Synergistic Autophagy Effect of miR-212-3p in Zoledronic Acid-Treated In Vitro and Orthotopic In Vivo Models and in Patient-Derived Osteosarcoma Cells

 , , ,
, , ,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
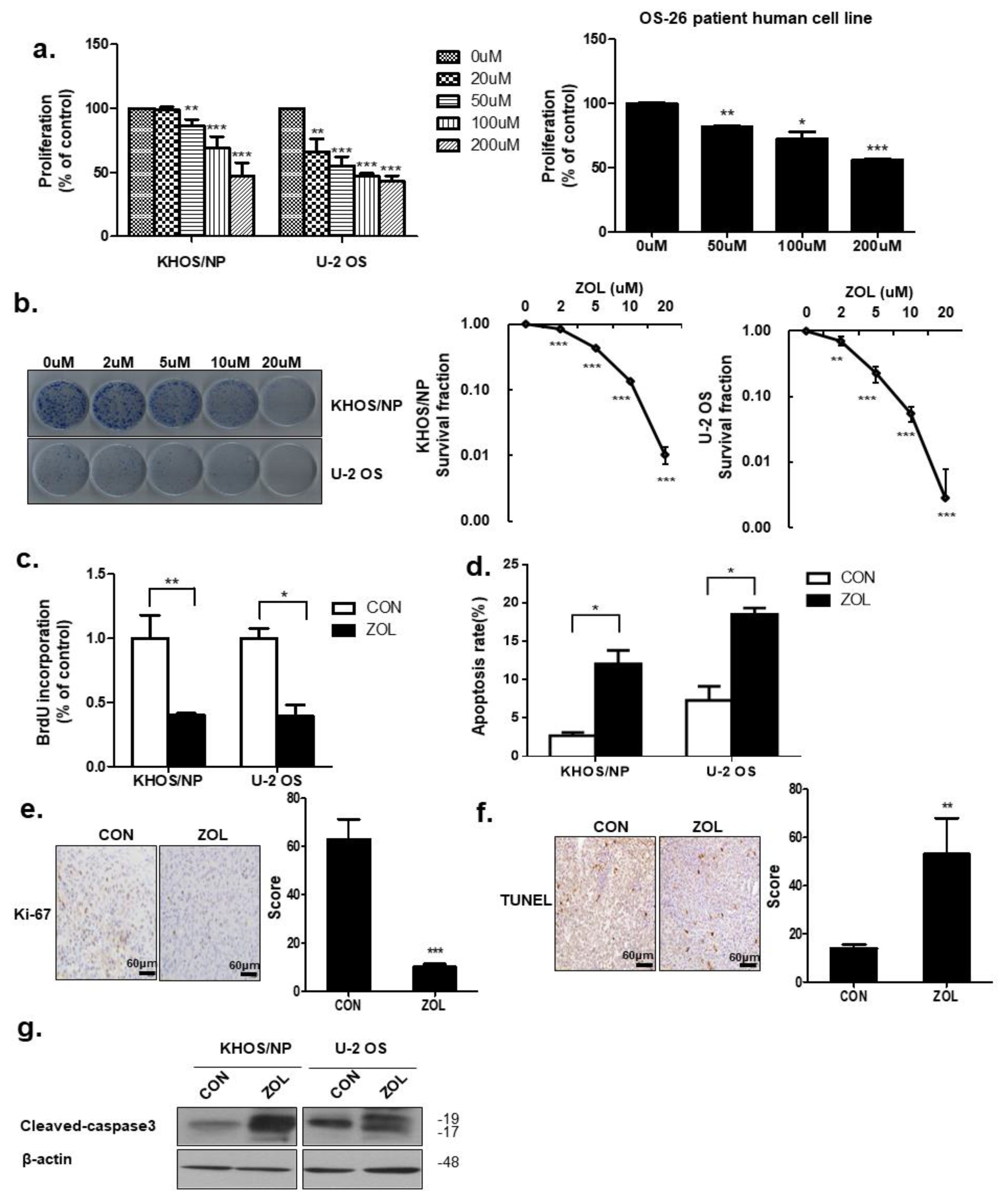
2.1. ZOL Decreased OS Cell Proliferation in a Dose-Dependent Manner
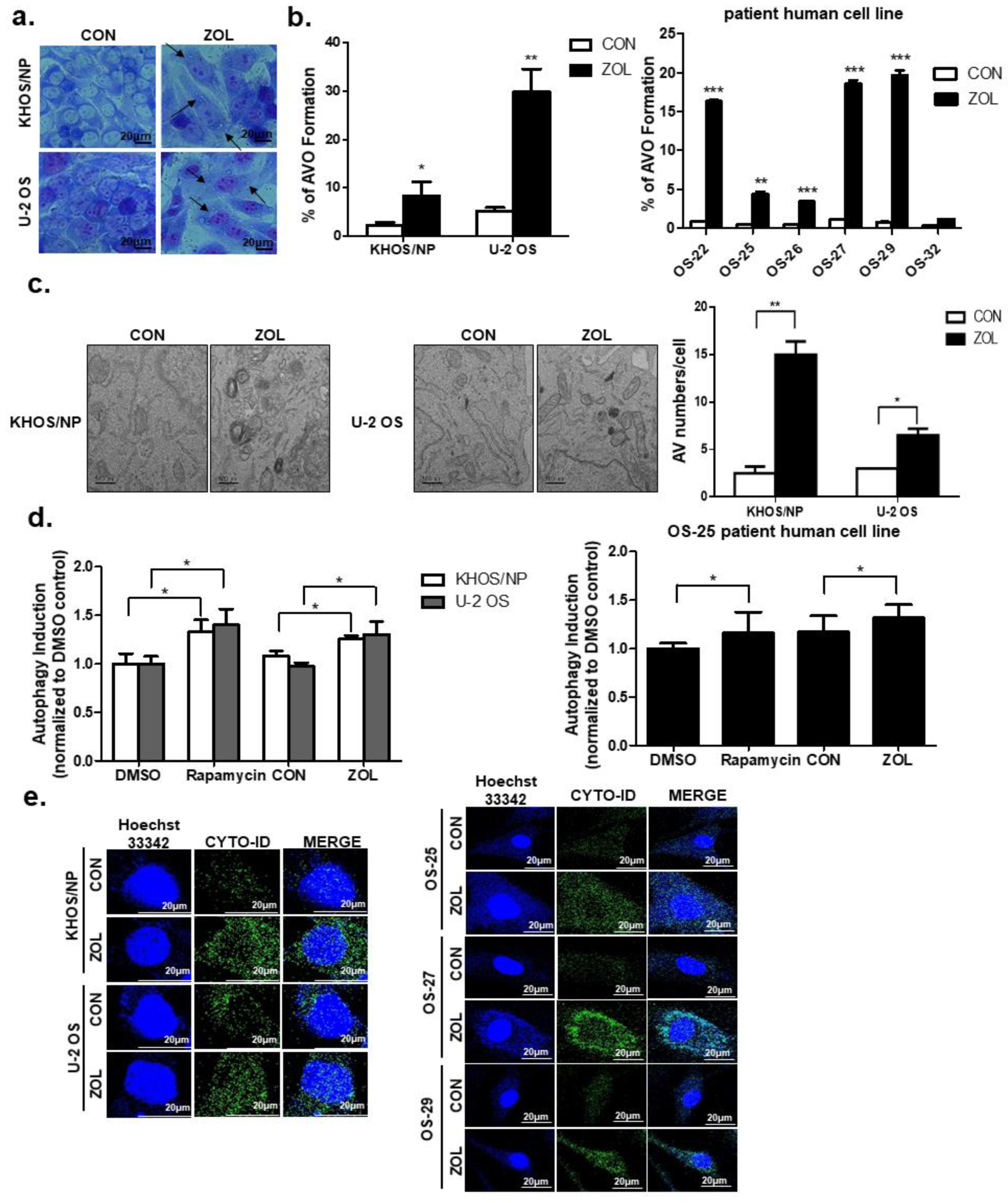
2.2. ZOL Induced Accumulation of Acidic Vacuoles (AVOs)
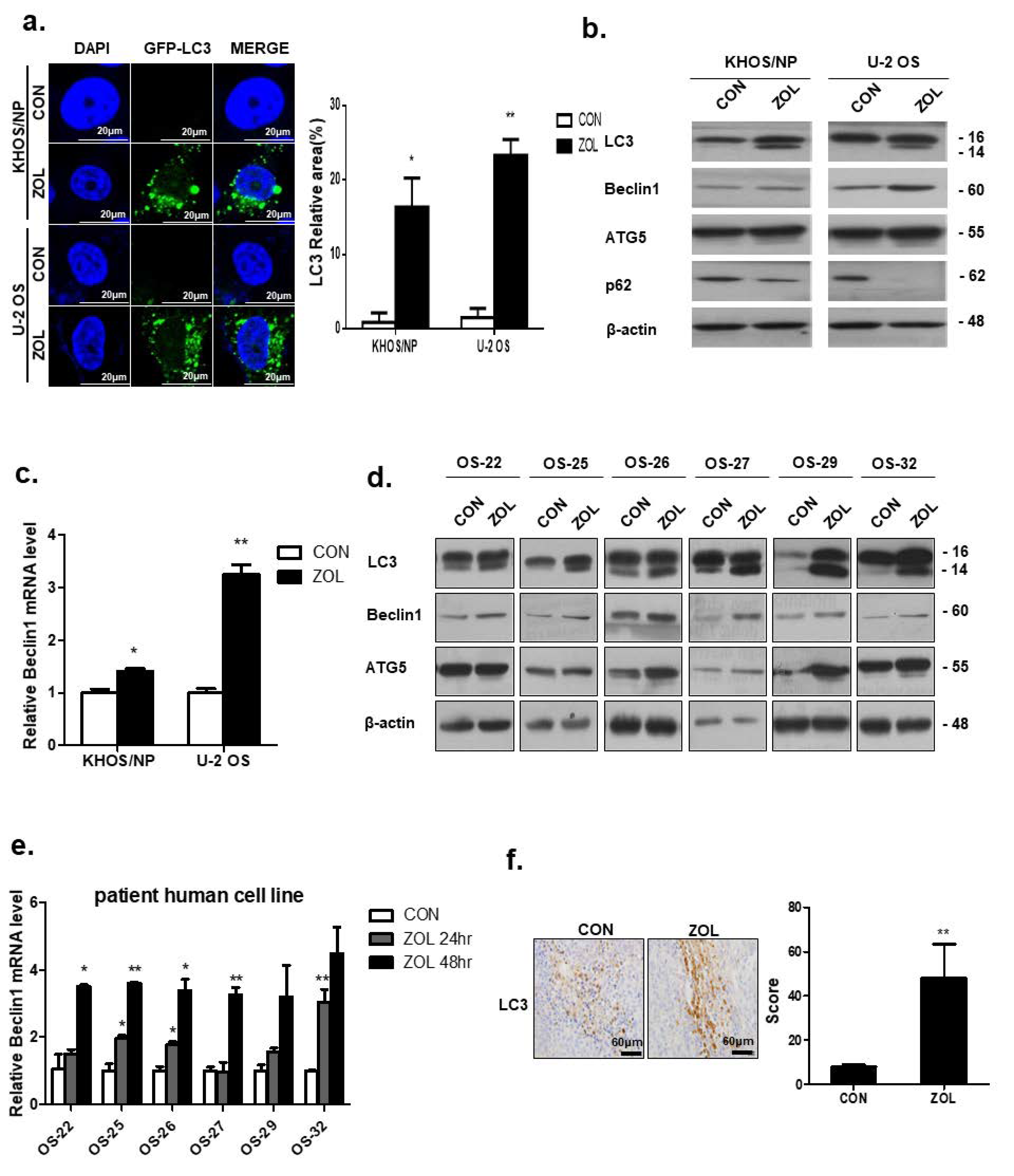
2.3. ZOL Treatment Induced Autophagy
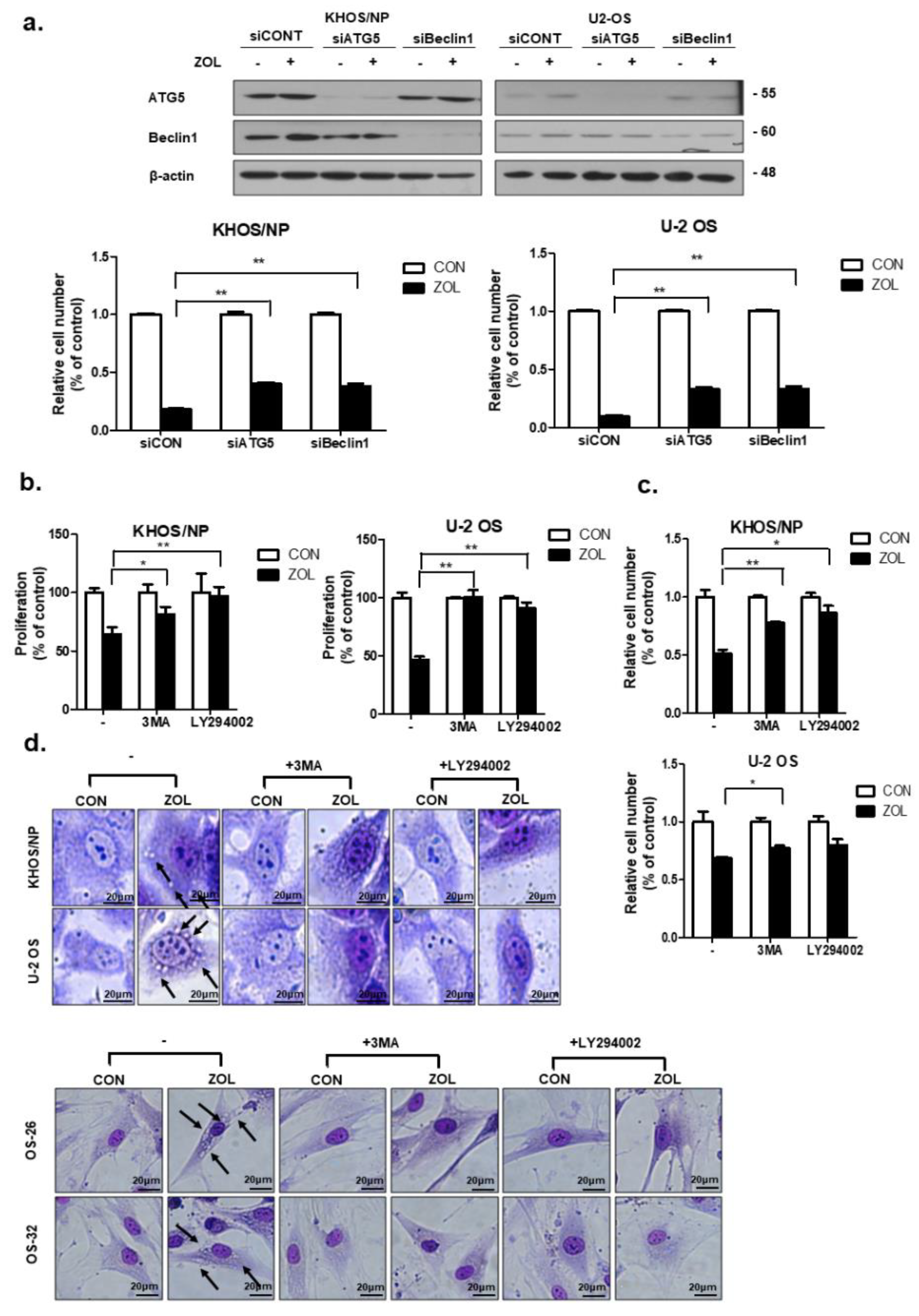
2.4. Inhibition of ZOL-Induced Autophagy Decreased Cell Death
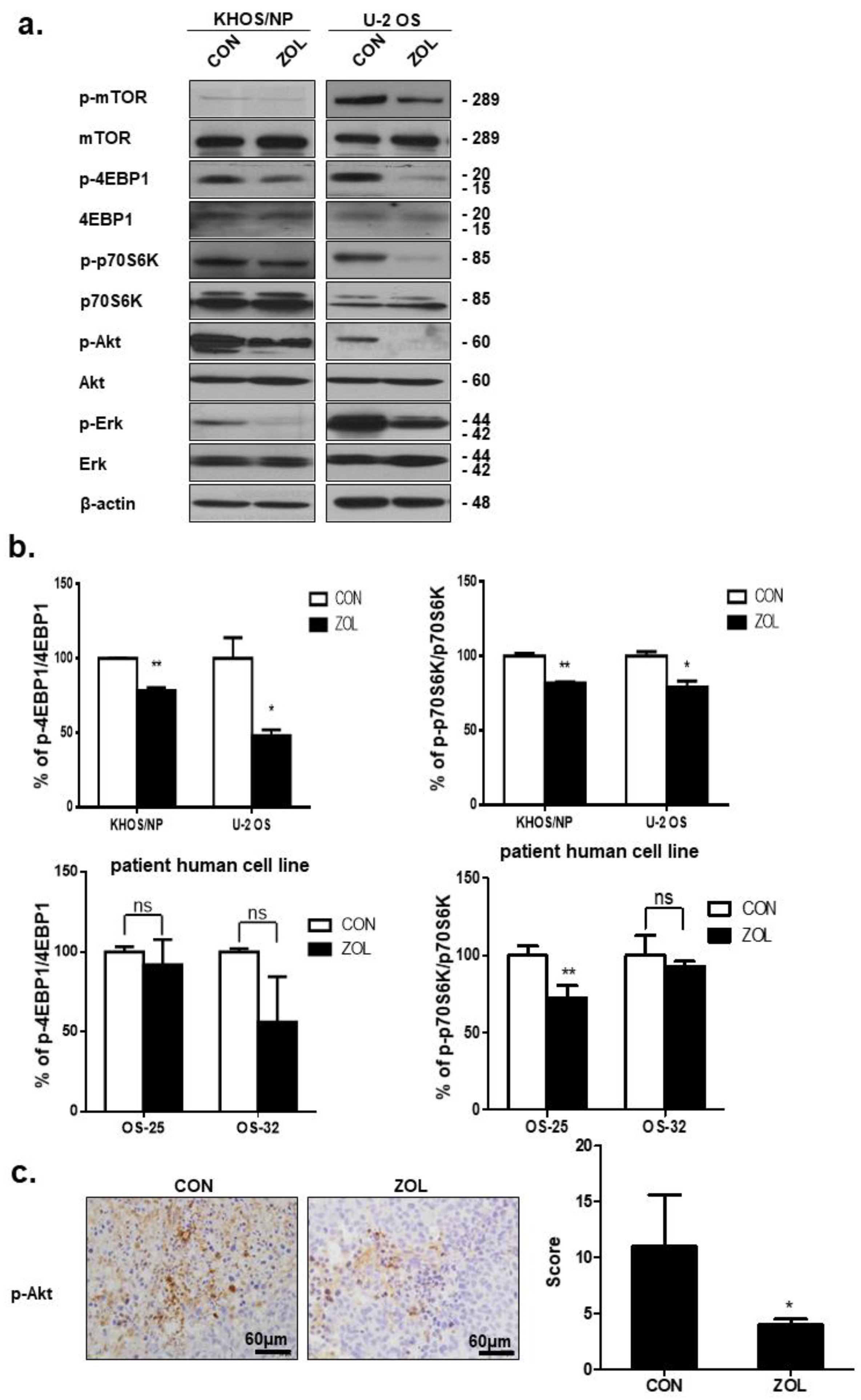
2.5. ZOL Suppressed the mTOR/p70S6K Signaling Pathway
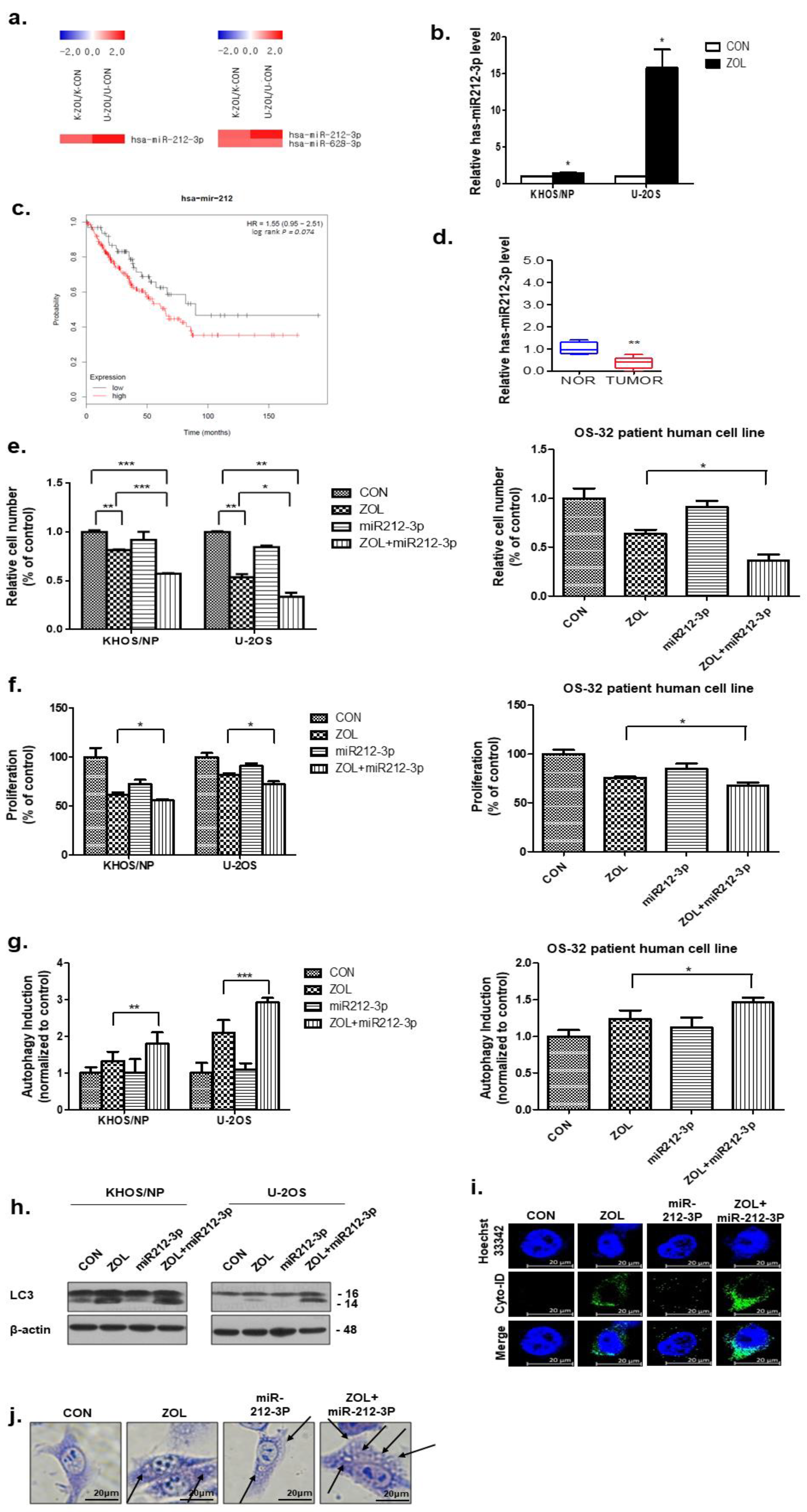
2.6. miR-212-3p Directly Targeted and, Thus, Positively Regulated Autophagy after ZOL Treatment
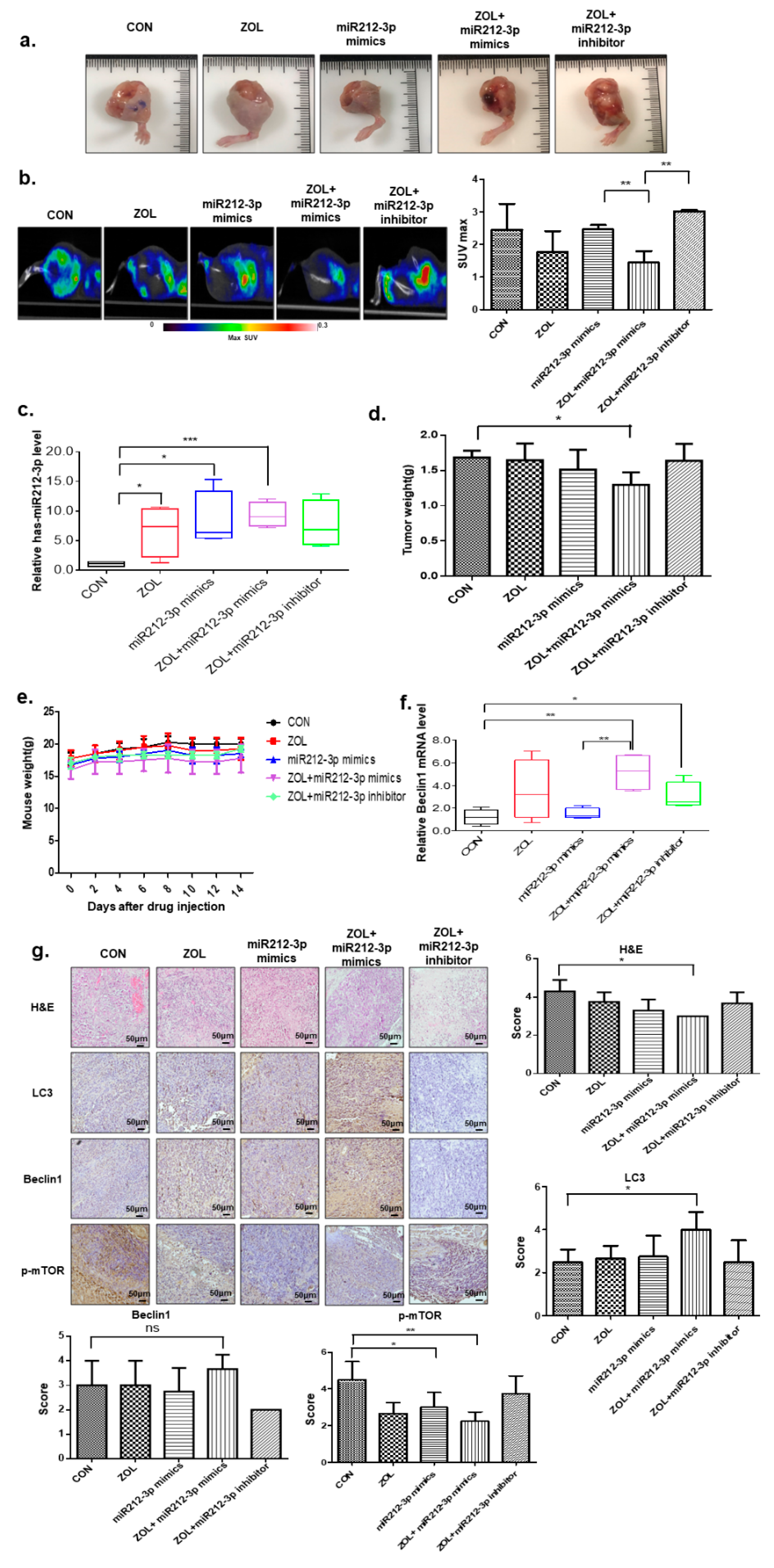
2.7. The Autophagic Effects of Combined miR-212-3p and ZOL Treatment in In Vivo Orthotopic Model
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Tissue Samples
4.2. Reagents
4.3. MTT Assay
4.4. Colony-Formation Assay
4.5. Detection of Apoptotic Cells by Annexin V Staining
4.6. Western Blotting
4.7. Morphology
4.8. 5-Bromo-2′-Deoxyuridine (BrdU)-Labeling Assay
4.9. Orthotopic Model and Histological Analysis
4.10. Quantification of Acidic Vacuoles (AVOs) by Acridine Orange (AO) Staining
4.11. Immunohistochemical Staining
4.12. Terminal Deoxynucleotidyl Transferase-Mediated dUTP Nick-End Labeling (TUNEL) Assays
4.13. Elisa Assay
4.14. miRNA and Transient Transfection
4.15. Quantitative Reverse-Transcriptase Polymerase Chain Reaction (qRT-PCR) Experiments
4.16. In Vivo Tumor Model and Administration of ZOL and miR-212-3p Mimic
4.17. Positron Emission Tomography (PET)/Computed Tomography (CT) Acquisition
4.18. Statistical Analysis
4.19. Data Availability
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| [18F]-FDG | 18F-fluoro-2-deoxy-d-glucose |
| AO | acridine orange |
| AVOs | acidic vacuoles |
