The Gasdermin E Gene Has Potential as a Pan-Cancer Biomarker, While Discriminating between Different Tumor Types


Abstract
:1. Introduction
2. Results
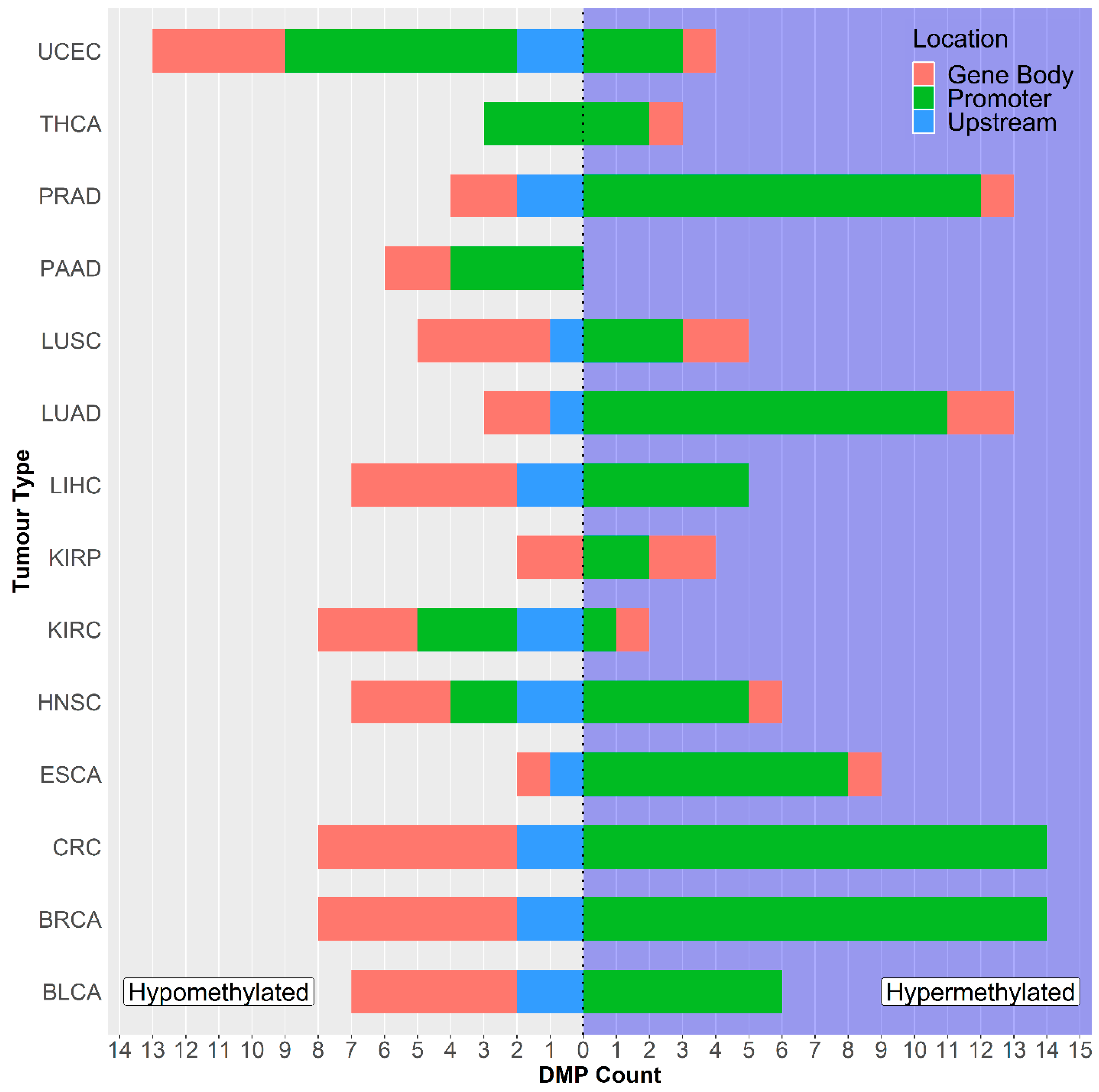
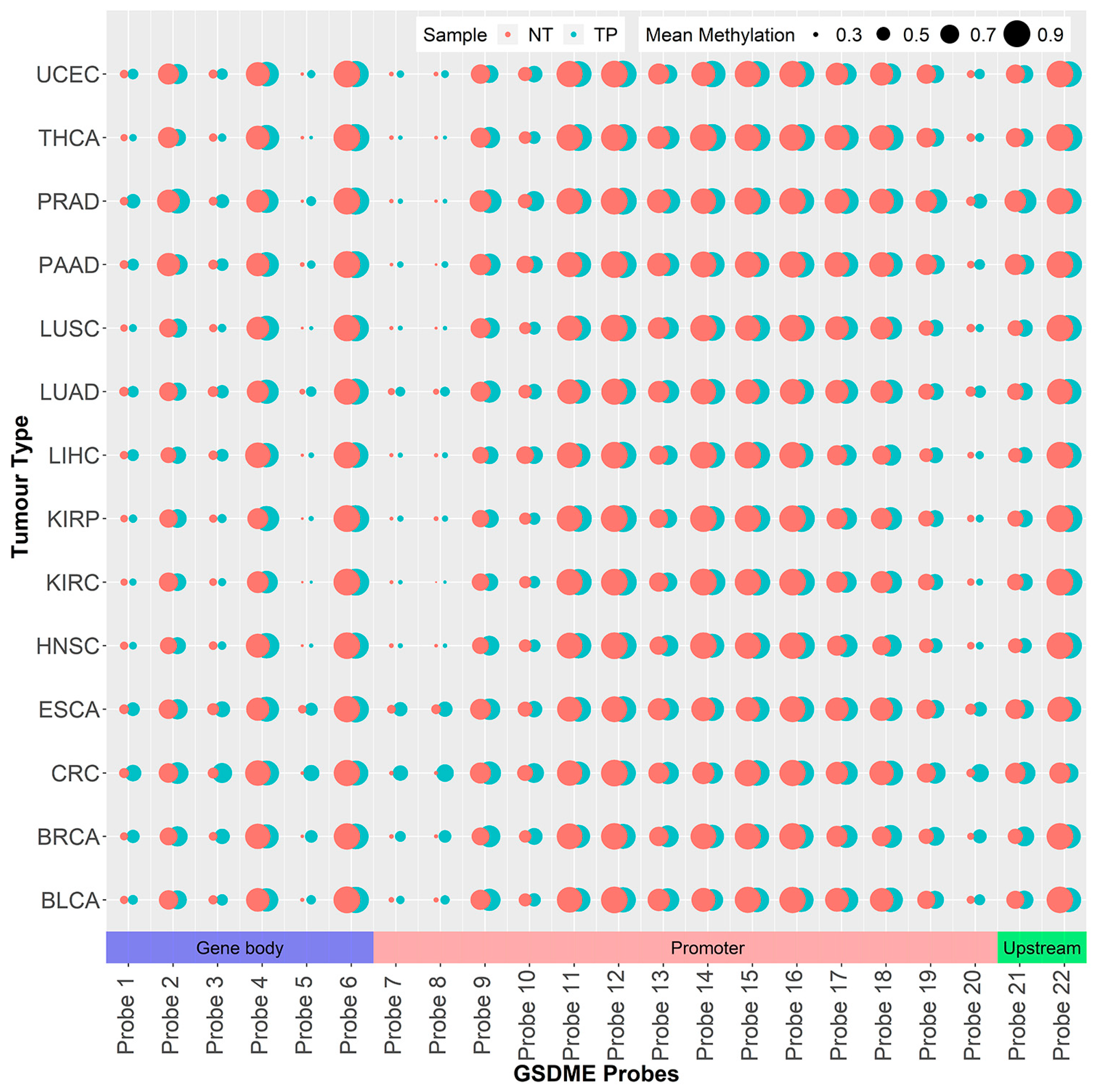
2.1. GSDME Differential Methylation Across 14 Tumor Types
2.2. GSDME Methylation as a Pan-Cancer Detection Biomarker
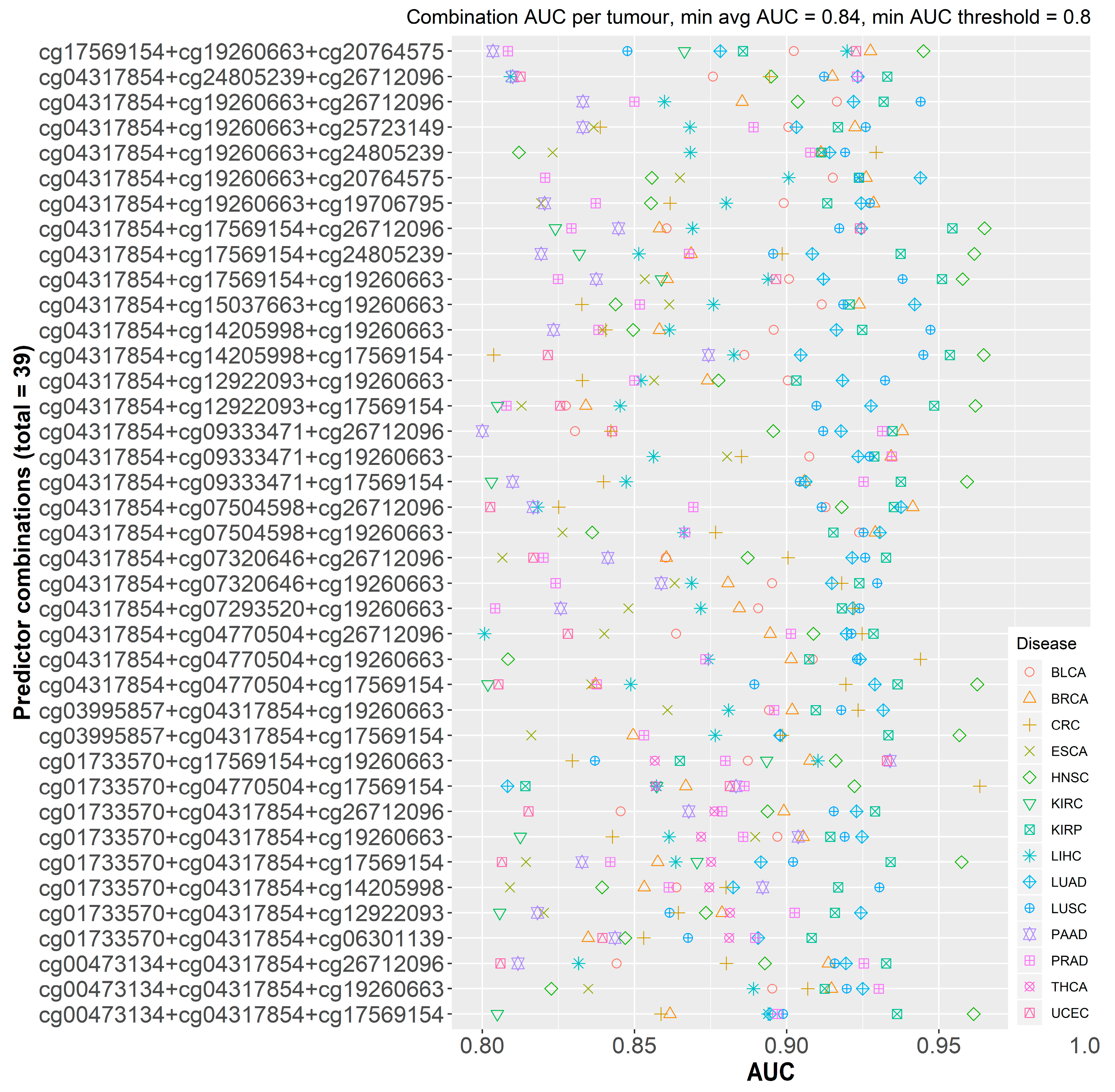
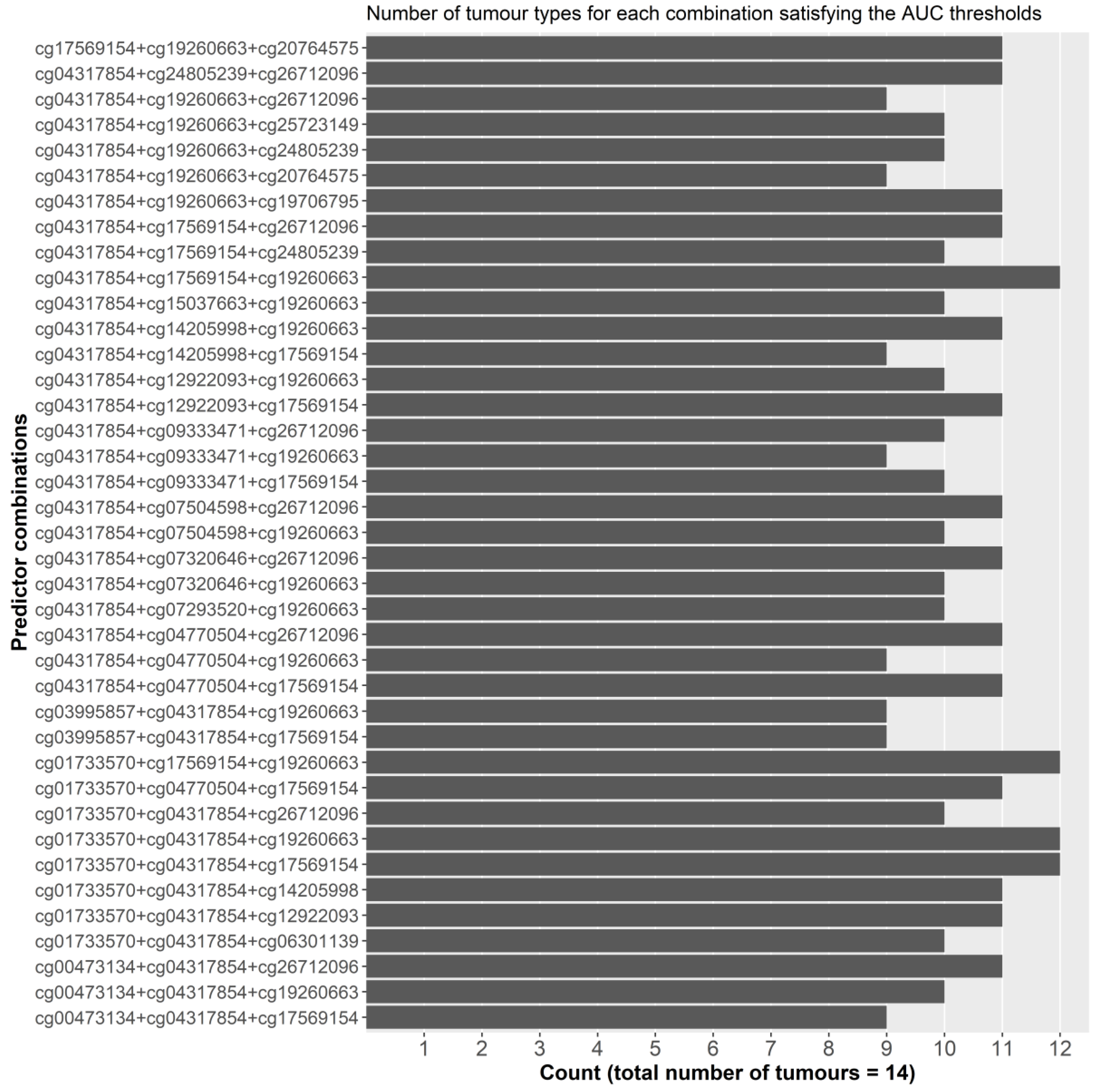
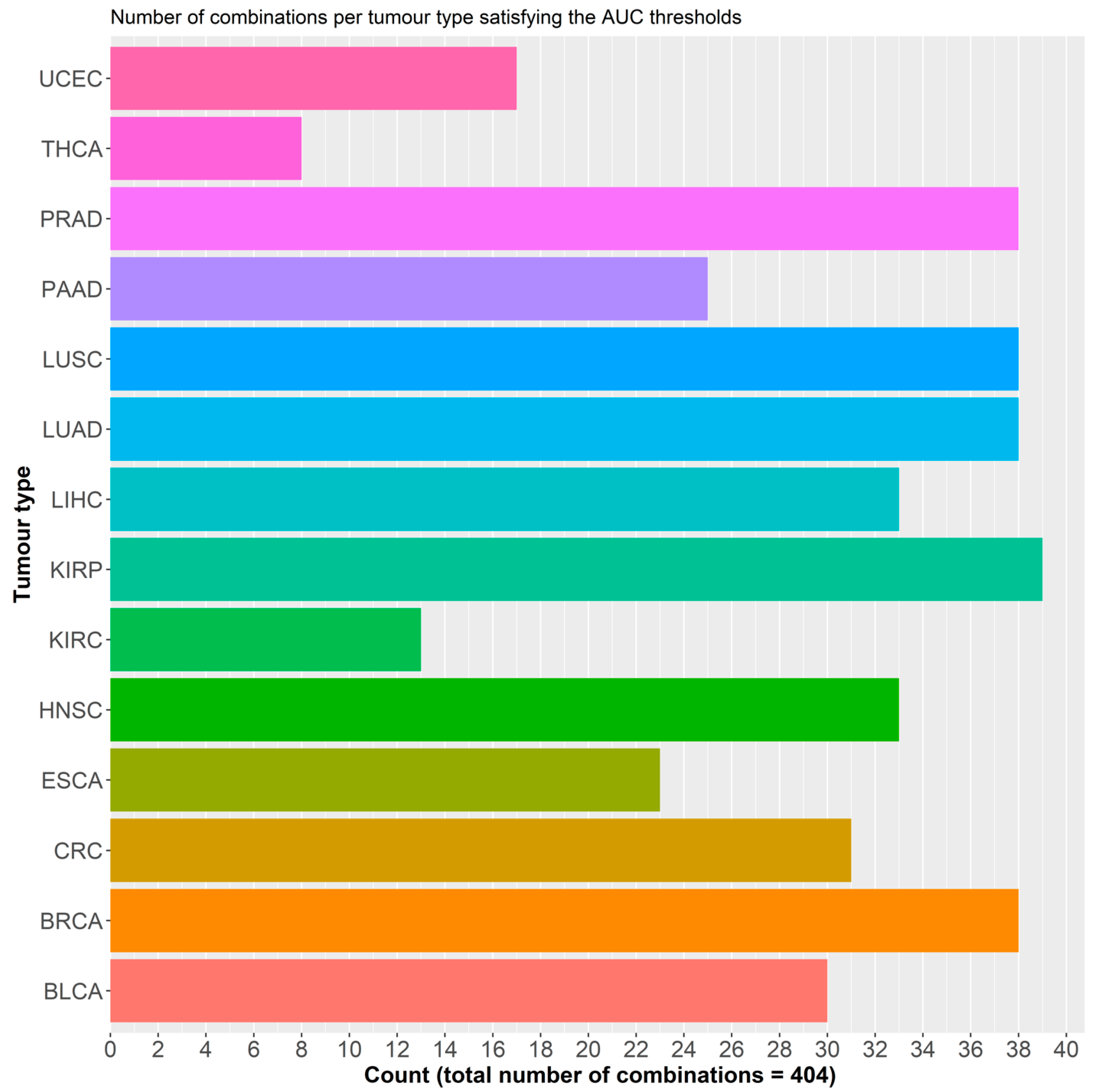
2.2.1. Initial Predictor Combination Selection
2.2.2. Individual Dataset Analysis
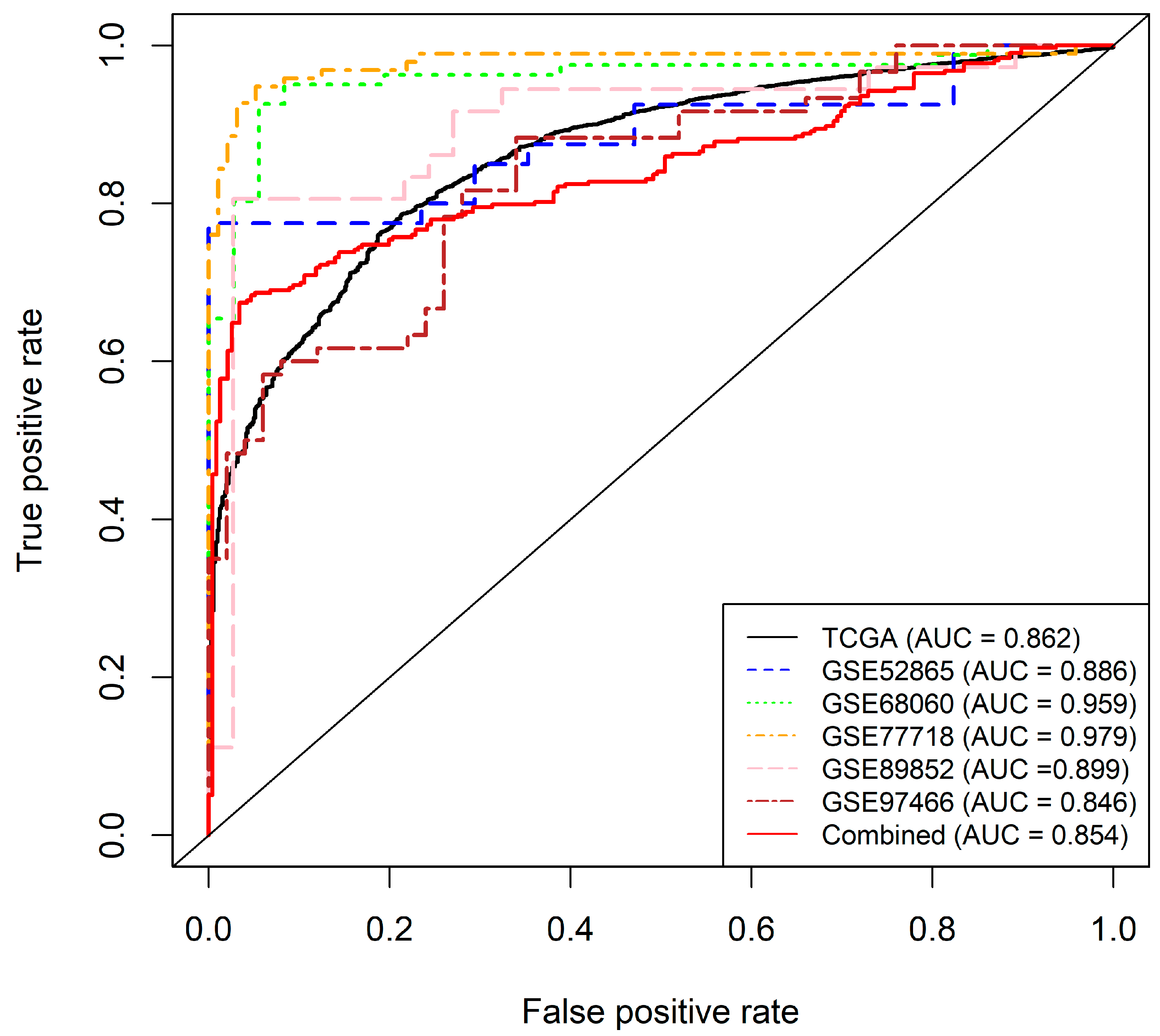
2.2.3. Final Model and Validation
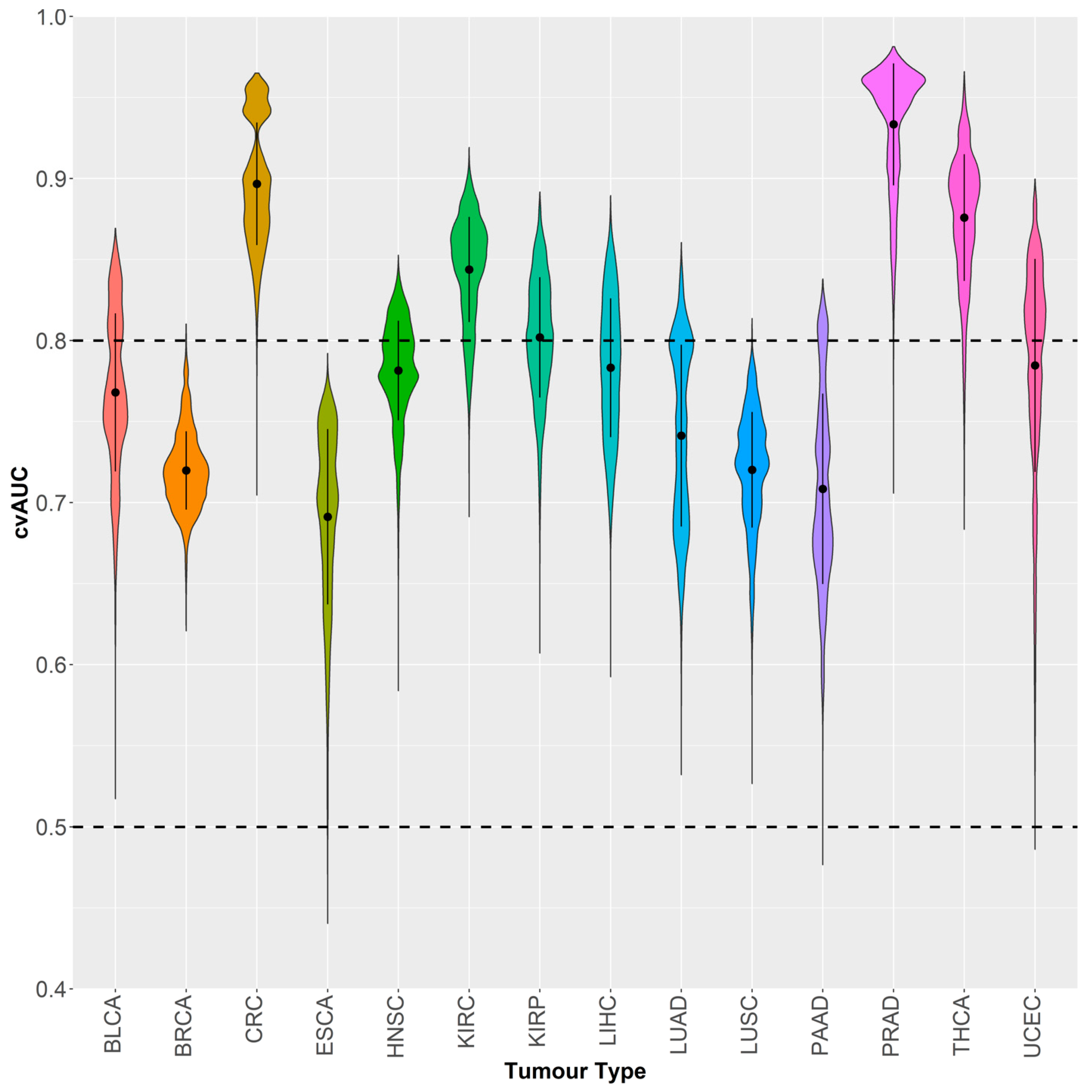
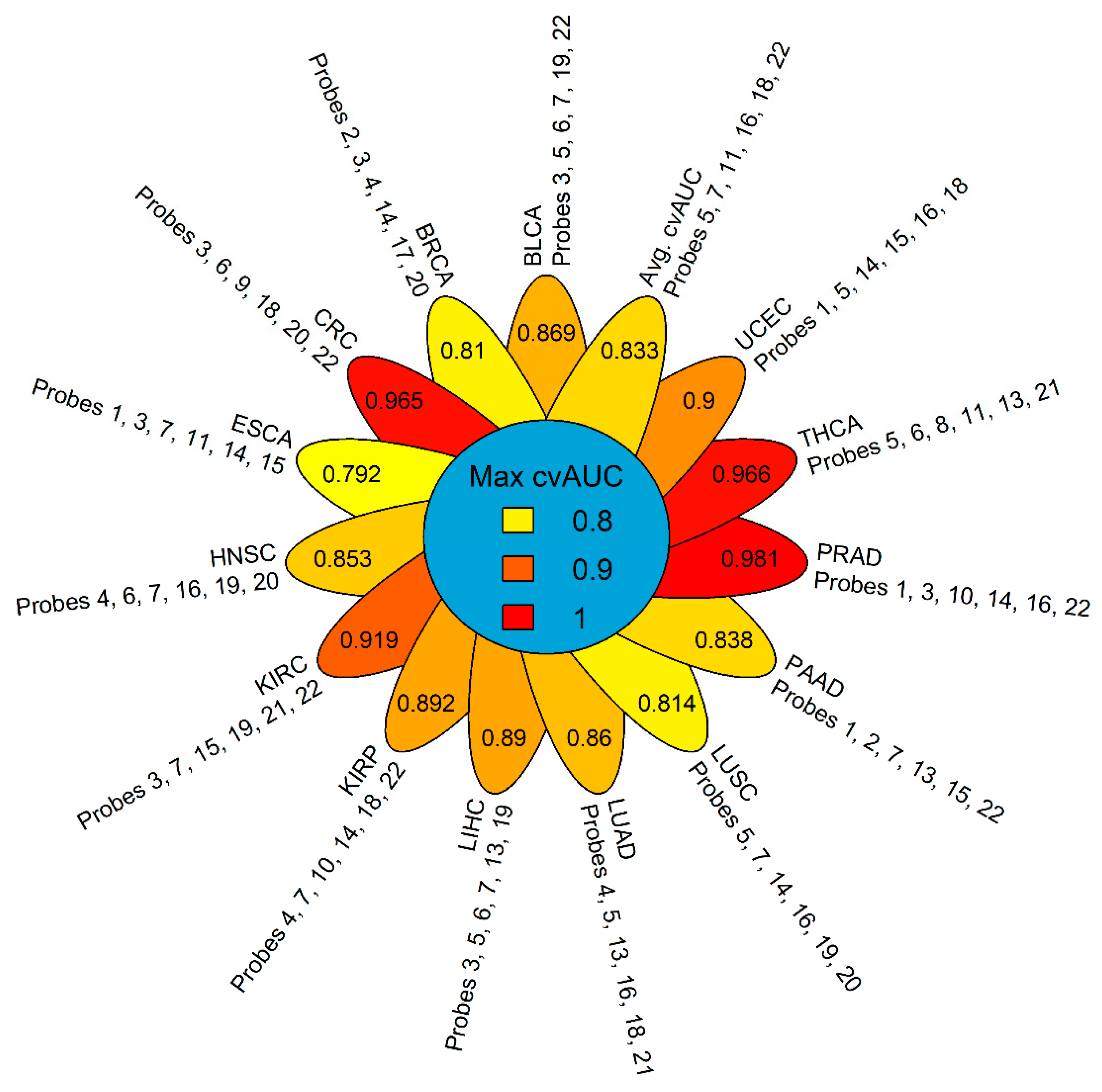
2.3. GSDME Methylation as a Tumor-Specific Biomarker
2.4. The Relation of GSDME Methylation to RNA-seq Expression and Clinicopathological Parameters
3. Discussion
4. Materials and Methods
4.1. Datasets and Study Population
4.2. Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- World Health Organization. International Agency For Research on Cancer GLOBOCAN 2012: Estimated Cancer Incidence, Mortality and Prevalence Worldwide in 2012. Lung Cancer; World Health Organization: Geneva, Switzerland, 2012. [Google Scholar]
- Luengo-Fernandez, R.; Leal, J.; Gray, A.; Sullivan, R. Economic burden of cancer across the European Union: A population-based cost analysis. Lancet Oncol. 2013, 14, 1165–1174. [Google Scholar] [CrossRef]
- McGuire, S. World Cancer Report 2014. Geneva, Switzerland: World Health Organization, International Agency for Research on Cancer, WHO Press, 2015. Adv. Nutr. Int. Rev. J. 2016, 7, 418–419. [Google Scholar] [CrossRef] [PubMed]
- Prager, G.W.; Braga, S.; Bystricky, B.; Qvortrup, C.; Criscitiello, C.; Esin, E.; Sonke, G.S.; Martínez, G.A.; Frenel, J.S.; Karamouzis, M.; et al. Global cancer control: Responding to the growing burden, rising costs and inequalities in access. ESMO Open 2018, 3, 285. [Google Scholar] [CrossRef] [PubMed]
- Aronson, J.K.; Ferner, R.E. Biomarkers—A general review. Curr. Protoc. Pharmacol. 2017, 76, 9–23. [Google Scholar] [PubMed]
- Liesenfeld, D.B.; Habermann, N.; Owen, R.W.; Scalbert, A.; Ulrich, C.M. Review of mass spectrometry-based metabolomics in cancer research. Cancer Epidemiol. Biomarkers Prev. 2013, 22, 2182–2201. [Google Scholar] [CrossRef]
- Rifai, N.; Gillette, M.A.; Carr, S.A. Protein biomarker discovery and validation: The long and uncertain path to clinical utility. Nat. Biotechnol. 2006, 24, 971–983. [Google Scholar] [CrossRef]
- Mikeska, T.; Craig, J.M. DNA methylation biomarkers: Cancer and beyond. Genes 2014, 5, 821–864. [Google Scholar] [CrossRef]
- Leygo, C.; Williams, M.; Jin, H.C.; Chan, M.W.Y.; Chu, W.K.; Grusch, M.; Cheng, Y.Y. DNA Methylation as a Noninvasive Epigenetic Biomarker for the Detection of Cancer. Dis. Markers 2017, 2017, 1–13. [Google Scholar] [CrossRef]
