version]
- Ji, M.; Wang, Z.; Chen, J.; Gu, L.; Chen, M.; Ding, Y.; Liu, T. Up-regulated ENO1 promotes the bladder cancer cell growth and proliferation via regulating beta-catenin. Biosci. Rep. 2019, 39. [Google Scholar] [CrossRef] [Green Version]
- Principe, M.; Borgoni, S.; Cascione, M.; Chattaragada, M.S.; Ferri-Borgogno, S.; Capello, M.; Bulfamante, S.; Chapelle, J.; Di Modugno, F.; Defilippi, P.; et al. Alpha-enolase (ENO1) controls alpha v/beta 3 integrin expression and regulates pancreatic cancer adhesion, invasion, and metastasis. J. Hematol. Oncol. 2017, 10, e16. [Google Scholar] [CrossRef] [Green Version]
- Qiao, H.; Wang, Y.; Zhu, B.; Jiang, L.; Yuan, W.; Zhou, Y.; Guan, Q. Enolase1 overexpression regulates the growth of gastric cancer cells and predicts poor survival. J. Cell Biochem. 2019, 120, 18714–18723. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.; Wang, L.; Liu, H.L. ENO1 Overexpression in Pancreatic Cancer Patients and Its Clinical and Diagnostic Significance. Gastroenterol. Res. Pract. 2018, e3842198. [Google Scholar] [CrossRef] [Green Version]
- Oslund, R.C.; Su, X.; Haugbro, M.; Kee, J.M.; Esposito, M.; David, Y.; Wang, B.; Ge, E.; Perlman, D.H.; Kang, Y.; et al. Bisphosphoglycerate mutase controls serine pathway flux via 3-phosphoglycerate. Nat. Chem. Biol. 2017, 13, 1081–1087. [Google Scholar] [CrossRef] [PubMed]
- Barajas, J.M.; Reyes, R.; Guerrero, M.J.; Jacob, S.T.; Motiwala, T.; Ghoshal, K. The role of miR-122 in the dysregulation of glucose-6-phosphate dehydrogenase (G6PD) expression in hepatocellular cancer. Sci. Rep. 2018, 8, e9105. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Xu, Z.; Zhu, Z.; Chen, A.; Fu, G.; Wang, Y.; Pan, H.; Jin, B. Modulation of G6PD affects bladder cancer via ROS accumulation and the AKT pathway in vitro. Int. J. Oncol. 2018, 53, 1703–1712. [Google Scholar] [CrossRef] [Green Version]
- Ju, H.Q.; Lu, Y.X.; Wu, Q.N.; Liu, J.; Zeng, Z.L.; Mo, H.Y.; Chen, Y.; Tian, T.; Wang, Y.; Kang, T.B.; et al. Disrupting G6PD-mediated Redox homeostasis enhances chemosensitivity in colorectal cancer. Oncogene 2017, 36, 6282–6292. [Google Scholar] [CrossRef] [Green Version]
- Shukla, S.K.; Purohit, V.; Mehla, K.; Gunda, V.; Chaika, N.V.; Vernucci, E.; King, R.J.; Abrego, J.; Goode, G.D.; Dasgupta, A.; et al. MUC1 and HIF-1alpha Signaling Crosstalk Induces Anabolic Glucose Metabolism to Impart Gemcitabine Resistance to Pancreatic Cancer. Cancer Cell 2017, 32, 71–87. [Google Scholar] [CrossRef] [Green Version]
- Tseng, C.W.; Kuo, W.H.; Chan, S.H.; Chan, H.L.; Chang, K.J.; Wang, L.H. Transketolase Regulates the Metabolic Switch to Control Breast Cancer Cell Metastasis via the alpha-Ketoglutarate Signaling Pathway. Cancer Res. 2018, 78, 2799–2812. [Google Scholar] [CrossRef] [Green Version]
- Hyuga, S.; Wada, H.; Eguchi, H.; Otsuru, T.; Iwgami, Y.; Yamada, D.; Noda, T.; Asaoka, T.; Kawamoto, K.; Gotoh, K.; et al. Expression of carbonic anhydrase IX is associated with poor prognosis through regulation of the epithelialmesenchymal transition in hepatocellular carcinoma. Int. J. Oncol. 2017, 51, 1179–1190. [Google Scholar] [CrossRef]
- Wakizaka, K.; Yokoo, H.; Kamiyama, T.; Kakisaka, T.; Ohira, M.; Tani, M.; Kato, K.; Fujii, Y.; Sugiyama, K.; Nagatsu, A.; et al. CD133 and epithelial cell adhesion molecule expressions in cholangiocarcinoma component are prognostic factors for combined hepatocellular-cholangiocarcinoma. Hepatol. Res. 2019. [Google Scholar] [CrossRef]
- Wang, R.; Li, Y.; Tsung, A.; Huang, H.; Du, Q.; Yang, M.; Deng, M.; Xiong, S.; Wang, X.; Zhang, L.; et al. iNOS promotes CD24(+)CD133(+) liver cancer stem cell phenotype through a TACE/ADAM17-dependent Notch signaling pathway. Proc. Natl. Acad. Sci. USA 2018, 115, 10127–10136. [Google Scholar] [CrossRef] [Green Version]
- Yokomichi, N.; Nishida, N.; Umeda, Y.; Taniguchi, F.; Yasui, K.; Toshima, T.; Mori, Y.; Nyuya, A.; Tanaka, T.; Yamada, T.; et al. Heterogeneity of Epigenetic and Epithelial Mesenchymal Transition Marks in Hepatocellular Carcinoma with Keratin 19 Proficiency. Liver Cancer 2019, 8, 239–254. [Google Scholar] [CrossRef]
- Bidkhori, G.; Benfeitas, R.; Klevstig, M.; Zhang, C.; Nielsen, J.; Uhlen, M.; Boren, J.; Mardinoglu, A. Metabolic network-based stratification of hepatocellular carcinoma reveals three distinct tumor subtypes. Proc. Natl. Acad. Sci. USA 2018, 115, 11874–11883. [Google Scholar] [CrossRef] [Green Version]
- Team, R.C. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2018. [Google Scholar]
- Gordon, A.; Glazko, G.; Qiu, X.; Yakovlev, A. Control of the mean number of false discoveries, Bonferroni and stability of multiple testing. Ann. Appl. Stat. 2007, 1, 179–190. [Google Scholar] [CrossRef] [Green Version]








© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, X.; Li, J.; Ghoshal, K.; Fernandez, S.; Li, L. Identification of a Subtype of Hepatocellular Carcinoma with Poor Prognosis Based on Expression of Genes within the Glucose Metabolic Pathway. Cancers 2019, 11, 2023. https://doi.org/10.3390/cancers11122023
Zhang X, Li J, Ghoshal K, Fernandez S, Li L. Identification of a Subtype of Hepatocellular Carcinoma with Poor Prognosis Based on Expression of Genes within the Glucose Metabolic Pathway. Cancers. 2019; 11(12):2023. https://doi.org/10.3390/cancers11122023
Chicago/Turabian StyleZhang, Xiaoli, Jin Li, Kalpana Ghoshal, Soledad Fernandez, and Lang Li. 2019. "Identification of a Subtype of Hepatocellular Carcinoma with Poor Prognosis Based on Expression of Genes within the Glucose Metabolic Pathway" Cancers 11, no. 12: 2023. https://doi.org/10.3390/cancers11122023