| BrdU | 5-bromo-2′-deoxyuridine |
| CT | computed tomography |
| FACS | fluorescence-activated cell sorting |
| i.p. | intraperitoneal |
| MTT | 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide |
| NSCLC | non-small cell lung cancer |
| OS | osteosarcoma |
| PBS | phosphate-buffered saline |
| PET | positron emission tomography |
| qRT-PCR | quantitative reverse-transcriptase polymerase chain reaction |
| SUV | standardized uptake value |
| TUNEL | terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling |
| VOI | volumes of interest |
| ZOL | zoledronic acid |
References
- Jaffe, N. Osteosarcoma: Review of the past, impact on the future. The American experience. Cancer Treat Res. 2009, 152, 239–262. [Google Scholar] [PubMed]
- Wu, C.-C.; Cheng, C.-H.; Lee, Y.-H.; Chang, I.-L.; Chen, H.-Y.; Hsieh, C.-P.; Chueh, P.-J. Ursolic acid triggers apoptosis in human osteosarcoma cells via caspase activation and the ERK1/2 MAPK pathway. J. Agric. Food Chem. 2016, 64, 4220–4226. [Google Scholar] [CrossRef] [PubMed]
- Clemons, M.J.; Dranitsaris, G.; Ooi, W.S.; Yogendran, G.; Sukovic, T.; Wong, B.Y.; Verma, S.; Pritchard, K.I.; Trudeau, M.; Cole, D.E. Phase II trial evaluating the palliative benefit of second-line zoledronic acid in breast cancer patients with either a skeletal-related event or progressive bone metastases despite first-line bisphosphonate therapy. J. Clin. Oncol. 2006, 24, 4895–4900. [Google Scholar] [CrossRef] [PubMed]
- Kohno, N.; Aogi, K.; Minami, H.; Nakamura, S.; Asaga, T.; Iino, Y.; Watanabe, T.; Goessl, C.; Ohashi, Y.; Takashima, S. Zoledronic acid significantly reduces skeletal complications compared with placebo in Japanese women with bone metastases from breast cancer: A randomized, placebo-controlled trial. J. Clin. Oncol. 2005, 23, 3314–3321. [Google Scholar] [CrossRef]
