Resistance to MET/VEGFR2 Inhibition by Cabozantinib Is Mediated by YAP/TBX5-Dependent Induction of FGFR1 in Castration-Resistant Prostate Cancer

 , , and
, , and 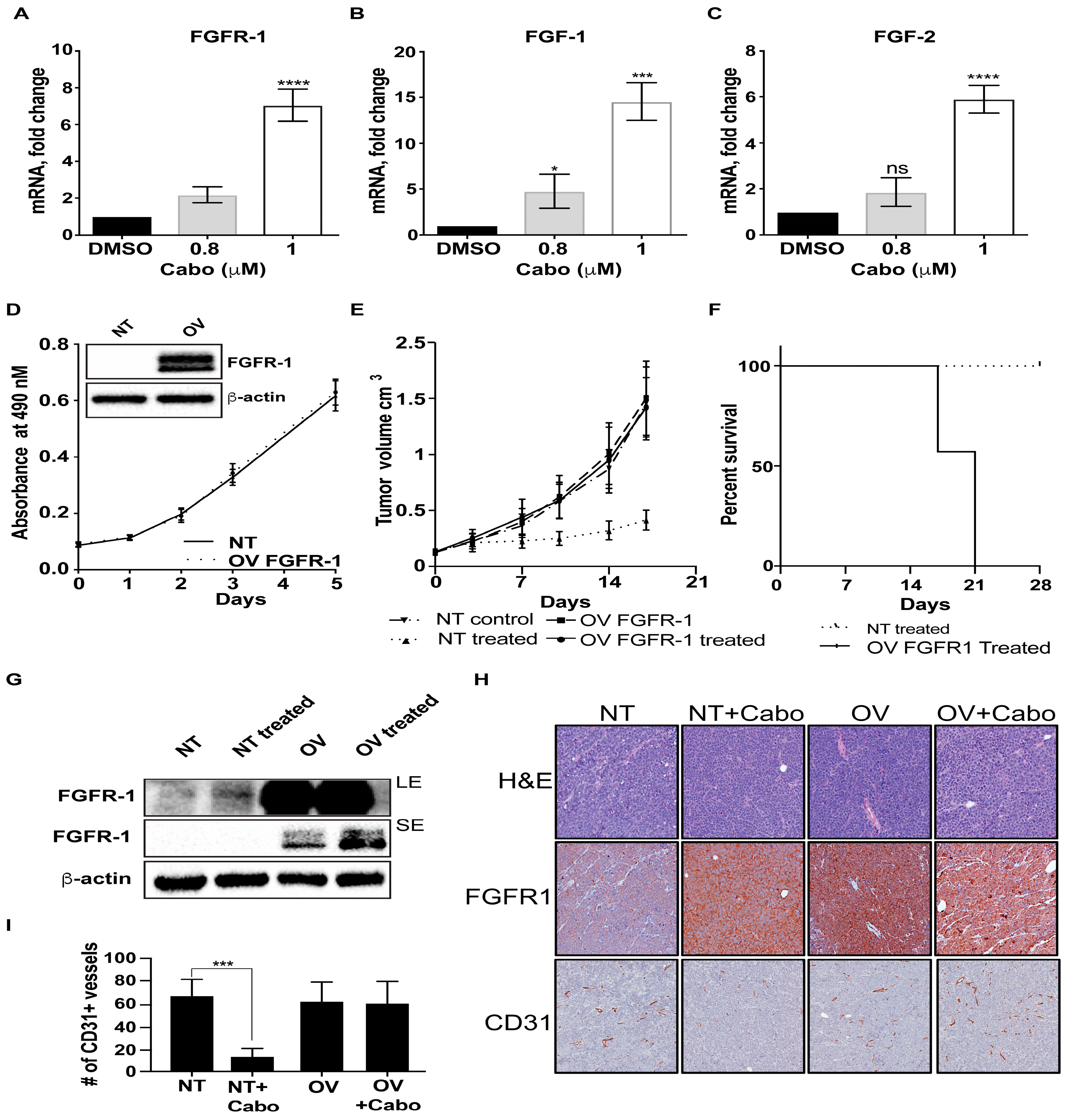{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
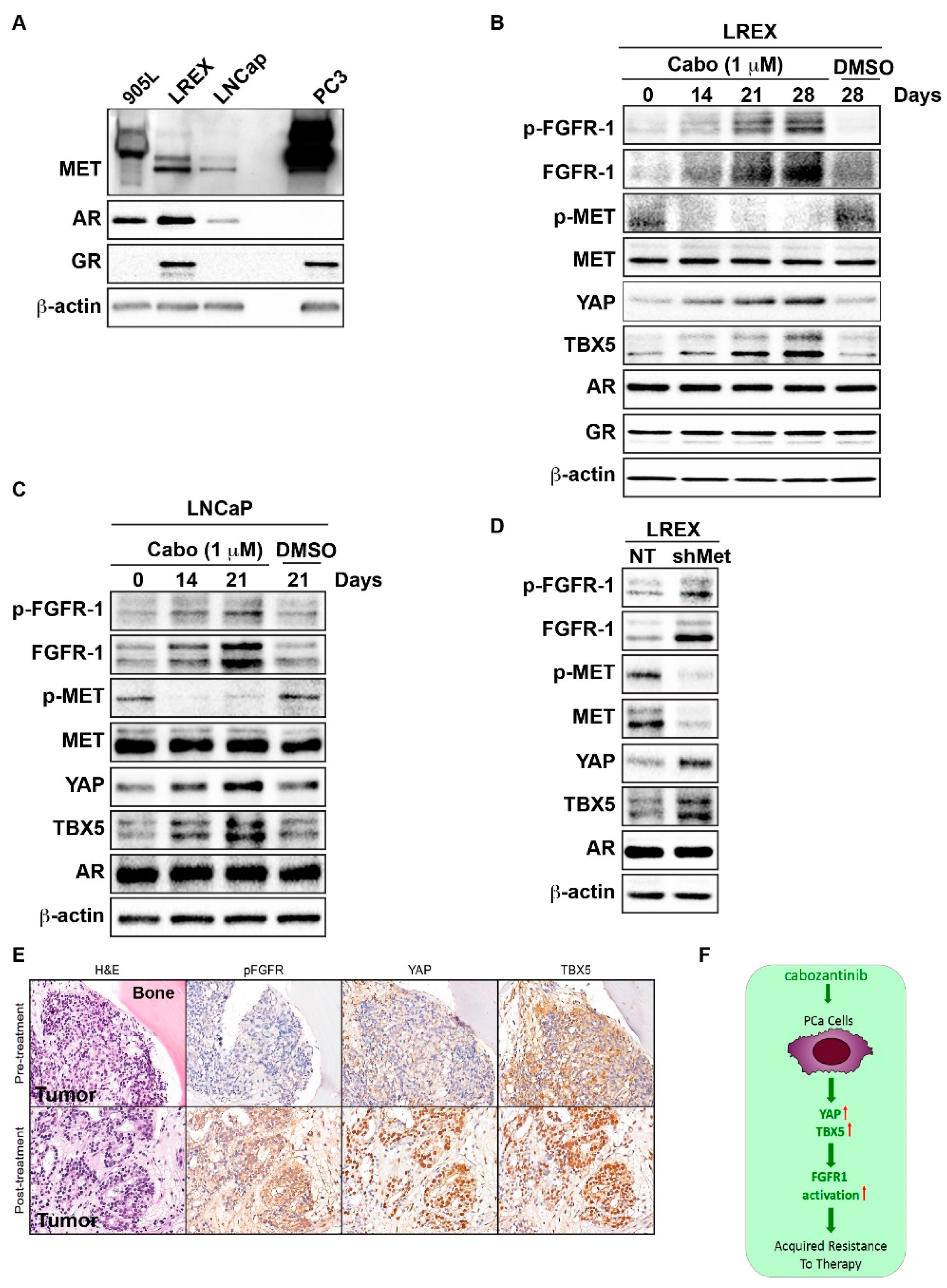
2.1. Increased FGFR1 Expression Mediates Acquired Resistance to MET Inhibition
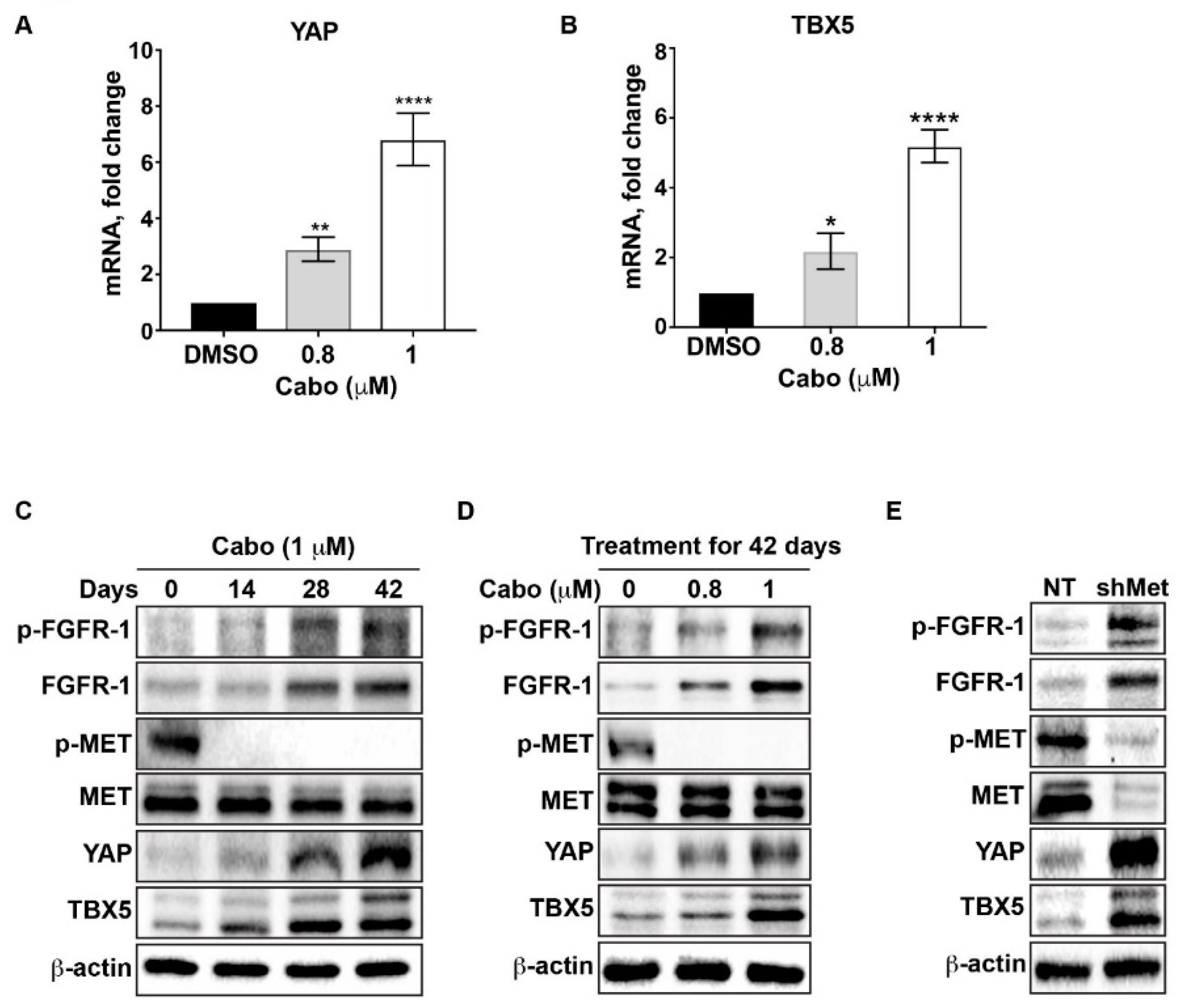
2.2. Cabozantinib Induces the Transcriptional Upregulation of YAP and TBX5
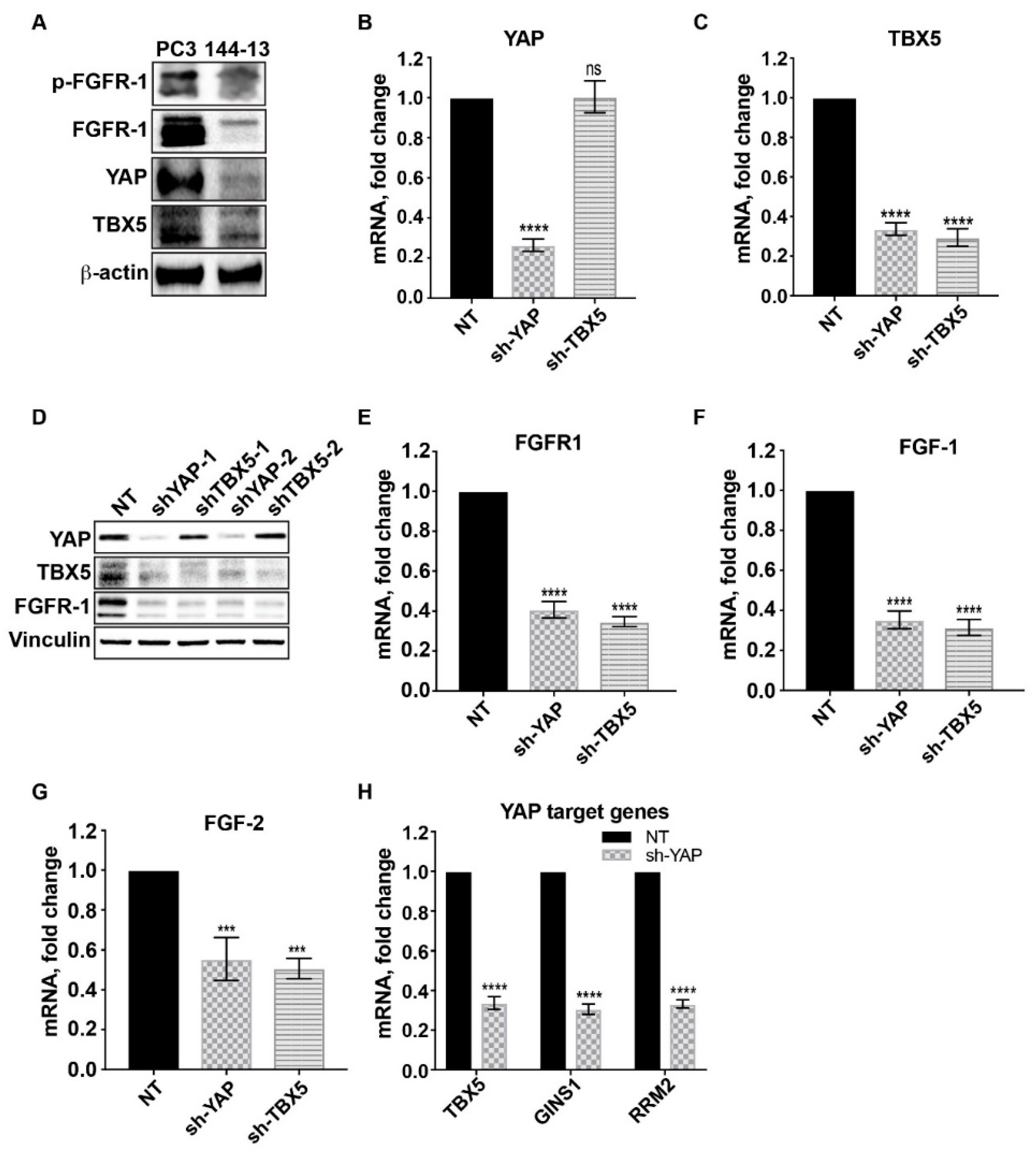
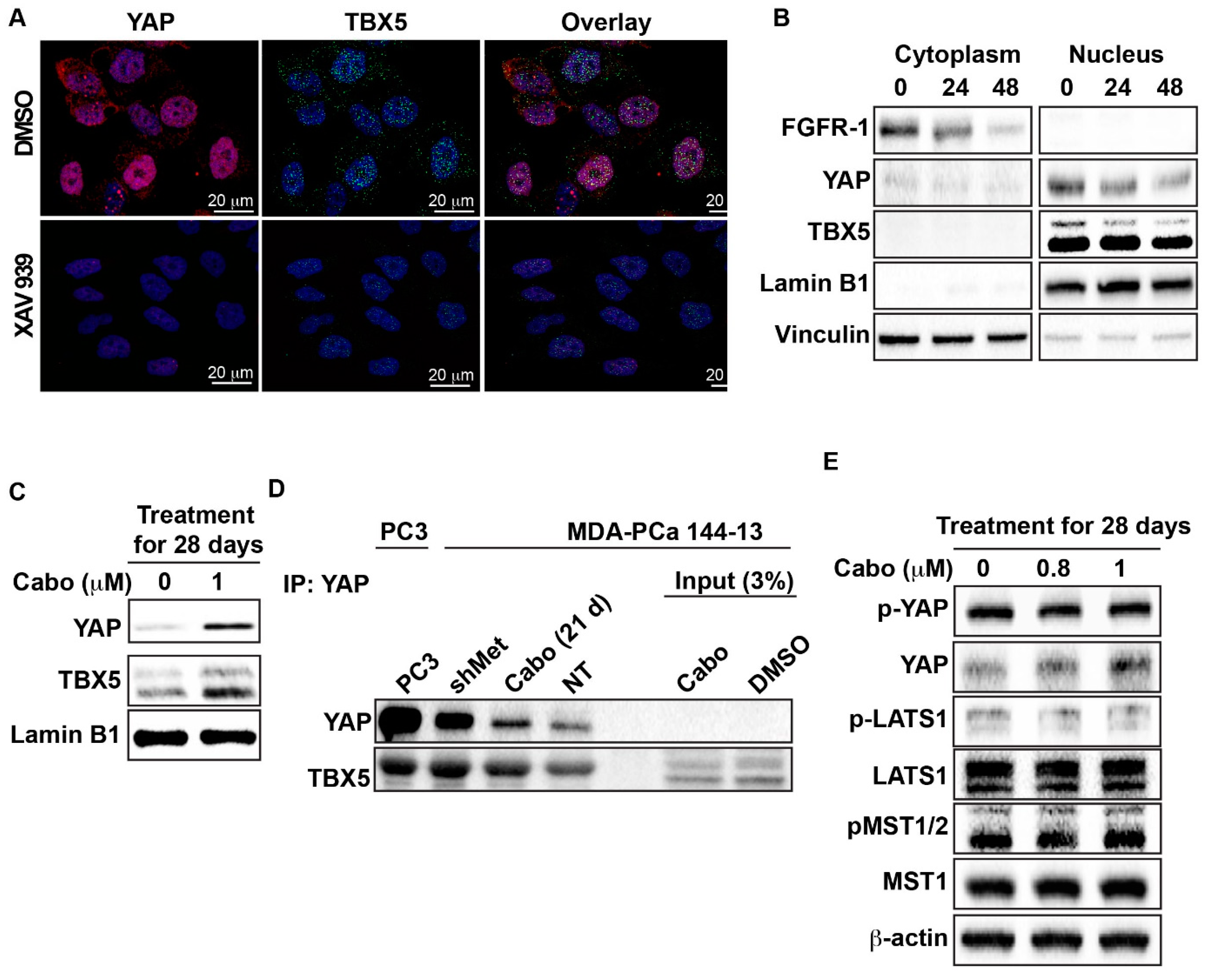
2.3. FGFR1, FGF1, and FGF2 Are Transcriptionally Upregulated Through a YAP/TBX5-Dependent Mechanism
2.4. Downregulation of MET Signaling is Associated With Upregulation of FGFR1 Expression in AR-Positive PCa Cells
2.5. Cabozantinib Induces FGFR1 Activation and Increases in YAP and TBX5 Expression in PCa Bone Metastasis Specimens from a Clinical Trial
3. Discussion
4. Materials and Methods
4.1. Cell Cultures
4.2. Reagents
4.3. Lentivirus-mediated YAP and TBX5 Silencing
4.4. FGFR1 Overexpression
4.5. Proliferation Assays
4.6. Quantitative PCR Analysis
4.7. Immunoblotting and Immunoprecipitation Assays
4.8. Animal Studies
4.9. Immunohistochemistry
4.10. Human Tissues
4.11. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| FDA | Food and Drug Administration |
| FGF1 | Fibroblast growth factor 1 |
| FGF2 | fibroblast growth factor 2 |
| FGFR1 | Fibroblast growth factor receptor 1 |
| GAPDH | Glyceraldehyde 3-phosphate dehydrogenase |
| IgG | Immunoglobulin G: IHC: Immuno HistoChemistry |
| LATS1 | Large tumor suppressor kinase 1 |
| MDA Pca | MD Anderson Prostate Cancer |
| MET | Mesenchymal to Epithelial Transition |
| MST1/2 | Mammalian Ste20-like kinases 1/2 |
| NOD/SCID | non-obese diabetic/severe combined immune deficiency |
| PBS | phosphate buffered saline |
| PCR | Polymerase chain reaction |
| qRT-PCR | Quantitative Real-Time Polymerase Chain Reaction |
| RPMO | Roswell Park Memorial Institute |