- Lao, V.V.; Grady, W.M. Epigenetics and colorectal cancer. Nat. Rev. Gastroenterol. Hepatol. 2011, 8, 686–700. [Google Scholar] [CrossRef]
- Delpu, Y.; Cordelier, P.; Cho, W.C.; Torrisani, J. DNA methylation and cancer diagnosis. Int. J. Mol. Sci. 2013, 14, 15029–15058. [Google Scholar] [CrossRef]
- Yang, M.; Park, J.Y. DNA methylation in promoter region as biomarkers in prostate cancer. Methods Mol. Biol. 2012, 863, 67–109. [Google Scholar] [PubMed]
- Guo, S.; Yan, F.; Xu, J.; Bao, Y.; Zhu, J.; Wang, X.; Wu, J.; Li, Y.; Pu, W.; Liu, Y.; et al. Identification and validation of the methylation biomarkers of non-small cell lung cancer (NSCLC). Clin. Epigenetics 2015, 7, 3. [Google Scholar] [CrossRef] [PubMed]
- Kulis, M.; Esteller, M. DNA Methylation and Cancer. Adv. Genet. 2010, 70, 27–56. [Google Scholar] [PubMed]
- Berdasco, M.; Esteller, M. Aberrant Epigenetic Landscape in Cancer: How Cellular Identity Goes Awry. Dev. Cell 2010, 19, 698–711. [Google Scholar] [CrossRef]
- Jones, P.A.; Baylin, S.B. The epigenomics of cancer. Cell 2007, 128, 683–692. [Google Scholar] [CrossRef]
- Sharma, S.; Kelly, T.K.; Jones, P.A. Epigenetics in cancer. Carcinogenesis 2009, 31, 27–36. [Google Scholar] [CrossRef]
- Ahn, J.B.; Chung, W.B.; Maeda, O.; Shin, S.J.; Kim, H.S.; Chung, H.C.; Kim, N.K.; Issa, J.P.J. DNA methylation predicts recurrence from resected stage III proximal colon cancer. Cancer 2011, 117, 1847–1854. [Google Scholar] [CrossRef]
- Javier Carmona, F.; Azuara, D.; Berenguer-Llergo, A.; Fernández, A.F.; Biondo, S.; De Oca, J.; Rodriguez-Moranta, F.; Salazar, R.; Villanueva, A.; Fraga, M.F.; et al. DNA methylation biomarkers for noninvasive diagnosis of colorectal cancer. Cancer Prev. Res. 2013, 6, 656–665. [Google Scholar] [CrossRef]
- Shivapurkar, N.; Gazdar, A. DNA Methylation Based Biomarkers in Non-Invasive Cancer Screening. Curr. Mol. Med. 2010, 10, 123–132. [Google Scholar] [CrossRef]
- Vedeld, H.M.; Andresen, K.; Eilertsen, I.A.; Nesbakken, A.; Seruca, R.; Gladhaug, I.P.; Thiis-Evensen, E.; Rognum, T.O.; Boberg, K.M.; Lind, G.E. The novel colorectal cancer biomarkers CDO1, ZSCAN18 and ZNF331 are frequently methylated across gastrointestinal cancers. Int. J. Cancer 2015, 136, 844–853. [Google Scholar] [CrossRef]
- Schmidt, B.; Liebenberg, V.; Dietrich, D.; Schlegel, T.; Kneip, C.; Seegebarth, A.; Flemming, N.; Seemann, S.; Distler, J.; Lewin, J.; et al. SHOX2 DNA Methylation is a Biomarker for the diagnosis of lung cancer based on bronchial aspirates. BMC Cancer 2010, 10, 600. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Li, G.; Zhang, J.; Zhou, Y.; Dang, S.; Chen, H.; Wu, Q.; Liu, M. Integrated analysis of genome-wide DNA methylation and gene expression profiles identifies potential novel biomarkers of rectal cancer. Oncotarget 2016, 7, 62547–62558. [Google Scholar] [CrossRef] [PubMed]