- Rosen, L.S.; Gordon, D.; Kaminski, M.; Howell, A.; Belch, A.; Mackey, J.; Apffelstaedt, J.; Hussein, M.; Coleman, R.E.; Reitsma, D.J. Zoledronic acid versus pamidronate in the treatment of skeletal metastases in patients with breast cancer or osteolytic lesions of multiple myeloma: A phase III, double-blind, comparative trial. Cancer J. 2001, 7, 377–387. [Google Scholar] [PubMed]
- Rosen, L.S.; Gordon, D.; Kaminski, M.; Howell, A.; Belch, A.; Mackey, J.; Apffelstaedt, J.; Hussein, M.A.; Coleman, R.E.; Reitsma, D.J. Long-term efficacy and safety of zoledronic acid compared with pamidronate disodium in the treatment of skeletal complications in patients with advanced multiple myeloma or breast carcinoma. Cancer 2003, 98, 1735–1744. [Google Scholar] [CrossRef]
- Hirbe, A.C.; Roelofs, A.J.; Floyd, D.H.; Deng, H.; Becker, S.N.; Lanigan, L.G.; Apicelli, A.J.; Xu, Z.; Prior, J.L.; Eagleton, M.C.J.B. The bisphosphonate zoledronic acid decreases tumor growth in bone in mice with defective osteoclasts. Bone 2009, 44, 908–916. [Google Scholar] [CrossRef]
- Moriceau, G.; Ory, B.; Mitrofan, L.; Riganti, C.; Blanchard, F.; Brion, R.; Charrier, C.; Battaglia, S.; Pilet, P.; Denis, M. Zoledronic acid potentiates mTOR inhibition and abolishes the resistance of osteosarcoma cells to RAD001 (Everolimus): Pivotal role of the prenylation process. Cancer Res. 2010, 70, 10329–10339. [Google Scholar] [CrossRef]
- Ory, B.; Heymann, M.F.; Kamijo, A.; Gouin, F.; Heymann, D.; Redini, F. Zoledronic acid suppresses lung metastases and prolongs overall survival of osteosarcoma-bearing mice. Cancer Interdiscip. Int. J. Am. Cancer Soc. 2005, 104, 2522–2529. [Google Scholar] [CrossRef]
- Conry, R.M.; Rodriguez, M.G.; Pressey, J.G. Zoledronic acid in metastatic osteosarcoma: Encouraging progression free survival in four consecutive patients. Clin. Sarcoma Res. 2016, 6, 6. [Google Scholar] [CrossRef]
- Wang, I.-T.; Chou, S.-C.; Lin, Y.-C. Zoledronic acid induces apoptosis and autophagy in cervical cancer cells. Tumor Biol. 2014, 35, 11913–11920. [Google Scholar] [CrossRef] [PubMed]
- Berenson, J.R. Recommendations for zoledronic acid treatment of patients with bone metastases. Oncologist 2005, 10, 52–62. [Google Scholar] [CrossRef] [PubMed]