| sh-c-Met | Short hairpin-C-MET |
| TKI | Tyrosine kinase inhibitors |
| TBX5 | T-Box Transcription Factor 5 |
| VEGFR2 | Vascular endothelial growth factor receptor 2 |
| YAP | Yes-associated protein 1 |
References
- Siegel, L.R.; Miller, K.D.; Jemal, A. Cancer statistics, 2019. CA Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kapoor, A.; Wu, C.; Shayegan, B.; Rybak, A.P. Contemporary agents in the management of metastatic castration-resistant prostate cancer. Can. Urol. Assoc. J. 2016, 10, E414–E423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drake, J.M.; Graham, N.A.; Lee, J.K.; Stoyanova, T.; Faltermeier, C.M.; Sud, S.; Titz, B.; Huang, J.; Pienta, K.J.; Graeber, T.G.; et al. Metastatic castration-resistant prostate cancer reveals intrapatient similarity and interpatient heterogeneity of therapeutic kinase targets. Proc. Natl. Acad. Sci. USA 2013, 110, E4762–E4769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, M.R.; Sweeney, C.J.; Corn, P.G.; Rathkopf, D.E.; Smith, D.C.; Hussain, M.; George, D.J.; Higano, C.S.; Harzstark, A.L.; Sartor, A.O.; et al. Cabozantinib in chemotherapy-pretreated metastatic castration-resistant prostate cancer: Results of a phase II nonrandomized expansion study. J. Clin. Oncol. 2014, 32, 3391–3399. [Google Scholar] [CrossRef] [Green Version]
- Smith, M.; De Bono, J.; Sternberg, C.; Le Moulec, S.; Oudard, S.; De Giorgi, U.; Krainer, M.; Bergman, A.; Hoelzer, W.; De Wit, R.; et al. Phase III Study of Cabozantinib in Previously Treated Metastatic Castration-Resistant Prostate Cancer: COMET-1. J. Clin. Oncol. 2016, 34, 3005–3013. [Google Scholar] [CrossRef]
- Von Manstein, V.; Yang, C.M.; Richter, D.; Delis, N.; Vafaizadeh, V.; Groner, B. Resistance of Cancer Cells to Targeted Therapies Through the Activation of Compensating Signaling Loops. Curr. Signal Transduct. Ther. 2013, 8, 193–202. [Google Scholar] [CrossRef]