- Meng, W.; Huebner, A.; Shabsigh, A.; Chakravarti, A.; Lautenschlaeger, T. Combined RASSF1A and RASSF2A Promoter Methylation Analysis as Diagnostic Biomarker for Bladder Cancer. Mol. Biol. Int. 2012, 2012, 701814. [Google Scholar] [CrossRef] [PubMed]
- Roperch, J.P.; Incitti, R.; Forbin, S.; Bard, F.; Mansour, H.; Mesli, F.; Baumgaertner, I.; Brunetti, F.; Sobhani, I. Aberrant methylation of NPY, PENK, and WIF1 as a promising marker for blood-based diagnosis of colorectal cancer. BMC Cancer 2013, 13, 566. [Google Scholar] [CrossRef]
- Chen, Y.Z.; Liu, D.; Zhao, Y.X.; Wang, H.T.; Gao, Y.; Chen, Y. Aberrant promoter methylation of the SFRP1 gene may contribute to colorectal carcinogenesis: A meta-analysis. Tumor Biol. 2014, 35, 9201–9210. [Google Scholar] [CrossRef]
- Payne, S.R. From discovery to the clinic: The novel DNA methylation biomarker mSEPT9 for the detection of colorectal cancer in blood. Epigenomics 2010, 2, 575–585. [Google Scholar] [CrossRef]
- Darwiche, K.; Zarogoulidis, P.; Baehner, K.; Welter, S.; Tetzner, R.; Wohlschlaeger, J.; Theegarten, D.; Nakajima, T.; Freitag, L. Assessment of SHOX2 methylation in EBUS-TBNA specimen improves accuracy in lung cancer staging. Ann. Oncol. 2013, 24, 2866–2870. [Google Scholar] [CrossRef]
- Chen, Y.; Li, J.; Yu, X.; Li, S.; Zhang, X.; Mo, Z.; Hu, Y. APC gene hypermethylation and prostate cancer: A systematic review and meta-analysis. Eur. J. Hum. Genet. 2013, 21, 929–935. [Google Scholar] [CrossRef]
- Wu, T.; Giovannucci, E.; Welge, J.; Mallick, P.; Tang, W.Y.; Ho, S.M. Measurement of GSTP1 promoter methylation in body fluids may complement PSA screening: A meta-analysis. Br. J. Cancer 2011, 105, 65–73. [Google Scholar] [CrossRef]
- Pan, J.; Chen, J.; Zhang, B.; Chen, X.; Huang, B.; Zhuang, J.; Mo, C.; Qiu, S. Association between RASSF1A Promoter Methylation and Prostate Cancer: A Systematic Review and Meta-Analysis. PLoS ONE 2013, 8, e75283. [Google Scholar] [CrossRef]
- Cohen, J.D.; Li, L.; Wang, Y.; Thoburn, C.; Afsari, B.; Danilova, L.; Douville, C.; Javed, A.A.; Wong, F.; Mattox, A.; et al. Detection and localization of surgically resectable cancers with a multi-analyte blood test. Science 2018, 359, 926–930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Laer, L.; Huizing, E.H.; Verstreken, M.; van Zuijlen, D.; Wauters, J.G.; Bossuyt, P.J.; Van de Heyning, P.; McGuirt, W.T.; Smith, R.J.; Willems, P.J.; et al. Nonsyndromic hearing impairment is associated with a mutation in DFNA5. Nat. Genet. 1998, 20, 194–197. [Google Scholar] [CrossRef] [PubMed]
- Rogers, C.; Fernandes-Alnemri, T.; Mayes, L.; Alnemri, D.; Cingolani, G.; Alnemri, E.S. Cleavage of DFNA5 by caspase-3 during apoptosis mediates progression to secondary necrotic/pyroptotic cell death. Nat. Commun. 2017, 8, 14128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akino, K.; Toyota, M.; Suzuki, H.; Imai, T.; Maruyama, R.; Kusano, M.; Nishikawa, N.; Watanabe, Y.; Sasaki, Y.; Abe, T.; et al. Identification of DFNA5 as a target of epigenetic inactivation in gastric cancer. Cancer Sci. 2006, 98, 88–95. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.S.; Chang, X.; Yamashita, K.; Nagpal, J.K.; Baek, J.H.; Wu, G.; Trink, B.; Ratovitski, E.A.; Mori, M.; Sidransky, D. Aberrant promoter methylation and tumor suppressive activity of the DFNA5 gene in colorectal carcinoma. Oncogene 2008, 27, 3624–3634. [Google Scholar] [CrossRef] [Green Version]