- Sewing, L.; Steinberg, F.; Schmidt, H.; Göke, R. The bisphosphonate zoledronic acid inhibits the growth of HCT-116 colon carcinoma cells and induces tumor cell apoptosis. Apoptosis 2008, 13, 782–789. [Google Scholar] [CrossRef] [PubMed]
- Marycz, K.; Kornicka, K.; Marędziak, M.; Golonka, P.; Nicpoń, J. Equine metabolic syndrome impairs adipose stem cells osteogenic differentiation by predominance of autophagy over selective mitophagy. J. Cell. Mol. Med. 2016, 20, 2384–2404. [Google Scholar] [CrossRef]
- Hippert, M.M.; O’Toole, P.S.; Thorburn, A. Autophagy in cancer: Good, bad, or both? Cancer Res. 2006, 66, 9349–9351. [Google Scholar] [CrossRef]
- Kang, R.; Zeh, H.; Lotze, M.; Tang, D. The Beclin 1 network regulates autophagy and apoptosis. Cell Death Differ. 2011, 18, 571. [Google Scholar] [CrossRef]
- O’Farrill, J.S.; Gordon, N. Autophagy in osteosarcoma. Adv. Exp. Med. Biol. 2014, 804, 147–160. [Google Scholar]
- Incoronato, M.; Garofalo, M.; Urso, L.; Romano, G.; Quintavalle, C.; Zanca, C.; Iaboni, M.; Nuovo, G.; Croce, C.M.; Condorelli, G. MiR-212 increases tumor necrosis factor–related apoptosis-inducing ligand sensitivity in non–small cell lung cancer by targeting the antiapoptotic protein PED. Cancer Res. 2010, 70, 3638–3646. [Google Scholar] [CrossRef]
- Jiping, Z.; Ming, F.; Lixiang, W.; Xiuming, L.; Yuqun, S.; Han, Y.; Zhifang, L.; Yundong, S.; Shili, L.; Chunyan, C.J. Micro RNA-212 inhibits proliferation of gastric cancer by directly repressing retinoblastoma binding protein 2. J. Cell. Biochem. 2013, 114, 2666–2672. [Google Scholar] [CrossRef]
- Liu, Y.; Bao, Z.; Tian, W.; Huang, G. miR-885-5p suppresses osteosarcoma proliferation, migration and invasion through regulation of β-catenin. Oncol. Lett. 2019, 17, 1996–2004. [Google Scholar] [CrossRef]