- Ahronian, L.G.; Corcoran, R.B. Strategies for monitoring and combating resistance to combination kinase inhibitors for cancer therapy. Genome Med. 2017, 9, 37. [Google Scholar] [CrossRef] [Green Version]
- Varkaris, A.; Corn, P.G.; Parikh, N.U.; Efstathiou, E.; Song, J.H.; Lee, Y.C.; Aparicio, A.; Hoang, A.G.; Gaur, S.; Thorpe, L.; et al. Integrating Murine and Clinical Trials with Cabozantinib to Understand Roles of MET and VEGFR2 as Targets for Growth Inhibition of Prostate Cancer. Clin. Cancer Res. 2016, 22, 107–121. [Google Scholar] [CrossRef] [Green Version]
- Acevedo, V.D.; Gangula, R.D.; Freeman, K.W.; Li, R.; Zhang, Y.; Wang, F.; Ayala, G.E.; Peterson, L.E.; Ittmann, M.; Spencer, D.M.; et al. Inducible FGFR-1 activation leads to irreversible prostate adenocarcinoma and an epithelial-to-mesenchymal transition. Cancer Cell 2007, 12, 559–571. [Google Scholar] [CrossRef] [Green Version]
- Yang, F.; Zhang, Y.; Ressler, S.J.; Ittmann, M.M.; Ayala, G.E.; Dang, T.D.; Wang, F.; Rowley, D.R. FGFR1 is essential for prostate cancer progression and metastasis. Cancer Res. 2013, 73, 3716–3724. [Google Scholar] [CrossRef] [Green Version]
- Yang, F.; Strand, D.W.; Rowley, D.R. Fibroblast growth factor-2 mediates transforming growth factor-beta action in prostate cancer reactive stroma. Oncogene 2008, 27, 450–459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, S.; Shao, L.; Yu, W.; Gavine, P.; Ittmann, M. Targeting fibroblast growth factor receptor signaling inhibits prostate cancer progression. Clin. Cancer Res. 2012, 18, 3880–3888. [Google Scholar] [CrossRef] [Green Version]
- Armstrong, K.; Ahmad, I.; Kalna, G.; Tan, S.S.; Edwards, J.; Robson, C.N.; Leung, H.Y. Upregulated FGFR1 expression is associated with the transition of hormone-naive to castrate-resistant prostate cancer. Br. J. Cancer 2011, 105, 1362–1369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murphy, T.; Darby, S.; Mathers, M.E.; Gnanapragasam, V.J. Evidence for distinct alterations in the FGF axis in prostate cancer progression to an aggressive clinical phenotype. J. Pathol. 2010, 220, 452–460. [Google Scholar] [CrossRef] [PubMed]