- Yokomizo, K.; Harada, Y.; Kijima, K.; Shinmura, K.; Sakata, M.; Sakuraba, K.; Kitamura, Y.; Shirahata, A.; Goto, T.; Mizukami, H.; et al. Methylation of the DFNA5 gene is frequently detected in colorectal cancer. Anticancer Res. 2012, 32, 1319–1322. [Google Scholar]
- Ibrahim, J.; Op de Beeck, K.; Fransen, E.; Croes, L.; Beyens, M.; Suls, A.; Vanden Berghe, W.; Peeters, M.; Camp, G. Methylation analysis of Gasdermin E shows great promise as a biomarker for colorectal cancer. Cancer Med. 2019, 8, 2133–2145. [Google Scholar] [CrossRef] [Green Version]
- Croes, L.; Beyens, M.; Fransen, E.; Ibrahim, J.; Vanden Berghe, W.; Suls, A.; Peeters, M.; Pauwels, P.; Van Camp, G.; Op de Beeck, K. Large-scale analysis of DFNA5 methylation reveals its potential as biomarker for breast cancer. Clin. Epigenetics 2018, 10, 51. [Google Scholar] [CrossRef]
- Croes, L.; de Beeck, K.O.; Pauwels, P.; Vanden Berghe, W.; Peeters, M.; Fransen, E.; Van Camp, G. DFNA5 promoter methylation a marker for breast tumorigenesis. Oncotarget 2017, 8, 31948–31958. [Google Scholar] [CrossRef] [Green Version]
- Rogers, C.; Erkes, D.A.; Nardone, A.; Aplin, A.E.; Fernandes-Alnemri, T.; Alnemri, E.S. Gasdermin pores permeabilize mitochondria to augment caspase-3 activation during apoptosis and inflammasome activation. Nat. Commun. 2019, 10, 1689. [Google Scholar] [CrossRef]
- Yu, J.; Li, S.; Qi, J.; Chen, Z.; Wu, Y.; Guo, J.; Wang, K.; Sun, X.; Zheng, J. Cleavage of GSDME by caspase-3 determines lobaplatin-induced pyroptosis in colon cancer cells. Cell Death Dis. 2019, 10, 193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, C.-C.; Li, C.-G.; Wang, Y.-F.; Xu, L.-H.; He, X.-H.; Zeng, Q.-Z.; Zeng, C.-Y.; Mai, F.-Y.; Hu, B.; Ouyang, D.-Y. Chemotherapeutic paclitaxel and cisplatin differentially induce pyroptosis in A549 lung cancer cells via caspase-3/GSDME activation. Apoptosis 1234, 24, 312–325. [Google Scholar] [CrossRef] [PubMed]
- Ding, W.; Chen, G.; Shi, T. Integrative analysis identifies potential DNA methylation biomarkers for pan-cancer diagnosis and prognosis. Epigenetics 2019, 14, 67–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoadley, K.A.; Yau, C.; Wolf, D.M.; Cherniack, A.D.; Tamborero, D.; Ng, S.; Leiserson, M.D.M.; Niu, B.; Mclellan, M.D.; Uzunangelov, V.; et al. Multi-platform analysis of 12 cancer types reveals molecular classification within and across tissues-of-origin NIH Public Access. Cell 2014, 158, 929–944. [Google Scholar] [CrossRef] [PubMed]
