Allometric Scaling Approaches for Predicting Human Pharmacokinetic of a Locked Nucleic Acid Oligonucleotide Targeting Cancer-Associated miR-221

 ,
,  , ,
, ,
Abstract
:1. Introduction
2. Results and Discussion
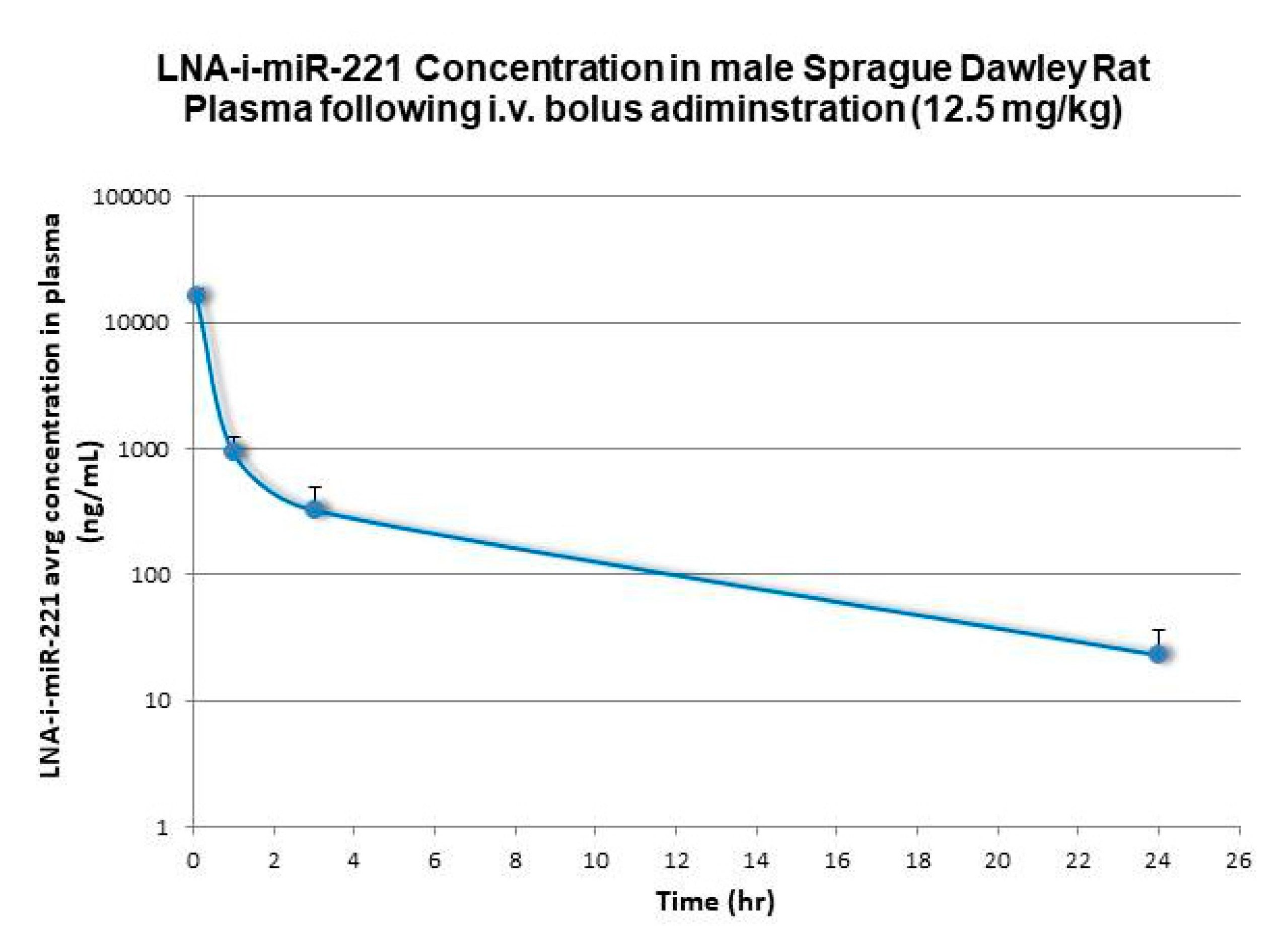
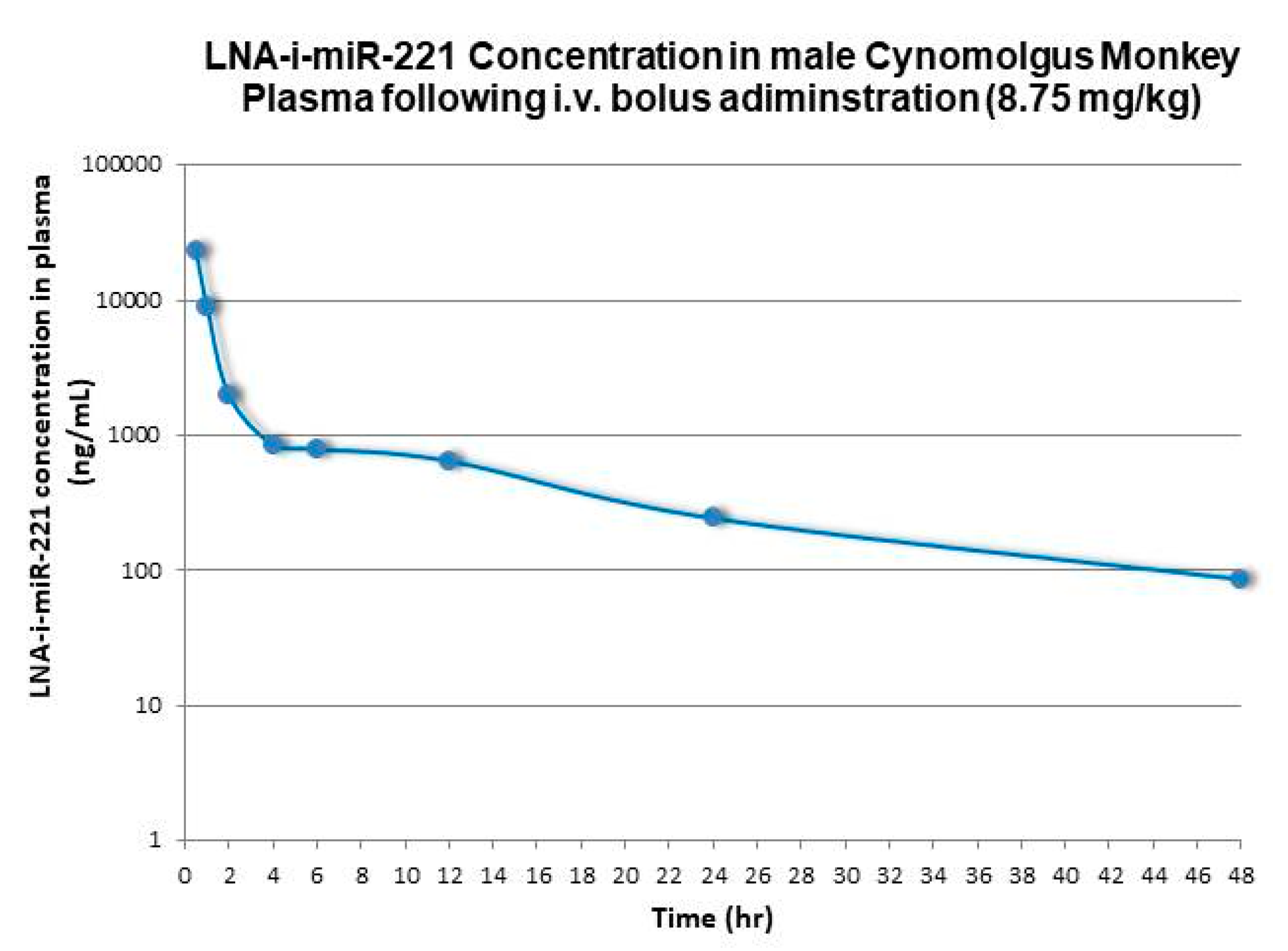
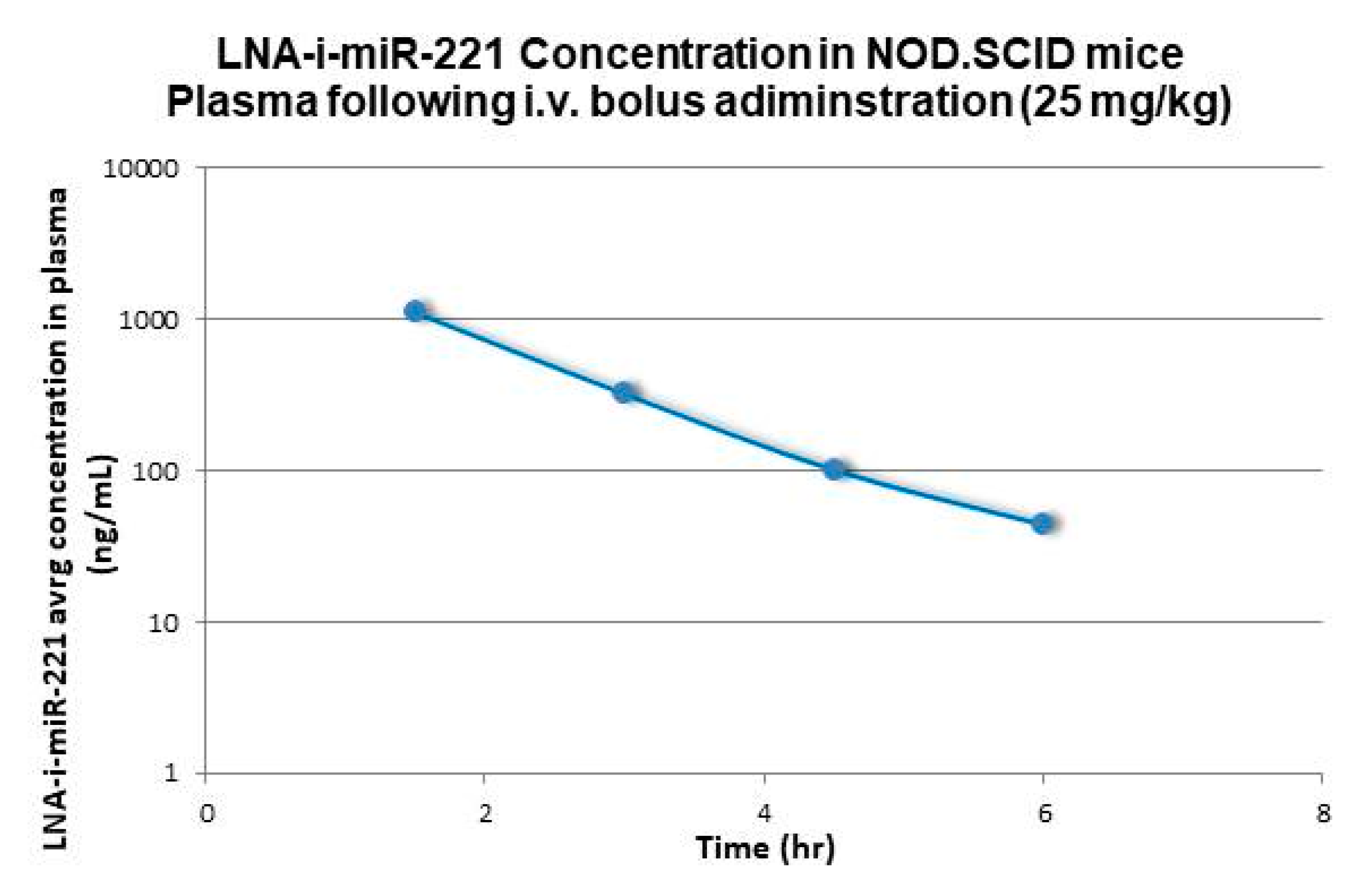
2.1. Pharmacokinetic Analysis
2.2. Allometric Scaling of Clearance Values