- Rizvi, S.; Yamada, D.; Hirsova, P.; Bronk, S.F.; Werneburg, N.W.; Krishnan, A.; Salim, W.; Zhang, L.; Trushina, E.; Truty, M.J.; et al. A Hippo and Fibroblast Growth Factor Receptor Autocrine Pathway in Cholangiocarcinoma. J. Biol. Chem. 2016, 291, 8031–8047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hua, G.; Lv, X.; He, C.; Remmenga, S.W.; Rodabough, K.J.; Dong, J.; Yang, L.; Lele, S.M.; Yang, P.; Zhou, J.; et al. YAP induces high-grade serous carcinoma in fallopian tube secretory epithelial cells. Oncogene 2016, 35, 2247–2265. [Google Scholar] [CrossRef] [Green Version]
- Rosenbluh, J.; Wang, X.; Hahn, W.C. Genomic insights into WNT/β-catenin signaling. Trends Pharmacol. Sci. 2014, 35, 103–109. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Li, N.; Li, X.; Tran, M.K.; Han, X.; Chen, J. Tankyrase Inhibitors Target YAP by Stabilizing Angiomotin Family Proteins. Cell Rep. 2015, 13, 524–532. [Google Scholar] [CrossRef] [Green Version]
- Rodon, J.; Postel-Vinay, S.; Hollebecque, A.; Nuciforo, P.; Azaro, A.; Cattan, V.; Marfai, L.; Sudey, I.; Brendel, K.; Delmas, A.; et al. First-in-human phase I study of oral S49076, a unique MET/AXL/FGFR inhibitor, in advanced solid tumours. Eur. J. Cancer 2017, 81, 142–150. [Google Scholar] [CrossRef]
- McDermott, U.; Pusapati, R.V.; Christensen, J.G.; Gray, N.S.; Settleman, J. Acquired resistance of non-small cell lung cancer cells to MET kinase inhibition is mediated by a switch to epidermal growth factor receptor dependency. Cancer Res. 2010, 70, 1625–1634. [Google Scholar] [CrossRef] [Green Version]
- Bahcall, M.; Sim, T.; Paweletz, C.P.; Patel, J.D.; Alden, R.S.; Kuang, Y.; Sacher, A.G.; Kim, N.D.; Lydon, C.A.; Awad, M.M.; et al. Acquired METD1228V Mutation and Resistance to MET Inhibition in Lung Cancer. Cancer Discov. 