- Stefansson, O.A.; Moran, S.; Gomez, A.; Sayols, S.; Arribas-Jorba, C.; Sandoval, J.; Hilmarsdottir, H.; Olafsdottir, E.; Tryggvadottir, L.; Jonasson, J.G.; et al. A DNA methylation-based definition of biologically distinct breast cancer subtypes. Mol. Oncol. 2015, 9, 555–568. [Google Scholar] [CrossRef]
- McInnes, T.; Zou, D.; Rao, D.S.; Munro, F.M.; Phillips, V.L.; McCall, J.L.; Black, M.A.; Reeve, A.E.; Guilford, P.J. Genome-wide methylation analysis identifies a core set of hypermethylated genes in CIMP-H colorectal cancer. BMC Cancer 2017, 17, 228. [Google Scholar] [CrossRef] [Green Version]
- Kuramoto, J.; Arai, E.; Tian, Y.; Funahashi, N.; Hiramoto, M.; Nammo, T.; Nozaki, Y.; Takahashi, Y.; Ito, N.; Shibuya, A.; et al. Genome-wide DNA methylation analysis during non-alcoholic steatohepatitis-related multistage hepatocarcinogenesis: Comparison with hepatitis virus-related carcinogenesis. Carcinogenesis 2017, 38, 261–270. [Google Scholar] [CrossRef]
- Mao, Z.; Cai, W.; Shao, X. Selecting significant genes by randomization test for cancer classification using gene expression data. J. Biomed. Inform. 2013, 46, 594–601. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.; Sun, Z.; Chen, J.; Jing, C. Raman Spectroscopy in Colorectal Cancer Diagnostics: Comparison of PCA-LDA and PLS-DA Models. J. Spectrosc. 2016, 2016, 1–6. [Google Scholar] [CrossRef]
- Lyng, F.M.; Traynor, D.; Nguyen, T.N.Q.; Meade, A.D.; Rakib, F.; Al-Saady, R.; Goormaghtigh, E.; Al-Saad, K.; Ali, M.H. Discrimination of breast cancer from benign tumours using Raman spectroscopy. PLoS ONE 2019, 14, e0212376. [Google Scholar]
- Chen, G.; Lin, X.; Lin, D.; Ge, X.; Feng, S.; Pan, J.; Lin, J.; Huang, Z.; Huang, X.; Chen, R. Identification of different tumor states in nasopharyngeal cancer using surface-enhanced Raman spectroscopy combined with Lasso-PLS-DA algorithm. RSC Adv. 2016, 6, 7760–7764. [Google Scholar] [CrossRef]
- Kim, M.S.; Lebron, C.; Nagpal, J.K.; Chae, Y.K.; Chang, X.; Huang, Y.; Chuang, T.; Yamashita, K.; Trink, B.; Ratovitski, E.A.; et al. Methylation of the DFNA5 increases risk of lymph node metastasis in human breast cancer. Biochem. Biophys. Res. Commun. 2008, 370, 38–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esteller, M. Aberrant DNA Methylation as a Cancer-Inducing Mechanism. Annu. Rev. Pharmacol. Toxicol. 2005, 45, 629–656. [Google Scholar] [CrossRef] [PubMed]
- Rao, X.; Evans, J.; Chae, H.; Pilrose, J.; Kim, S.; Yan, P.; Huang, R.-L.; Lai, H.-C.; Lin, H.; Liu, Y.; et al. CpG island shore methylation regulates caveolin-1 expression in breast cancer. Oncogene 2013, 32, 4519–4528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ogoshi, K.; Hashimoto, S.; Nakatani, Y.; Qu, W.; Oshima, K.; Tokunaga, K.; Sugano, S.; Hattori, M.; Morishita, S.; Matsushima, K. Genome-wide profiling of DNA methylation in human cancer cells. Genomics 2011, 98, 280–287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Irizarry, R.A.; Ladd-Acosta, C.; Wen, B.; Wu, Z.; Montano, C.; Onyango, P.; Cui, H.; Gabo, K.; Rongione, M.; Webster, M.; et al. The human colon cancer methylome shows similar hypo- and hypermethylation at conserved tissue-specific CpG island shores. Nat. Genet. 