Staphylococcus aureus α-Toxin: Nearly a Century of Intrigue

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Historic Studies
3. Properties of α-Toxin
3.1. Toxin Structure and Regulation of Production

3.2. Host Cell Binding
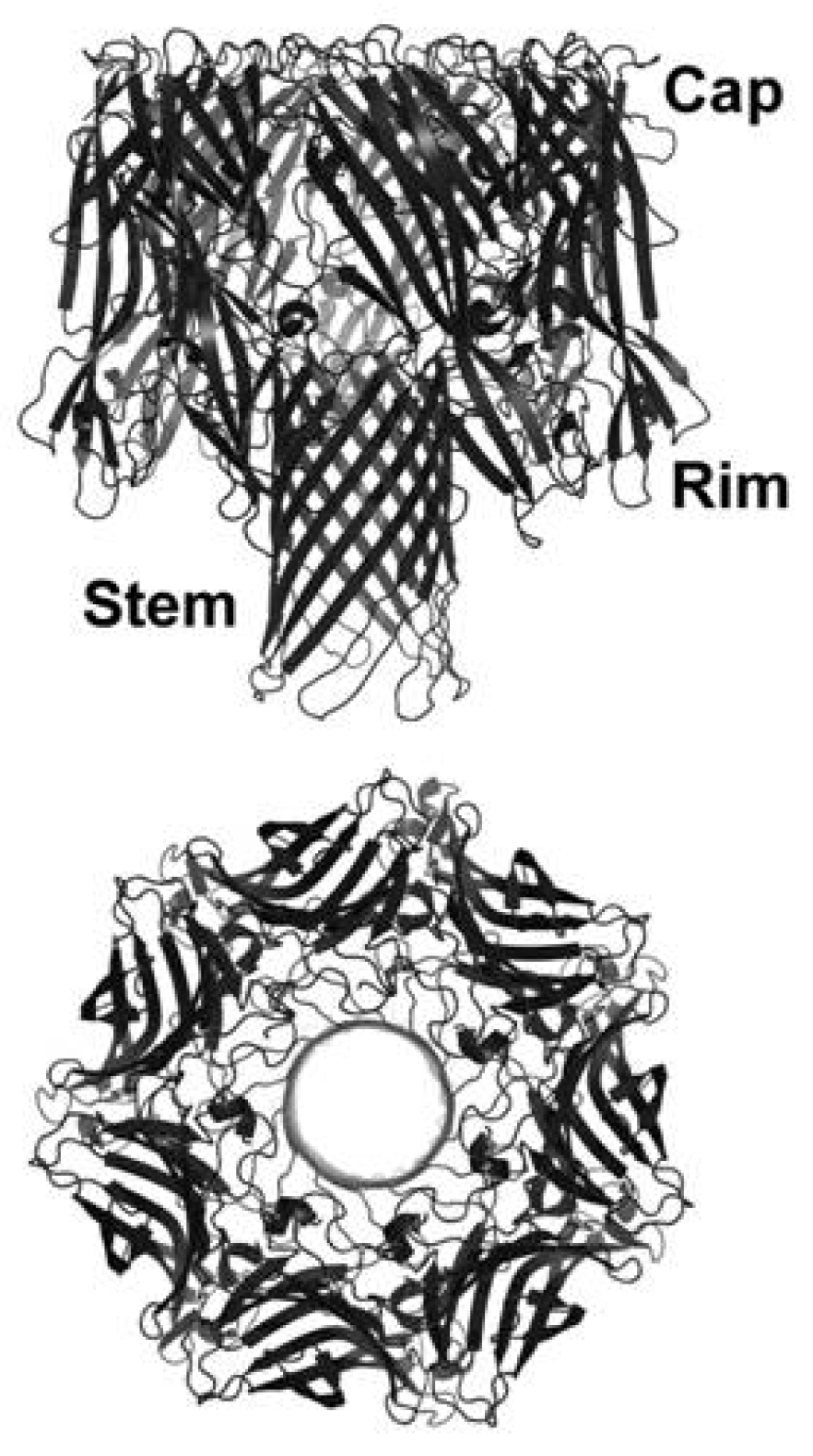
3.3. Oligomerization and Pore Formation
4. Contribution of α-Toxin to S. aureus Disease

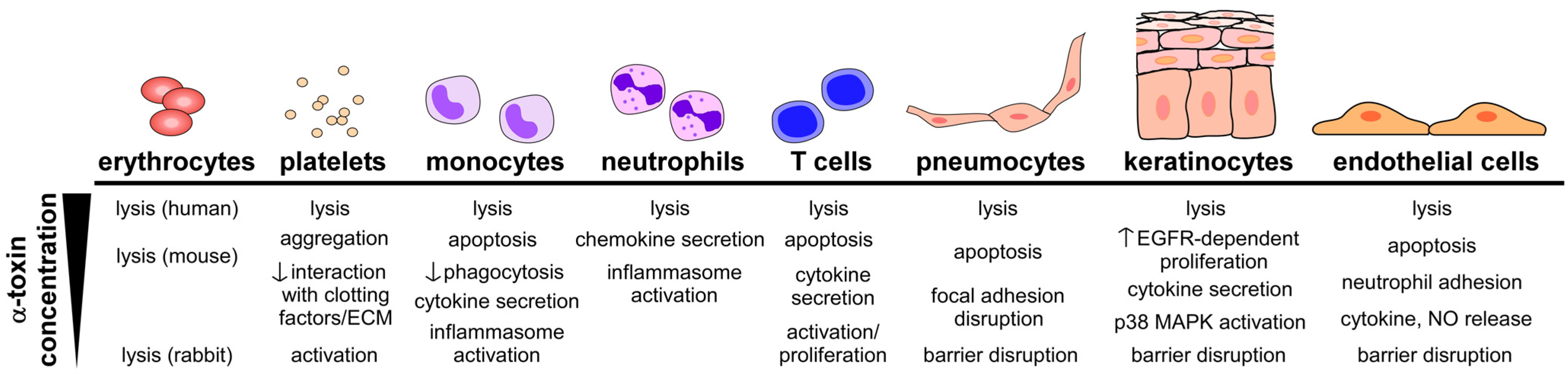
4.1. Toxin-Induced Tissue Injury

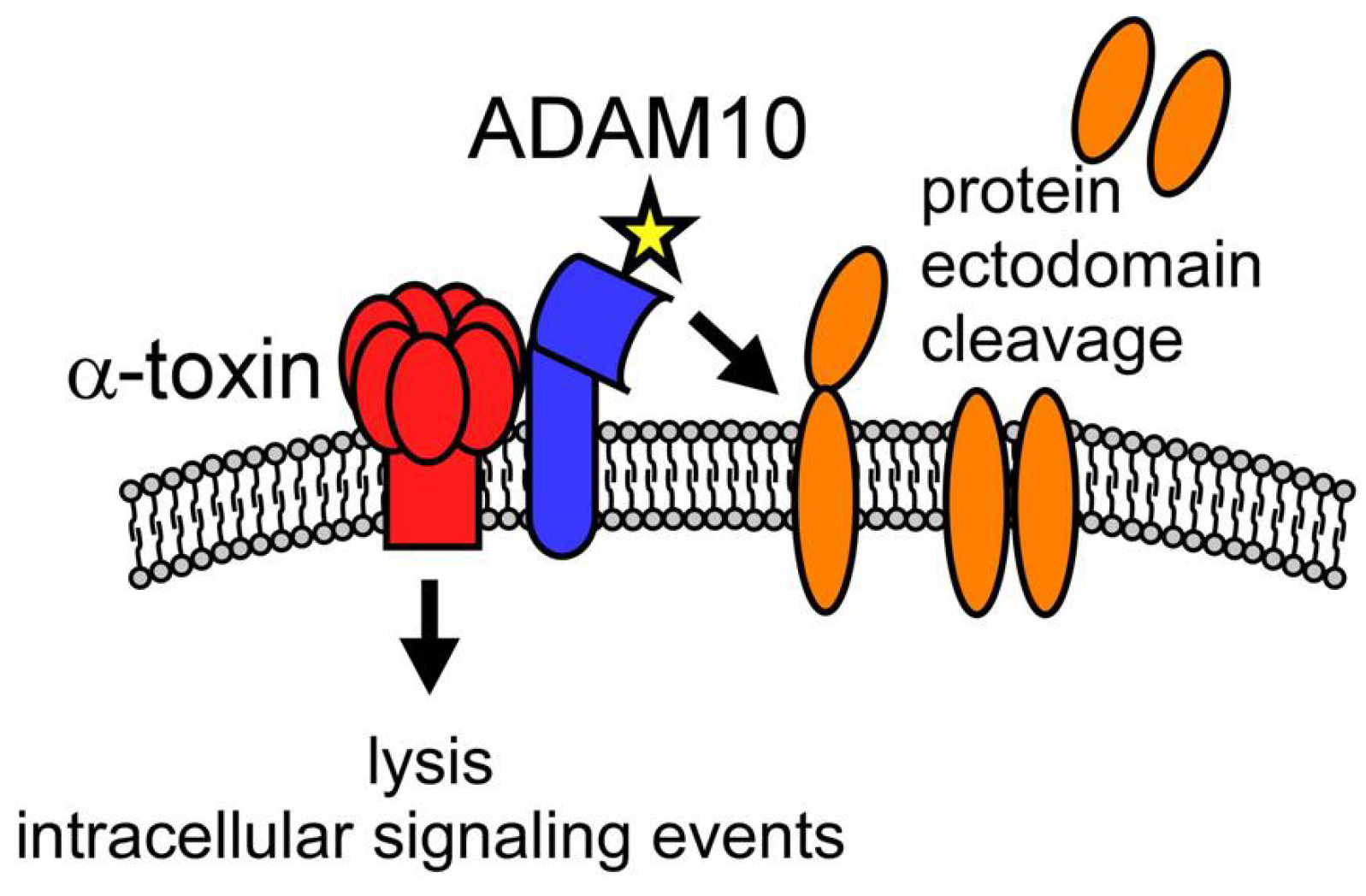
4.2. Toxin-Induced Immunomodulation
5. Conclusions and Future Directions
Conflict of Interest
Acknowledgements
References
- Bhakdi, S.; Tranum-Jensen, J. Alpha-toxin of Staphylococcus aureus. Microbiol. Rev. 1991, 55, 733–751. [Google Scholar]
- Van der Goot, F.G.E. Pore Forming Toxins; Springer Verlag: Berlin, Germany, 2001. [Google Scholar]
- Prevost, G.; Mourey, L.; Colin, D.; Monteil, H.; Dalla Serra, M.; Menestrina, G. Alpha-helix and Beta-barrel Pore-forming Toxins (Leucocidins, alpha-, gamma-, and delta-cytolysins) of Staphylococcus aureus. In The Comprehensive Sourcebook of Bacterial Toxins; Alouf, J.E., Freer, J.H., Eds.; Academic Press: London, UK, 2005; pp. 590–607. [Google Scholar]
- Parker, M.W.; Feil, S.C. Pore-forming protein toxins: From structure to function. Prog. Biophys. Mol. Biol. 2005, 88, 91–142. [Google Scholar] [CrossRef]
- Song, L.; Hobaugh, M.R.; Shustak, C.; Cheley, S.; Bayley, H.; Gouaux, J.E. Structure of staphylococcal alpha-hemolysin, a heptameric transmembrane pore. Science 1996, 274, 1859–1866. [Google Scholar] [CrossRef]
- Gouaux, J.E.; Braha, O.; Hobaugh, M.R.; Song, L.; Cheley, S.; Shustak, C.; Bayley, H. Subunit stoichiometry of staphylococcal alpha-hemolysin in crystals and on membranes: A heptameric transmembrane pore. Proc. Natl. Acad. Sci. USA 1994, 91, 12828–12831. [Google Scholar] [CrossRef]
- Bhakdi, S.; Walev, I.; Hussmann, M.; Valeva, A. Staphylococcal Alpha-Toxin. In Microbial Protein Toxins; Schmitt, M.J., Schaffrath, R., Eds.; Springer-Verlag: Berlin, Germany, 2005. [Google Scholar]