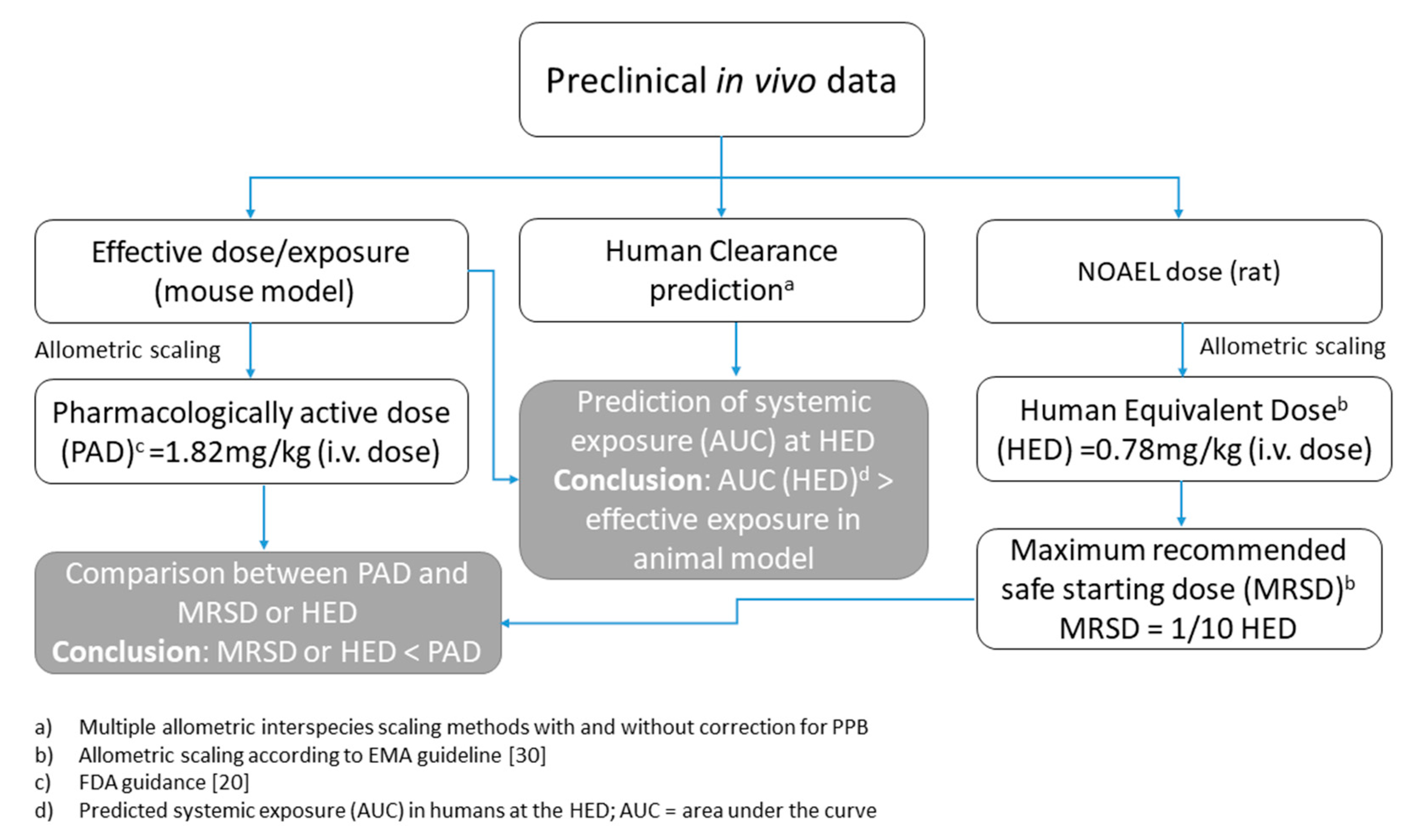
2.3. Calculation of Human Equivalent Dose (HED)
2.4. Allometric Scaling of PK Parameters
2.5. Two-Species Scaling
2.6. One-Species Scaling
2.7. Plasma Protein Binding and CLpu
- (i)
- specificity, linearity, range and lower limit of quantification (LOQ),
- (ii)
- within-batch precision and accuracy,
- (iii)
- matrix effect,
- (iv)
- stability in human, monkey, and rat plasma at 37 °C.