2009, 41, 178–186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maunakea, A.K.; Chepelev, I.; Cui, K.; Zhao, K. Intragenic DNA methylation modulates alternative splicing by recruiting MeCP2 to promote exon recognition. Cell Res. 2013, 23, 1256–1269. [Google Scholar] [CrossRef] [Green Version]
- Kulis, M.; Queirós, A.C.; Beekman, R.; Martín-Subero, J.I. Intragenic DNA methylation in transcriptional regulation, normal differentiation and cancer. Biochim. Biophys. Acta Gene Regul. Mech. 2013, 1829, 1161–1174. [Google Scholar] [CrossRef]
- Neri, F.; Rapelli, S.; Krepelova, A.; Incarnato, D.; Parlato, C.; Basile, G.; Maldotti, M.; Anselmi, F.; Oliviero, S. Intragenic DNA methylation prevents spurious transcription initiation. Nature 2017, 543, 72–77. [Google Scholar] [CrossRef]
- Shirahata, A.; Hibi, K. Serum vimentin methylation as a potential marker for colorectal cancer. Anticancer Res. 2014, 34, 4121–4126. [Google Scholar]
- Kirby, M.K.; Ramaker, R.C.; Roberts, B.S.; Lasseigne, B.N.; Gunther, D.S.; Burwell, T.C.; Davis, N.S.; Gulzar, Z.G.; Absher, D.M.; Cooper, S.J.; et al. Genome-wide DNA methylation measurements in prostate tissues uncovers novel prostate cancer diagnostic biomarkers and transcription factor binding patterns. BMC Cancer 2017, 17, 273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hao, X.; Luo, H.; Krawczyk, M.; Wei, W.; Wang, W.; Wang, J.; Flagg, K.; Hou, J.; Zhang, H.; Yi, S.; et al. DNA methylation markers for diagnosis and prognosis of common cancers. Proc. Natl. Acad. Sci. USA 2017, 114, 7414–7419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, L.-L.; Li, Y.-M. Current noninvasive tests for colorectal cancer screening: An overview of colorectal cancer screening tests. World J. Gastrointest. Oncol. 2016, 8, 793. [Google Scholar] [CrossRef] [PubMed]
- Semaan, A.; van Ellen, A.; Meller, S.; Bergheim, D.; Branchi, V.; Lingohr, P.; Goltz, D.; Kalff, J.C.; Kristiansen, G.; Matthaei, H.; et al. SEPT9 and SHOX2 DNA methylation status and its utility in the diagnosis of colonic adenomas and colorectal adenocarcinomas. Clin. Epigenetics 2016, 8, 100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papadopoulos, N.; Springer, S.; Koch, W.M.; Bettegowda, C.; Gourin, C.G.; Diaz, L.A.; Kiess, A.; Mulvey, C.L.; Sidransky, D.; Wang, Y.; et al. Detection of somatic mutations and HPV in the saliva and plasma of patients with head and neck squamous cell carcinomas. Sci. Transl. Med. 2015, 7, 293ra104. [Google Scholar]