- Haugwitz, U.; Bobkiewicz, W.; Han, S.R.; Beckmann, E.; Veerachato, G.; Shaid, S.; Biehl, S.; Dersch, K.; Bhakdi, S.; Husmann, M. Pore-forming Staphylococcus aureus alpha-toxin triggers epidermal growth factor receptor-dependent proliferation. Cell Microbiol. 2006, 8, 1591–1600. [Google Scholar] [CrossRef]
- Craven, R.R.; Gao, X.; Allen, I.C.; Gris, D.; Bubeck Wardenburg, J.; McElvania-Tekippe, E.; Ting, J.P.; Duncan, J.A. Staphylococcus aureus alpha-hemolysin activates the nlrp3-inflammasome in human and mouse monocytic cells. PLoS One 2009, 4, e7446. [Google Scholar] [CrossRef]
- Cho, J.S.; Guo, Y.; Ramos, R.I.; Hebroni, F.; Plaisier, S.B.; Xuan, C.; Granick, J.L.; Matsushima, H.; Takashima, A.; Iwakura, Y.; et al. Neutrophil-derived il-1beta is sufficient for abscess formation in immunity against Staphylococcus aureus in mice. PLoS Pathog. 2012, 8, e1003047. [Google Scholar] [CrossRef]
- Inoshima, I.; Inoshima, N.; Wilke, G.A.; Powers, M.E.; Frank, K.M.; Wang, Y.; Bubeck Wardenburg, J. A Staphylococcus aureus pore-forming toxin subverts the activity of adam10 to cause lethal infection in mice. Nat. Med. 2011, 17, 1310–1314. [Google Scholar] [CrossRef]
- Inoshima, N.; Wang, Y.; Wardenburg, J.B. Genetic requirement for adam10 in severe Staphylococcus aureus skin infection. J. Invest. Dermatol. 2012, 132, 1513–1516. [Google Scholar] [CrossRef]
- Powers, M.E.; Kim, H.K.; Wang, Y.; Bubeck Wardenburg, J. Adam10 mediates vascular injury induced by Staphylococcus aureus alpha-hemolysin. J. Infect. Dis. 2012, 206, 352–356. [Google Scholar] [CrossRef]
- Burnet, F.M. The exotoxins of Staphylococcus pyogenes aureus. J. Pathol. Bacteriol. 1929, 32, 717–734. [Google Scholar] [CrossRef]
- Burnet, F.M. The production of staphylococcal toxin. J. Pathol. Bacteriol. 1930, 33, 1–16. [Google Scholar] [CrossRef]
- Cassidy, P.S.; Harshman, S. The binding of staphylococcal 125I-alpha-toxin (b) to erythrocytes. J. Biol. Chem. 1973, 248, 5545–5546. [Google Scholar]