- Walter, B.A.; Valera, V.A.; Pinto, P.A.; Merino, M.J. Comprehensive microRNA profiling of prostate cancer. J. Cancer 2013, 4, 350. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.; Nong, K.; Wu, B.; Dong, B.; Bai, Y.; Zhu, H.; Wang, W.; Huang, X.; Yuan, Z.; Ai, K. miR-212 promotes pancreatic cancer cell growth and invasion by targeting the hedgehog signaling pathway receptor patched-1. J. Exp. Clin. Cancer Res. 2014, 33, 54. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.H.; Kim, M.-S.; Lee, K.-H.; Koh, J.-S.; Jung, W.-G.; Kong, C.-B. Zoledronic acid is an effective radiosensitizer in the treatment of osteosarcoma. Oncotarget 2016, 7, 70869. [Google Scholar] [CrossRef] [PubMed]
- Kadowaki, M.; Karim, M.R. Cytosolic LC3 ratio as a quantitative index of macroautophagy. Methods Enzymol. 2009, 452, 199–213. [Google Scholar]
- Tanida, I.; Ueno, T.; Kominami, E. LC3 conjugation system in mammalian autophagy. Int. J. Biochem. Cell Biol. 2004, 36, 2503–2518. [Google Scholar] [CrossRef]
- Moretti, L.; Yang, E.S.; Kim, K.W.; Lu, B. Autophagy signaling in cancer and its potential as novel target to improve anticancer therapy. Drug Resist. Updates 2007, 10, 135–143. [Google Scholar] [CrossRef]
- Settembre, C.; Di Malta, C.; Polito, V.A.; Arencibia, M.G.; Vetrini, F.; Erdin, S.; Erdin, S.U.; Huynh, T.; Medina, D.; Colella, P. TFEB links autophagy to lysosomal biogenesis. Science 2011, 332, 1429–1433. [Google Scholar] [CrossRef]
- Martinez-Lopez, N.; Athonvarangkul, D.; Mishall, P.; Sahu, S.; Singh, R. Autophagy proteins regulate ERK phosphorylation. Nat. Commun. 2013, 4, 2799. [Google Scholar] [CrossRef]
- Janku, F.; McConkey, D.J.; Hong, D.S.; Kurzrock, R. Autophagy as a target for anticancer therapy. Nat. Rev. Clin. Oncol. 2011, 8, 528. [Google Scholar] [CrossRef]
- Bincoletto, C.; Bechara, A.; Pereira, G.; Santos, C.; Antunes, F.; da-Silva, J.P.; Muler, M.; Gigli, R.; Monteforte, P.; Hirata, H. Interplay between apoptosis and autophagy, a challenging puzzle: New perspectives on antitumor chemotherapies. Chem. Biol. Interact. 2013, 206, 279–288. [Google Scholar] [CrossRef]