- Dawson, S.-J.; Tsui, D.W.; Murtaza, M.; Biggs, H.; Rueda, O.M.; Chin, S.-F.; Dunning, M.J.; Gale, D.; Forshew, T.; Mahler-Araujo, B.; et al. Analysis of Circulating Tumor DNA to Monitor Metastatic Breast Cancer. N. Engl. J. Med. 2013, 368, 1199–1209. [Google Scholar] [CrossRef] [Green Version]
- Haber, D.A.; Velculescu, V.E. Blood-based analyses of cancer: Circulating tumor cells and circulating tumor DNA. Cancer Discov. 2014, 4, 650–661. [Google Scholar] [CrossRef]
- Bettegowda, C.; Sausen, M.; Leary, R.J.; Kinde, I.; Wang, Y.; Agrawal, N.; Bartlett, B.R.; Wang, H.; Luber, B.; Alani, R.M.; et al. Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci. Transl. Med. 2014, 6, 224ra24. [Google Scholar] [CrossRef] [Green Version]
- Cree, I.A.; Uttley, L.; Buckley Woods, H.; Kikuchi, H.; Reiman, A.; Harnan, S.; Whiteman, B.L.; Philips, S.T.; Messenger, M.; Cox, A.; et al. The evidence base for circulating tumour DNA blood-based biomarkers for the early detection of cancer: A systematic mapping review. BMC Cancer 2017, 17, 697. [Google Scholar] [CrossRef]
- Cohen, J.D.; Javed, A.A.; Thoburn, C.; Wong, F.; Tie, J.; Gibbs, P.; Schmidt, C.M.; Yip-Schneider, M.T.; Allen, P.J.; Schattner, M.; et al. Combined circulating tumor DNA and protein biomarker-based liquid biopsy for the earlier detection of pancreatic cancers. Proc. Natl. Acad. Sci. USA 2017, 114, 10202–10207. [Google Scholar] [CrossRef] [Green Version]
- Parashar, S.; Cheishvili, D.; Mahmood, N.; Arakelian, A.; Tanvir, I.; Khan, H.A.; Kremer, R.; Mihalcioiu, C.; Szyf, M.; Rabbani, S.A. DNA methylation signatures of breast cancer in peripheral T-cells. BMC Cancer 2018, 18, 574. [Google Scholar] [CrossRef] [PubMed]
- Gu, S.; Lin, S.; Ye, D.; Qian, S.; Jiang, D.; Zhang, X.; Li, Q.; Yang, J.; Ying, X.; Li, Z.; et al. Genome-wide methylation profiling identified novel differentially hypermethylated biomarker MPPED2 in colorectal cancer. Clin. Epigenetics 2019, 11, 41. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Deng, H.; Xin, S.; Zhang, K.; Shi, R.; Bao, X. Prognostic and predictive value of three DNA methylation signatures in lung adenocarcinoma. Front. Genet. 2019, 10, 349. [Google Scholar] [CrossRef] [PubMed]
- Fan, S.; Tang, J.; Li, N.; Zhao, Y.; Ai, R.; Zhang, K.; Wang, M.; Du, W.; Wang, W. Integrative analysis with expanded DNA methylation data reveals common key regulators and pathways in cancers. npj Genomic Med. 2019, 4, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- R Core Team R: A Language and Environment for Statistical Computing. 2017. Available online: https://www.r-project.org/ (accessed on 1 November 2019).
- World Health Organization Global Health Observatory Data Repository. Available online: https://www.who.int/ (accessed on 12 January 2018).









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Dataset Name (TCGA Abbreviation) | Number of NT | Number of TP | Total |
|---|---|---|---|
| Bladder urothelial carcinoma (BLCA) | 21 | 418 | 439 |
| Breast carcinoma (BRCA) | 96 | 791 | 887 |
| Esophageal carcinoma (ESCA) | 16 | 185 | 201 |