- Cassidy, P.; Harshman, S. Studies on the binding of staphylococcal 125I-labeled alpha-toxin to rabbit erythrocytes. Biochemistry 1976, 15, 2348–2355. [Google Scholar] [CrossRef]
- Hildebrand, A.; Pohl, M.; Bhakdi, S. Staphylococcus aureus alpha-toxin. Dual mechanism of binding to target cells. J. Biol. Chem. 1991, 266, 17195–17200. [Google Scholar]
- Siegel, I.; Cohen, S. Action of staphylococcal toxin on human platelets. J. Infect. Dis. 1964, 114, 488–502. [Google Scholar] [CrossRef]
- Bubeck Wardenburg, J.; Patel, R.J.; Schneewind, O. Surface proteins and exotoxins are required for the pathogenesis of Staphylococcus aureus pneumonia. Infect. Immun. 2007, 75, 1040–1044. [Google Scholar] [CrossRef]
- Grimminger, F.; Rose, F.; Sibelius, U.; Meinhardt, M.; Potzsch, B.; Spriestersbach, R.; Bhakdi, S.; Suttorp, N.; Seeger, W. Human endothelial cell activation and mediator release in response to the bacterial exotoxins Escherichia coli hemolysin and staphylococcal alpha-toxin. J. Immunol. 1997, 159, 1909–1916. [Google Scholar]
- Nygaard, T.K.; Pallister, K.B.; DuMont, A.L.; DeWald, M.; Watkins, R.L.; Pallister, E.Q.; Malone, C.; Griffith, S.; Horswill, A.R.; Torres, V.J.; et al. Alpha-toxin induces programmed cell death of human T cells, B cells, and monocytes during USA300 infection. PLoS One 2012, 7, e36532. [Google Scholar]
- Manohar, M.; Maheswaran, S.K.; Frommes, S.P.; Lindorfer, R.K. Platelet damaging factor, a fifth activity of staphylococcal alpha-toxin. J. Bacteriol. 1967, 94, 224–231. [Google Scholar]
- Bhakdi, S.; Muhly, M.; Mannhardt, U.; Hugo, F.; Klapettek, K.; Mueller-Eckhardt, C.; Roka, L. Staphylococcal alpha toxin promotes blood coagulation via attack on human platelets. J. Exp. Med. 1988, 168, 527–542. [Google Scholar] [CrossRef]
- Holtfreter, S.; Nguyen, T.T.H.; Wertheim, H.; Steil, L.; Kusch, H.; Truong, Q.P.; Engelmann, S.; Hecker, M.; Volker, U.; van Belkum, A.; et al. Human immune proteome in experimental colonization with Staphylococcus aureus. Clin. Vaccine Immunol. 2009, 16, 1607–1614. [Google Scholar] [CrossRef]