- Shintani, T.; Klionsky, D.J. Autophagy in health and disease: A double-edged sword. Science 2004, 306, 990–995. [Google Scholar] [CrossRef] [PubMed]
- Lorin, S.; Hamaï, A.; Mehrpour, M.; Codogno, P. Autophagy Regulation and its Role in Cancer. Semin. Cancer Biol. 2013, 23, 361–379. [Google Scholar] [CrossRef] [PubMed]
- Eisenberg-Lerner, A.; Bialik, S.; Simon, H.-U.; Kimchi, A. Life and death partners: Apoptosis, autophagy and the cross-talk between them. Cell Death Differ. 2009, 16, 966. [Google Scholar] [CrossRef] [PubMed]
- Kumar, D.; Shankar, S.; Srivastava, R.K. Rottlerin-induced autophagy leads to the apoptosis in breast cancer stem cells: Molecular mechanisms. Mol. Cancer 2013, 12, 171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Műzes, G.; Sipos, F. Anti-tumor immunity, autophagy and chemotherapy. World J. Gastroenterol. WJG 2012, 18, 6537. [Google Scholar] [CrossRef] [PubMed]
- He, H.; Ni, J.; Huang, J. Molecular mechanisms of chemoresistance in osteosarcoma. Oncol. Lett. 2014, 7, 1352–1362. [Google Scholar] [CrossRef] [Green Version]
- Hu, W.; Chen, S.-S.; Zhang, J.-L.; Lou, X.-E.; Zhou, H.-J. Dihydroartemisinin induces autophagy by suppressing NF-κB activation. Cancer Lett. 2014, 343, 239–248. [Google Scholar] [CrossRef]
- Zhou, J.; Zhang, W.; Liang, B.; Casimiro, M.C.; Whitaker-Menezes, D.; Wang, M.; Lisanti, M.P.; Lanza-Jacoby, S.; Pestell, R.G.; Wang, C. PPARγ activation induces autophagy in breast cancer cells. Int. J. Biochem. Cell Biol. 2009, 41, 2334–2342. [Google Scholar] [CrossRef] [Green Version]
- Aoki, H.; Takada, Y.; Kondo, S.; Sawaya, R.; Aggarwal, B.; Kondo, Y. Evidence that curcumin suppresses the growth of malignant gliomas in vitro and in vivo through induction of autophagy: Role of Akt and ERK signaling pathways. Mol. Pharmacol. 2007, 72, 29–39. [Google Scholar] [CrossRef] [Green Version]
- Hu, C.; Zou, M.-J.; Zhao, L.; Lu, N.; Sun, Y.-J.; Gou, S.-H.; Xi, T.; Guo, Q.-L. E Platinum, a newly synthesized platinum compound, induces autophagy via inhibiting phosphorylation of mTOR in gastric carcinoma BGC-823 cells. Toxicol. Lett. 2012, 210, 78–86. [Google Scholar] [CrossRef]