2.8. Protein Binding
3. Methods
3.1. Chemicals and Materials
3.2. Ultrafiltration Procedure
3.2.1. LNA-i-miR-221 Protein Binding in Human, Rat, and Monkey
3.2.2. Non-Specific Binding of LNA-i-miR-221 to Amicon Plastic
3.3. Analytical Conditions
4. Conclusions
5. Patents
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Di Martino, M.T.; Rossi, M.; Caracciolo, D.; Gulla, A.; Tagliaferri, P.; Tassone, P. Mir-221/222 are promising targets for innovative anticancer therapy. Expert Opin. Ther. Targets 2016, 20, 1099–1108. [Google Scholar] [CrossRef] [PubMed]
- Gallo Cantafio, M.E.; Nielsen, B.S.; Mignogna, C.; Arbitrio, M.; Botta, C.; Frandsen, N.M.; Rolfo, C.; Tagliaferri, P.; Tassone, P.; Di Martino, M.T. Pharmacokinetics and Pharmacodynamics of a 13-mer LNA-inhibitor-miR-221 in Mice and Non-human Primates. Mol. Ther. Nucleic Acids 2016, 5, e326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Martino, M.T.; Gulla, A.; Cantafio, M.E.; Lionetti, M.; Leone, E.; Amodio, N.; Guzzi, P.H.; Foresta, U.; Conforti, F.; Cannataro, M.; et al. In vitro and in vivo anti-tumor activity of miR-221/222 inhibitors in multiple myeloma. Oncotarget 2013, 4, 242–255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Martino, M.T.; Gulla, A.; Gallo Cantafio, M.E.; Altomare, E.; Amodio, N.; Leone, E.; Morelli, E.; Lio, S.G.; Caracciolo, D.; Rossi, M.; et al. In vitro and in vivo activity of a novel locked nucleic acid (LNA)-inhibitor-miR-221 against multiple myeloma cells. PLoS ONE 2014, 9, e89659. [Google Scholar] [CrossRef] [PubMed]
- Santolla, M.F.; Lappano, R.; Cirillo, F.; Rigiracciolo, D.C.; Sebastiani, A.; Abonante, S.; Tassone, P.; Tagliaferri, P.; Di Martino, M.T.; Maggiolini, M.; et al. miR-221 stimulates breast cancer cells and cancer-associated fibroblasts (CAFs) through selective interference with the A20/c-Rel/CTGF signaling. J. Exp. Clin. Cancer Res. CR 2018, 37, 94. [Google Scholar] [CrossRef] [PubMed]
- Gulla, A.; Di Martino, M.T.; Gallo Cantafio, M.E.; Morelli, E.; Amodio, N.; Botta, C.; Pitari, M.R.; Lio, S.G.; Britti, D.; Stamato, M.A.; et al. A 13 mer LNA-i-miR-221 Inhibitor Restores Drug Sensitivity in Melphalan-Refractory Multiple Myeloma Cells. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2016, 22, 1222–1233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, R.Z.; Grundy, J.S.; Geary, R.S. Clinical pharmacokinetics of second generation antisense oligonucleotides. Expert Opin. Drug Metab. Toxicol. 2013, 9, 169–182. [Google Scholar] [CrossRef]
- Franzoni, S.; Vezzelli, A.; Turtoro, A.; Solazzo, L.; Greco, A.; Tassone, P.; Di Martino, M.T.; Breda, M. Development and validation of a bioanalytical method for quantification of LNA-i-miR-221, a 13-mer oligonucleotide, in rat plasma using LC-MS/MS. J. Pharm. Biomed. Anal. 2018, 150, 300–307. [Google Scholar] [CrossRef]
- Geary, R.S. Antisense oligonucleotide pharmacokinetics and metabolism. Expert Opin. Drug Metab. Toxicol. 2009, 5, 381–391. [Google Scholar] [CrossRef]
- Huang, Q.; Riviere, J.E. The application of allometric scaling principles to predict pharmacokinetic parameters across species. Expert Opin. Drug Metab. Toxicol. 2014, 10, 1241–1253. [Google Scholar] [CrossRef]
- Shin, J.W.; Seol, I.C.; Son, C.G. Interpretation of Animal Dose and Human Equivalent Dose for Drug Development. J. Korean Orient. Med. 2010, 31, 1–7. [Google Scholar]
- Boxenbaum, H. Interspecies scaling, allometry, physiological time, and the ground plan of pharmacokinetics. J. Pharmacokinet. Biopharm. 1982, 10, 201–227. [Google Scholar] [CrossRef] [PubMed]
- Heimbach, T.; Lakshminarayana, S.B.; Hu, W.; He, H. Practical anticipation of human efficacious doses and pharmacokinetics using in vitro and preclinical in vivo data. AAPS J. 2009, 11, 602–614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rowland, M. Protein binding and drug clearance. Clin. Pharmacokinet. 1984, 9, 10–17. [Google Scholar] [CrossRef] [PubMed]