- Kolata, J.; Bode, L.G.; Holtfreter, S.; Steil, L.; Kusch, H.; Holtfreter, B.; Albrecht, D.; Hecker, M.; Engelmann, S.; van Belkum, A.; et al. Distinctive patterns in the human antibody response to Staphylococcus aureus bacteremia in carriers and non-carriers. Proteomics 2011, 11, 3914–3927. [Google Scholar]
- Fritz, S.A.; Tiemann, K.M.; Hogan, P.G.; Epplin, E.K.; Rodriguez, M.; Al-Zubeidi, D.N.; Bubeck Wardenburg, J.; Hunstad, D.A. A serologic correlate of protective immunity against community-onset Staphylococcus aureus infection. Clin. Infect. Dis. 2013, 56, 1554–1561. [Google Scholar] [CrossRef]
- Bubeck Wardenburg, J.; Bae, T.; Otto, M.; Deleo, F.R.; Schneewind, O. Poring over pores: Alpha-hemolysin and panton-valentine leukocidin in Staphylococcus aureus pneumonia. Nat. Med. 2007, 13, 1405–1406. [Google Scholar]
- Frank, K.M.; Zhou, T.; Moreno-Vinasco, L.; Hollett, B.; Garcia, J.G.; Bubeck Wardenburg, J. Host response signature to Staphylococcus aureus alpha-hemolysin implicates pulmonary th17 response. Infect. Immun. 2012, 80, 3161–3169. [Google Scholar] [CrossRef]
- Kennedy, A.D.; Bubeck Wardenburg, J.; Gardner, D.J.; Long, D.; Whitney, A.R.; Braughton, K.R.; Schneewind, O.; DeLeo, F.R. Targeting of alpha-hemolysin by active or passive immunization decreases severity of USA300 skin infection in a mouse model. J. Infect. Dis. 2010, 202, 1050–1058. [Google Scholar] [CrossRef]
- Kebaier, C.; Chamberland, R.R.; Allen, I.C.; Gao, X.; Broglie, P.M.; Hall, J.D.; Jania, C.; Doerschuk, C.M.; Tilley, S.L.; Duncan, J.A. Staphylococcus aureus alpha-hemolysin mediates virulence in a murine model of severe pneumonia through activation of the nlrp3 inflammasome. J. Infect. Dis. 2012, 205, 807–817. [Google Scholar] [CrossRef]
- Kielian, T.; Cheung, A.; Hickey, W.F. Diminished virulence of an alpha-toxin mutant of Staphylococcus aureus in experimental brain abscesses. Infect. Immun. 2001, 69, 6902–6911. [Google Scholar] [CrossRef]