- Dalby, K.; Tekedereli, I.; Lopez-Berestein, G.; Ozpolat, B. Targeting the pro-death and pro-survival functions of autophagy as novel therapeutic strategies in cancer. Autophagy 2010, 6, 322–329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Chen, P.; Hong, H.; Wang, L.; Zhou, Y.; Lang, Y. JNK pathway mediates curcumin-induced apoptosis and autophagy in osteosarcoma MG63 cells. Exp. Ther. Med. 2017, 14, 593–599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, J.; Cheng, C.; Verstovsek, S.; Chen, Q.; Jin, Y.; Cao, Q. The BH3-mimetic GX15-070 induces autophagy, potentiates the cytotoxicity of carboplatin and 5-fluorouracil in esophageal carcinoma cells. Cancer Lett. 2010, 293, 167–174. [Google Scholar] [CrossRef] [PubMed]
- Ramalinga, M.; Roy, A.; Srivastava, A.; Bhattarai, A.; Harish, V.; Suy, S.; Collins, S.; Kumar, D. MicroRNA-212 negatively regulates starvation induced autophagy in prostate cancer cells by inhibiting SIRT1 and is a modulator of angiogenesis and cellular senescence. Oncotarget 2015, 6, 34446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corsini, L.R.; Bronte, G.; Terrasi, M.; Amodeo, V.; Fanale, D.; Fiorentino, E.; Cicero, G.; Bazan, V.; Russo, A. The role of microRNAs in cancer: Diagnostic and prognostic biomarkers and targets of therapies. Expert Opin. Ther. Targets 2012, 16, S103–S109. [Google Scholar] [CrossRef] [PubMed]
- Trang, P.; Wiggins, J.F.; Daige, C.L.; Cho, C.; Omotola, M.; Brown, D.; Weidhaas, J.B.; Bader, A.G.; Slack, F.J. Systemic delivery of tumor suppressor microRNA mimics using a neutral lipid emulsion inhibits lung tumors in mice. Mol. Ther. 2011, 19, 1116–1122. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.S.; Erkeland, S.J.; Pester, R.E.; Chen, C.Y.; Ebert, M.S.; Sharp, P.A.; Jacks, T. Suppression of non-small cell lung tumor development by the let-7 microRNA family. Proc. Natl. Acad. Sci. USA 2008, 105, 3903–3908. [Google Scholar] [CrossRef] [Green Version]