2016, 6, 1334–1341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heist, R.S.; Sequist, L.V.; Borger, D.; Gainor, J.F.; Arellano, R.S.; Le, L.P.; Dias-Santagata, D.; Clark, J.W.; Engelman, J.A.; Shaw, A.T.; et al. Acquired Resistance to Crizotinib in NSCLC with MET Exon 14 Skipping. J. Thorac. Oncol. 2016, 11, 1242–1245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oddo, D.; Siravegna, G.; Gloghini, A.; Vernieri, C.; Mussolin, B.; Morano, F.; Crisafulli, G.; Berenato, R.; Corti, G.; Volpi, C.C.; et al. Emergence of MET hyper-amplification at progression to MET and BRAF inhibition in colorectal cancer. Br. J. Cancer 2017, 117, 347–352. [Google Scholar] [CrossRef] [PubMed]
- Kentsis, A.; Reed, C.; Rice, K.L.; Sanda, T.; Rodig, S.J.; Tholouli, E.; Christie, A.; Valk, P.J.; Delwel, R.; Ngo, V.; et al. Autocrine activation of the MET receptor tyrosine kinase in acute myeloid leukemia. Nat. Med. 2012, 18, 1118–1122. [Google Scholar] [CrossRef]
- Ware, K.E.; Hinz, T.K.; Kleczko, E.; Singleton, K.R.; Marek, L.A.; Helfrich, B.A.; Cummings, C.T.; Graham, D.K.; Astling, D.; Tan, A.C.; et al. A mechanism of resistance to gefitinib mediated by cellular reprogramming and the acquisition of an FGF2-FGFR1 autocrine growth loop. Oncogenesis 2013, 2, e39. [Google Scholar] [CrossRef] [Green Version]
- Harbinski, F.; Craig, V.J.; Sanghavi, S.; Jeffery, D.; Liu, L.; Sheppard, K.A.; Wagner, S.; Stamm, C.; Buness, A.; Chatenay-Rivauday, C.; et al. Rescue screens with secreted proteins reveal compensatory potential of receptor tyrosine kinases in driving cancer growth. Cancer Discov. 2012, 2, 948–959. [Google Scholar] [CrossRef] [Green Version]
- Kim, B.; Wang, S.; Lee, J.M.; Jeong, Y.; Ahn, T.; Son, D.S.; Park, H.W.; Yoo, H.S.; Song, Y.J.; Lee, E.; et al. Synthetic lethal screening reveals FGFR as one of the combinatorial targets to overcome resistance to Met-targeted therapy. Oncogene 2015, 34, 1083–1093. [Google Scholar] [CrossRef] [Green Version]