- O’Callaghan, R.J.; Callegan, M.C.; Moreau, J.M.; Green, L.C.; Foster, T.J.; Hartford, O.M.; Engel, L.S.; Hill, J.M. Specific roles of alpha-toxin and beta-toxin during Staphylococcus aureus corneal infection. Infect. Immun. 1997, 65, 1571–1578. [Google Scholar]
- Arbuthnott, J.P. Staphylococcal Alpha-toxin. In Microbial Toxins; Montie, T.C., Kadis, S., Ajl, S.I., Eds.; Academic Press: New York, NY, USA, 1970; Volume III, pp. 189–236. [Google Scholar]
- Wiseman, G.M. The hemolysins of Staphylococcus aureus. Bacteriol. Rev. 1975, 39, 317–344. [Google Scholar]
- Gouaux, E. Alpha-hemolysin from Staphylococcus aureus: An archetype of beta-barrel, channel-forming toxins. J. Struct. Biol. 1998, 121, 110–122. [Google Scholar] [CrossRef]
- De Christmas, M.J. Recherches expérimentales sur la suppuration. Ann. Inst. Pasteur 1888, 2, 469. [Google Scholar]
- Von Leber, T. Uber die entstehung der entzundung und die wirkung der entzundungserregenden schadlichkeiten. Fortschr. Med. 1888, 6, 460. [Google Scholar]
- Breiger, L.; Fraenkel, C. Untersuchungen uber bacteriengifte. Berlin Klin. Wochschr. 1890, 27, 241. [Google Scholar]
- Rodet, A.; Courmont, J. Produits du staphylocoque pyogène. Bull. Med. 1892, 23, 84. [Google Scholar]
- Van de Velde, H. Mécanisme de la virulence du staphylocoque pyogène. Cellule 1894, 10, 401. [Google Scholar]
- Neisser, M.; Wechsberg, F. Ueber das staphylotoxin. Z. Hyg. Infektionskrankh 1901, 36, 299. [Google Scholar] [CrossRef]
- Kraus, R.; Pribram, E. Ueber staphylokokkentoxin und dessen antitoxin. Wien. Klin. Wochschr. 1906, 17, 493. [Google Scholar]
- Royal Commission of Inquiry into Fatalities at Bundaberg, Report of the Royal Commission of Inquiry into Fatalities at Bundaberg, Together with Appendices; Green, H.J. (Ed.) Government Printer: Melbourne, Australia, 1928.
- Elek, S.D. Staphylococcus Pyogenes; E. and S. Livingstone, Ltd.: Edinburgh, UK, 1959. [Google Scholar]
- Glenny, A.T.; Stevens, M.F. Staphylococcus toxins and antitoxins. J. Pathol. Bacteriol. 1935, 40, 201–210. [Google Scholar] [CrossRef]