- Laschi, M.; Bernardini, G.; Geminiani, M.; Manetti, F.; Mori, M.; Spreafico, A.; Campanacci, D.; Capanna, R.; Schenone, S.; Botta, M. Differentially activated Src kinase in chemo--naïve human primary osteosarcoma cells and effects of a Src kinase inhibitor. Biofactors 2017, 43, 801–811. [Google Scholar] [CrossRef]
- Gingras, A.-C.; Gygi, S.P.; Raught, B.; Polakiewicz, R.D.; Abraham, R.T.; Hoekstra, M.F.; Aebersold, R.; Sonenberg, N. Regulation of 4E-BP1 phosphorylation: A novel two-step mechanism. Genes Dev. 1999, 13, 1422–1437. [Google Scholar] [CrossRef]
- Kim, E.H.; Jo, Y.; Sai, S.; Park, M.-J.; Kim, J.-Y.; Kim, J.S.; Lee, Y.-J.; Cho, J.-M.; Kwak, S.-Y.; Jeong, Y.K. Tumor-treating fields induce autophagy by blocking the Akt2/miR29b axis in glioblastoma cells. Oncogene 2019, 38, 6630–6646. [Google Scholar] [CrossRef]







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oh, J.Y.; Kim, E.H.; Lee, Y.-J.; Sai, S.; Lim, S.H.; Park, J.W.; Chung, H.K.; Kim, J.; Vares, G.; Takahashi, A.; et al. Synergistic Autophagy Effect of miR-212-3p in Zoledronic Acid-Treated In Vitro and Orthotopic In Vivo Models and in Patient-Derived Osteosarcoma Cells. Cancers 2019, 11, 1812. https://doi.org/10.3390/cancers11111812
Oh JY, Kim EH, Lee Y-J, Sai S, Lim SH, Park JW, Chung HK, Kim J, Vares G, Takahashi A, et al. Synergistic Autophagy Effect of miR-212-3p in Zoledronic Acid-Treated In Vitro and Orthotopic In Vivo Models and in Patient-Derived Osteosarcoma Cells. Cancers. 2019; 11(11):1812. https://doi.org/10.3390/cancers11111812
Chicago/Turabian StyleOh, Ju Yeon, Eun Ho Kim, Yeon-Joo Lee, Sei Sai, Sun Ha Lim, Jang Woo Park, Hye Kyung Chung, Joon Kim, Guillaume Vares, Akihisa Takahashi, and et al. 2019. "Synergistic Autophagy Effect of miR-212-3p in Zoledronic Acid-Treated In Vitro and Orthotopic In Vivo Models and in Patient-Derived Osteosarcoma Cells" Cancers 11, no. 11: 1812. https://doi.org/10.3390/cancers11111812