- Faltermeier, C.M.; Drake, J.M.; Clark, P.M.; Smith, B.A.; Zong, Y.; Volpe, C.; Mathis, C.; Morrissey, C.; Castor, B.; Huang, J.; et al. Functional screen identifies kinases driving prostate cancer visceral and bone metastasis. Proc. Natl. Acad. Sci. USA 2016, 113, E172–E181. [Google Scholar] [CrossRef] [Green Version]
- Aparicio, A.; Tzelepi, V.; Araujo, J.C.; Guo, C.C.; Liang, S.; Troncoso, P.; Logothetis, C.J.; Navone, N.M.; Maity, S.N. Neuroendocrine prostate cancer xenografts with large-cell and small-cell features derived from a single patient’s tumor: Morphological, immunohistochemical, and gene expression profiles. Prostate 2011, 71, 846–856. [Google Scholar] [CrossRef] [Green Version]
- Zhu, C.; Li, L.; Zhao, B. The regulation and function of YAP transcription co-activator. Acta Biochim. Biophys. Sin. 2015, 47, 16–28. [Google Scholar] [CrossRef] [Green Version]
- Wu, H.; Xiao, Y.; Zhang, S.; Ji, S.; Wei, L.; Fan, F.; Geng, J.; Tian, J.; Sun, X.; Qin, F.; et al. The Ets transcription factor GABP is a component of the hippo pathway essential for growth and antioxidant defense. Cell Rep. 2013, 3, 1663–1677. [Google Scholar] [CrossRef] [Green Version]
- Danovi, S.A.; Rossi, M.; Gudmundsdottir, K.; Yuan, M.; Melino, G.; Basu, S. Yes-associated protein (YAP) is a critical mediator of c-Jun-dependent apoptosis. Cell Death Differ. 2008, 15, 217–219. [Google Scholar] [CrossRef] [Green Version]
- Konsavage, W.M.; Kyler, S.L.; Rennoll, S.A.; Jin, G.; Yochum, G.S. Wnt/β-catenin signaling regulates Yes-associated protein (YAP) gene expression in colorectal carcinoma cells. J. Biol. Chem. 2012, 287, 11730–11739. [Google Scholar] [CrossRef] [Green Version]
- Liu, A.M.; Poon, R.T.; Luk, J.M. MicroRNA-375 targets Hippo-signaling effector YAP in liver cancer and inhibits tumor properties. Biochem. Biophys. Res. Commun. 2010, 394, 623–627. [Google Scholar] [CrossRef] [PubMed]