- Cooper, L.Z.; Madoff, M.A.; Weinstein, L. Heat stability and species range of purified staphylococcal alpha-toxin. J. Bacteriol. 1966, 91, 1686–1692. [Google Scholar]
- Bernheimer, A.W.; Avigad, L.S.; Grushoff, P. Lytic effects of staphylococcal alpha-toxin and delta-hemolysin. J. Bacteriol. 1968, 96, 487–491. [Google Scholar]
- Cooper, L.Z.; Madoff, M.A.; Weinstein, L. Hemolysis of rabbit erythrocytes by purified staphylococcal alpha-toxin. I. Kinetics of the lytic reaction. J. Bacteriol. 1964, 87, 127–135. [Google Scholar]
- Kumar, S.; Lindorfer, R.K. The characterization of staphylococcal toxins. I. The electrophoretic migration of the alpha hemolytic, dermonecrotic, lethal, and leucocidal activities of crude toxin. J. Exp. Med. 1962, 115, 1095–1106. [Google Scholar] [CrossRef]
- Bernheimer, A.W.; Schwartz, L.L. Isolation and composition of staphylococcal alpha toxin. J. Gen. Microbiol. 1963, 30, 455–468. [Google Scholar] [CrossRef]
- Weissmann, G.; Sessa, G.; Bernheimer, A.W. Staphylococcal alpha-toxin: Effects on artificial lipid spherules. Science 1966, 154, 772–774. [Google Scholar]
- Thelestam, M.; Mollby, R.; Wadstrom, T. Effects of staphylococcal alpha-, beta-, delta-, and gamma-hemolysins on human diploid fibroblasts and hela cells: Evaluation of a new quantitative as say for measuring cell damage. Infect. Immun. 1973, 8, 938–946. [Google Scholar]
- Thelestam, M.; Mollby, R. Sensitive assay for detection of toxin-induced damage to the cytoplasmic membrane of human diploid fibroblasts. Infect. Immun. 1975, 12, 225–232. [Google Scholar]
- Thelestam, M.; Mollby, R. Determination of toxin-induced leakage of different-size nucleotides through the plasma membrane of human diploid fibroblasts. Infect. Immun. 1975, 11, 640–648. [Google Scholar]
- Fussle, R.; Bhakdi, S.; Sziegoleit, A.; Tranum-Jensen, J.; Kranz, T.; Wellensiek, H.J. On the mechanism of membrane damage by Staphylococcus aureus alpha-toxin. J. Cell Biol. 1981, 91, 83–94. [Google Scholar] [CrossRef]
- Freer, J.H.; Arbuthnott, J.P.; Bernheimer, A.W. Interaction of staphylococcal alpha-toxin with artificial and natural membranes. J. Bacteriol. 1968, 95, 1153–1168. [Google Scholar]