- Mahmood, I. Interspecies Pharmacokinetic Scaling: Principles And Application of Allometric Scaling; Hardcover: Danvers, MA, USA, 2005. [Google Scholar]
- Tang, H.; Hussain, A.; Leal, M.; Mayersohn, M.; Fluhler, E. Interspecies prediction of human drug clearance based on scaling data from one or two animal species. Drug Metab. Dispos. Biol. Fate Chem. 2007, 35, 1886–1893. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, T.A.; Geary, R.S.; Levin, A.A. Plasma protein binding of an antisense oligonucleotide targeting human ICAM-1 (ISIS 2302). Oligonucleotides 2006, 16, 169–180. [Google Scholar] [CrossRef]
- Yu, R.Z.; Kim, T.W.; Hong, A.; Watanabe, T.A.; Gaus, H.J.; Geary, R.S. Cross-species pharmacokinetic comparison from mouse to man of a second-generation antisense oligonucleotide, ISIS 301012, targeting human apolipoprotein B-100. Drug Metab. Dispos. Biol. Fate Chem. 2007, 35, 460–468. [Google Scholar] [CrossRef] [Green Version]
- Mahmood, I. Pharmacokinetic allometric scaling of oligonucleotides. Nucleic Acid Ther. 2011, 21, 315–321. [Google Scholar] [CrossRef]
- FDA. Guidance for Industry “Estimating the Maximum Safe Starting Dose in Initial Clinical Trials for Therapeutics in Adult Healthy Volunteers”; U.S. Department of Health and Human Services Food and Drug Administration Center for Drug Evaluation and Research (CDER) Pharmacology and Toxicology: Rockville, MD, USA, 2005.
- Frazier, K.S. Antisense oligonucleotide therapies: The promise and the challenges from a toxicologic pathologist’s perspective. Toxicol. Pathol. 2015, 43, 78–89. [Google Scholar] [CrossRef]
- Geary, R.S.; Leeds, J.M.; Henry, S.P.; Monteith, D.K.; Levin, A.A. Antisense oligonucleotide inhibitors for the treatment of cancer: 1. Pharmacokinetic properties of phosphorothioate oligodeoxynucleotides. Anti-Cancer Drug Des. 1997, 12, 383–393. [Google Scholar]
- Leeds, J.M.; Henry, S.P.; Truong, L.; Zutshi, A.; Levin, A.A.; Kornbrust, D. Pharmacokinetics of a potential human cytomegalovirus therapeutic, a phosphorothioate oligonucleotide, after intravitreal injection in the rabbit. Drug Metab. Dispos. Biol. Fate Chem. 1997, 25, 921–926. [Google Scholar] [PubMed]
- Yu, R.Z.; Zhang, H.; Geary, R.S.; Graham, M.; Masarjian, L.; Lemonidis, K.; Crooke, R.; Dean, N.M.; Levin, A.A. Pharmacokinetics and pharmacodynamics of an antisense phosphorothioate oligonucleotide targeting Fas mRNA in mice. J. Pharmacol. Exp. Ther. 2001, 296, 388–395. [Google Scholar] [PubMed]
- Geary, R.S.; Yu, R.Z.; Levin, A.A. Pharmacokinetics of phosphorothioate antisense oligodeoxynucleotides. Curr. Opin. Investig. Drugs 2001, 2, 562–573. [Google Scholar] [PubMed]
- Agrawal, S.; Tan, W.; Cai, Q.; Xie, X.; Zhang, R. In vivo pharmacokinetics of phosphorothioate oligonucleotides containing contiguous guanosines. Antisense Nucleic Acid Drug Dev. 1997, 7, 245–249. [Google Scholar] [CrossRef]
- Phillips, J.A.; Craig, S.J.; Bayley, D.; Christian, R.A.; Geary, R.; Nicklin, P.L. Pharmacokinetics, metabolism, and elimination of a 20-mer phosphorothioate oligodeoxynucleotide (CGP 69846A) after intravenous and subcutaneous administration. Biochem. Pharmacol. 1997, 54, 657–668. [Google Scholar] [CrossRef]
- Cossum, P.A.; Sasmor, H.; Dellinger, D.; Truong, L.; Cummins, L.; Owens, S.R.; Markham, P.M.; Shea, J.P.; Crooke, S. Disposition of the 14C-labeled phosphorothioate oligonucleotide ISIS 2105 after intravenous administration to rats. J. Pharmacol. Exp. Ther. 1993, 267, 1181–1190. [Google Scholar]
- London, UK European Medicines Agency. Available online: http://www.emea.europa.eu/ (accessed on 19 July 2007).