| Head and Neck squamous cell carcinoma (HNSC) | 50 | 528 | 578 |
| Kidney renal clear cell carcinoma (KIRC) | 160 | 324 | 484 |
| Kidney renal papillary cell carcinoma (KIRP) | 45 | 275 | 320 |
| Liver hepatocellular carcinoma (LIHC) | 50 | 377 | 427 |
| Lung adenocarcinoma (LUAD) | 32 | 473 | 505 |
| Lung squamous cell carcinoma (LUSC) | 42 | 370 | 412 |
| Pancreatic adenocarcinoma (PAAD) | 10 | 184 | 194 |
| Prostate adenocarcinoma (PRAD) | 50 | 502 | 552 |
| Thyroid carcinoma (THCA) | 56 | 507 | 563 |
| Uterine Corpus Endometrial Carcinoma (UCEC) | 46 | 438 | 484 |
| Colorectal carcinoma (CRC) | 45 | 411 | 456 |
| Total | 719 | 5783 | 6502 |
| Probe Abbreviation | Probe Name (Illumina) | Genomic Coordinate * | Location | Chromosome |
|---|---|---|---|---|
| Probe 1 | CpG17790129 | 24738572 | Gene body | 7 |
| Probe 2 | CpG14205998 | 24748668 | Gene body | 7 |
| Probe 3 | CpG04317854 | 24762562 | Gene body | 7 |
| Probe 4 | CpG12922093 | 24767644 | Gene body | 7 |
| Probe 5 | CpG17569154 | 24781545 | Gene body | 7 |
| Probe 6 | CpG19260663 | 24791121 | Gene body | 7 |
| Probe 7 | CpG09333471 | 24796355 | Putative Promoter | 7 |
| Probe 8 | CpG00473134 | 24796494 | Putative Promoter | 7 |
| Probe 9 | CpG03995857 | 24796553 | Putative Promoter | 7 |
| Probe 10 | CpG07320646 | 24796981 | Putative Promoter | 7 |
| Probe 11 | CpG07293520 | 24797192 | Putative Promoter | 7 |
| Probe 12 | CpG04770504 | 24797363 | Putative Promoter | 7 |
| Probe 13 | CpG24805239 | 24797486 | Putative Promoter | 7 |
| Probe 14 | CpG01733570 | 24797656 | Putative Promoter | 7 |
| Probe 15 | CpG25723149 | 24797680 | Putative Promoter | 7 |
| Probe 16 | CpG22804000 | 24797691 | Putative Promoter | 7 |
| Probe 17 | CpG07504598 | 24797786 | Putative Promoter | 7 |
| Probe 18 | CpG15037663 | 24797835 | Putative Promoter | 7 |
| Probe 19 | CpG19706795 | 24797839 | Putative Promoter | 7 |
| Probe 20 | CpG20764575 | 24797884 | Putative Promoter | 7 |
| Probe 21 | CpG06301139 | 24798175 | Upstream Region | 7 |
| Probe 22 | CpG26712096 | 24798855 | Upstream Region | 7 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ibrahim, J.; Op de Beeck, K.; Fransen, E.; Peeters, M.; Van Camp, G. The Gasdermin E Gene Has Potential as a Pan-Cancer Biomarker, While Discriminating between Different Tumor Types. Cancers 2019, 11, 1810. https://doi.org/10.3390/cancers11111810
Ibrahim J, Op de Beeck K, Fransen E, Peeters M, Van Camp G. The Gasdermin E Gene Has Potential as a Pan-Cancer Biomarker, While Discriminating between Different Tumor Types. Cancers. 2019; 11(11):1810. https://doi.org/10.3390/cancers11111810
Chicago/Turabian StyleIbrahim, Joe, Ken Op de Beeck, Erik Fransen, Marc Peeters, and Guy Van Camp. 2019. "The Gasdermin E Gene Has Potential as a Pan-Cancer Biomarker, While Discriminating between Different Tumor Types" Cancers 11, no. 11: 1810. https://doi.org/10.3390/cancers11111810