- Nishikawa, E.; Osada, H.; Okazaki, Y.; Arima, C.; Tomida, S.; Tatematsu, Y.; Taguchi, A.; Shimada, Y.; Yanagisawa, K.; Yatabe, Y. miR-375 is activated by ASH1 and inhibits YAP1 in a lineage-dependent manner in lung cancer. Cancer Res. 2011, 71, 6165–6173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Selth, L.A.; Das, R.; Townley, S.L.; Coutinho, I.; Hanson, A.R.; Centenera, M.M.; Stylianou, N.; Sweeney, K.; Soekmadji, C.; Jovanovic, L.; et al. A ZEB1-miR-375-YAP1 pathway regulates epithelial plasticity in prostate cancer. Oncogene 2017, 36, 24–34. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Lieberman, R.; Pan, J.; Zhang, Q.; Du, M.; Zhang, P.; Nevalainen, M.; Kohli, M.; Shenoy, N.K.; Meng, H.; et al. miR-375 induces docetaxel resistance in prostate cancer by targeting SEC23A and YAP1. Mol. Cancer 2016, 15, 70. [Google Scholar] [CrossRef] [Green Version]
- Schneider, J.A.; Logan, S.K. Revisiting the role of Wnt/β-catenin signaling in prostate cancer. Mol. Cell. Endocrinol. 2018, 462, 3–8. [Google Scholar] [CrossRef]
- Kachakova, D.; Mitkova, A.; Popov, E.; Popov, I.; Vlahova, A.; Dikov, T.; Christova, S.; Mitev, V.; Slavov, C.; Kaneva, R. Combinations of serum prostate-specific antigen and plasma expression levels of let-7c, miR-30c, miR-141, and miR-375 as potential better diagnostic biomarkers for prostate cancer. DNA Cell Biol. 2015, 34, 189–200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheng, X.; Li, W.B.; Wang, D.L.; Chen, K.H.; Cao, J.J.; Luo, Z.; He, J.; Li, M.C.; Liu, W.J.; Yu, C. YAP is closely correlated with castration-resistant prostate cancer, and downregulation of YAP reduces proliferation and induces apoptosis of PC-3 cells. Mol. Med. Rep. 2015, 12, 4867–4876. [Google Scholar] [CrossRef] [Green Version]
- Aherne, N.J.; Rangaswamy, G.; Thirion, P. Prostate Cancer in a Male with Holt-Oram Syndrome: First Clinical Association of the TBX5 Mutation. Case Rep. Urol. 2013, 2013, 405343. [Google Scholar] [PubMed]