- EMA; Commitee for Medical Products for Human Use (CHMP). Guideline on Strategies to Identify and Mitigate Risks for First-In Human Clinical Trials with Investigational Medical Products; EMEA; CHMP: London, UK, 19 July 2007. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Dose (i.v. Bolus) | C0/Dose | AUC 1 | AUC/Dose | CL | Terminal Half-Life | Vd Terminal |
|---|---|---|---|---|---|---|---|
| mg/kg | ng/mL/mg | (h·ng/mL) | (h·ng/mL)/mg | mL/min/kg | h | mL/Kg | |
| Rat | 12.5 | 4462 | 27192 | 2160 | 7.9 | 3.9 | 2626 |
| Mouse 2 | 25 | 82 | 3226 | 129 | 129 | 1.5 | 16443 |
| Monkey | 8.75 | 7017 | 49300 | 5634 | 3.0 | 12.8 | 3286 |
| Species | Average Body Weights (kg) Used for Allometric Scaling |
|---|---|
| Mouse | 0.025 |
| Rat | 0.25 |
| Monkey (Cynomolgus) | 3.75 |
| Human | 70 |
| b | −0.362198 |
| a | 0.6789 |
| CLhuman (mL/min/kg) | 1.02 |
| CLhuman (mL/min) | 71.7 |
| b | 0.65 |
| arat–monkey | 0.6789 |
| CLhuman (mL/min/kg) | 1.1 |
| CLhuman (mL/min) | 76 |
| Species Used for Scaling | Human CLp (mL/min/kg) | Human CLp (mL/min) |
|---|---|---|
| Single Species Allometry | ||
| rat | 1.9 | 135 |
| monkey | 1.4 | 100 |
| Total Plasma Clearance | AUC/Dose | AUC for 0.78 ‡ mg/kg i.v. Dose | AUC for 1.82 ‡ mg/kg i.v. Dose | AUC for 5.0 ‡ mg/kg i.v. Dose | |
|---|---|---|---|---|---|
| Allometric Method Used for Prediction | mL/min/kg | h·ng/mL·(mg Dose)−1 | h·ng/mL | h·ng/mL | h·ng/mL |
| Direct scaling two-species (r, mk) (Equation (1a)) | 1.0 | 16,264 | 12,686 | 29,600 | 81,318 |
| Tang et al. method two-species (r, mk) (Equation (3)) | 1.1 | 15,443 | 12,046 | 28,106 | 77,215 |
| One-species (r) allometric scaling (Equation (4)) | 1.9 | 8643 | 6741 | 15,730 | 43,214 |
| One-species (mk) allometric scaling (Equation (4)) | 1.4 | 11,711 | 9135 | 21,315 | 58,557 |
| Geometric mean | Geometric mean | Geometric mean | Geometric mean | Geometric mean | |
| 1.3 | 12,627 | 9849 | 22,981 | 63,135 |
| Total Plasma Clearance | AUC/Dose | AUC for 0.78 ‡ mg/kg i.v. Dose | AUC for 1.82 ‡ mg/kg i.v. Dose | AUC for 5.0 ‡ mg/kg i.v. Dose | |
|---|---|---|---|---|---|
| Allometric Method Used for Prediction | mL/min/kg | h·ng/mL·(mg Dose)−1 | h·ng/mL | h·ng/mL | h·ng/mL |
| Direct scaling two-species (r, mk) (Equation (1a)) | 1.0 | 16,386 | 12781 | 29,823 | 81,931 |
| Tang et al. method two-species (r, mk) (Equation (3)) | 1.15 | 14,462 | 11280 | 26,320 | 72,309 |
| One-species (r) allometric scaling (Equation (4)) | 2.2 | 7749 | 6044 | 14,103 | 38,743 |
| One-species (mk) allometric scaling (Equation (4)) | 1.5 | 11,106 | 8662 | 20,212 | 55,528 |
| Geometric mean | Geometric mean | Geometric mean | Geometric mean | Geometric mean | |
| 1.4 | 11950 | 9321 | 21,749 | 59,750 |
| Species | LNA-i-miR-221 Concentration | |||
|---|---|---|---|---|
| 1 µM | 10 µM | Mean PPB | Mean fu | |
| Human | 98.6 ± 0.32 | 98.5 ± 0.09 | 98.55 | 0.0145 |
| Monkey | 98.2 ± 0.39 | 99.05 ± 0.39 | 98.63 | 0.0138 |
| Rat | 98.5 ± 0.17 | 98.9 ± 0.17 | 98.70 | 0.0130 |
| Species | Sex | Supplier | Anticoagulant |
|---|---|---|---|
| Rat a (Sprague–Dawley) | Male Pool of 25 animals | Citoxlab | K2EDTA |
| Monkey b (Cynomolgus) | Male Pool of 10 animals | Citoxlab | K2EDTA |
| Human c | Male Pool of 10 donors | Biopredic (PLA152A050) | K2EDTA |
| Sex | λ | t1/2 | C0 | AUC0–∞ | AUC0–∞/Dose | AUC Extrapolated | Vz | Cl | |
|---|---|---|---|---|---|---|---|---|---|
| 1/h | h | ng/mL | h·ng/mL | % | mL/kg | mL/min/kg | |||
| F | 0.174 | 3.99 | 43,094 | 23,980 | 1918 | 0.869 | 3000 | 8.7 | |
| 0.187 | 3.70 | 47,033 | 24,961 | 1997 | 0.603 | 2672 | 8.3 | ||
| 0.187 | 3.70 | 64,272 | 31,455 | 2516 | 0.561 | 2120 | 6.6 | ||
| mean | 0.183 | 3.80 | 51,467 | 26,799 | 2144 | 0.678 | 2597 | 7.9 | |
| SD | 0.008 | 0.168 | 11,264 | 4062 | 325 | 0.167 | 445 | 1.1 | |
| CV% | 4 | 4 | 22 | 15 | 15 | 25 | 17 | 0.2 | |
| M | 0.178 | 3.90 | 49,251 | 24,364 | 1949 | 0.724 | 2890 | 8.6 | |
| 0.184 | 3.76 | 86,432 | 34,091 | 2727 | 0.490 | 1990 | 6.1 | ||
| 0.175 | 3.95 | 44,593 | 23,120 | 1850 | 0.792 | 3083 | 9.0 | ||
| mean | 0.179 | 3.87 | 60,092 | 27,192 | 2175 | 0.669 | 2654 | 7.9 | |
| SD | 0.005 | 0.099 | 22,930 | 6008 | 481 | 0.158 | 583 | 1.6 | |
| CV% | 3 | 3 | 38 | 22 | 22 | 24 | 22 | 0.3 | |
| M + F | mean | 0.181 | 3.83 | 55,779 | 26,995 | 2160 | 0.673 | 2626 | 7.9 |
| SD | 0.006 | 0.131 | 16,834 | 4592 | 367 | 0.145 | 465 | 1.2 | |
| CV% | 3 | 3 | 30 | 17 | 17 | 22 | 18 | 0.0 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Di Martino, M.T.; Arbitrio, M.; Fonsi, M.; Erratico, C.A.; Scionti, F.; Caracciolo, D.; Tagliaferri, P.; Tassone, P. Allometric Scaling Approaches for Predicting Human Pharmacokinetic of a Locked Nucleic Acid Oligonucleotide Targeting Cancer-Associated miR-221. Cancers 2020, 12, 27. https://doi.org/10.3390/cancers12010027
Di Martino MT, Arbitrio M, Fonsi M, Erratico CA, Scionti F, Caracciolo D, Tagliaferri P, Tassone P. Allometric Scaling Approaches for Predicting Human Pharmacokinetic of a Locked Nucleic Acid Oligonucleotide Targeting Cancer-Associated miR-221. Cancers. 2020; 12(1):27. https://doi.org/10.3390/cancers12010027
Chicago/Turabian StyleDi Martino, Maria Teresa, Mariamena Arbitrio, Massimiliano Fonsi, Claudio Alberto Erratico, Francesca Scionti, Daniele Caracciolo, Pierosandro Tagliaferri, and Pierfrancesco Tassone. 2020. "Allometric Scaling Approaches for Predicting Human Pharmacokinetic of a Locked Nucleic Acid Oligonucleotide Targeting Cancer-Associated miR-221" Cancers 12, no. 1: 27. https://doi.org/10.3390/cancers12010027