- Tzelepi, V.; Zhang, J.; Lu, J.F.; Kleb, B.; Wu, G.; Wan, X.; Hoang, A.; Efstathiou, E.; Sircar, K.; Navone, N.M. Modeling a lethal prostate cancer variant with small-cell carcinoma features. Clin. Cancer Res. 2012, 18, 666–677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wan, X.; Corn, P.G.; Yang, J.; Palanisamy, N.; Starbuck, M.W.; Efstathiou, E.; Li Ning Tapia, E.M.; Zurita, A.J.; Aparicio, A.; Ravoori, M.K.; et al. Prostate cancer cell-stromal cell crosstalk via FGFR1 mediates antitumor activity of dovitinib in bone metastases. Sci. Transl. Med. 2014, 6, 252ra122. [Google Scholar] [CrossRef] [Green Version]
- Windham, T.C.; Parikh, N.U.; Siwak, D.R.; Summy, J.M.; McConkey, D.J.; Kraker, A.J.; Gallick, G.E. Src activation regulates anoikis in human colon tumor cell lines. Oncogene 2002, 21, 7797–7807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomayko, M.M.; Reynolds, C.P. Determination of subcutaneous tumor size in athymic (nude) mice. Cancer Chemother. Pharmacol. 1989, 24, 148–154. [Google Scholar] [CrossRef] [PubMed]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Koinis, F.; Corn, P.; Parikh, N.; Song, J.; Vardaki, I.; Mourkioti, I.; Lin, S.-H.; Logothetis, C.; Panaretakis, T.; Gallick, G. Resistance to MET/VEGFR2 Inhibition by Cabozantinib Is Mediated by YAP/TBX5-Dependent Induction of FGFR1 in Castration-Resistant Prostate Cancer. Cancers 2020, 12, 244. https://doi.org/10.3390/cancers12010244
Koinis F, Corn P, Parikh N, Song J, Vardaki I, Mourkioti I, Lin S-H, Logothetis C, Panaretakis T, Gallick G. Resistance to MET/VEGFR2 Inhibition by Cabozantinib Is Mediated by YAP/TBX5-Dependent Induction of FGFR1 in Castration-Resistant Prostate Cancer. Cancers. 2020; 12(1):244. https://doi.org/10.3390/cancers12010244
Chicago/Turabian StyleKoinis, Filippos, Paul Corn, Nila Parikh, Jian Song, Ioulia Vardaki, Ioanna Mourkioti, Sue-Hwa Lin, Christopher Logothetis, Theocharis Panaretakis, and Gary Gallick. 2020. "Resistance to MET/VEGFR2 Inhibition by Cabozantinib Is Mediated by YAP/TBX5-Dependent Induction of FGFR1 in Castration-Resistant Prostate Cancer" Cancers 12, no. 1: 244. https://doi.org/10.3390/cancers12010244