- Kehoe, M.; Duncan, J.; Foster, T.; Fairweather, N.; Dougan, G. Cloning, expression, and mapping of the Staphylococcus aureus alpha-hemolysin determinant in Escherichia coli k-12. Infect. Immun. 1983, 41, 1105–1111. [Google Scholar]
- Fairweather, N.; Kennedy, S.; Foster, T.J.; Kehoe, M.; Dougan, G. Expression of a cloned Staphylococcus aureus alpha-hemolysin determinant in Bacillus subtilis and Staphylococcus aureus. Infect. Immun. 1983, 41, 1112–1117. [Google Scholar]
- Gray, G.S.; Kehoe, M. Primary sequence of the alpha-toxin gene from Staphylococcus aureus Wood 46. Infect. Immun. 1984, 46, 615–618. [Google Scholar]
- Tweten, R.K.; Christianson, K.K.; Iandolo, J.J. Transport and processing of staphylococcal alpha-toxin. J. Bacteriol. 1983, 156, 524–528. [Google Scholar]
- Tobkes, N.; Wallace, B.A.; Bayley, H. Secondary structure and assembly mechanism of an oligomeric channel protein. Biochemistry 1985, 24, 1915–1920. [Google Scholar] [CrossRef]
- Arbuthnott, J.P.; Freer, J.H.; Bernheimer, A.W. Physical states of staphylococcal alpha-toxin. J. Bacteriol. 1967, 94, 1170–1177. [Google Scholar]
- Remsen, C.C.; Watson, S.W.; Bernheimer, A.W. Evidence for an ordered arrangement in erythrocyte membranes. Biochem. Biophys. Res. Commun. 1970, 40, 1297–1304. [Google Scholar] [CrossRef]
- Freer, J.H.; Arbuthnott, J.P.; Billcliffe, B. Effects of staphylococcal-toxin on the structure of erythrocyte membranes: A biochemical and freeze-etching study. J. Gen. Microbiol. 1973, 75, 321–332. [Google Scholar] [CrossRef]
- Arbuthnott, J.P.; Freer, J.H.; Billcliffe, B. Lipid-induced polymerization of staphylococcal-toxin. J. Gen. Microbiol. 1973, 75, 309–319. [Google Scholar] [CrossRef]
- Arbuthnott, J.P.; Freer, J.H.; McNiven, A.C. Physical properties of staphylococcal alpha-toxin and aspects of alpha-toxin membrane interactions. Contrib. Microbiol. Immunol. 1973, 1, 285–297. [Google Scholar]
- Hugo, F.; Sinner, A.; Reichwein, J.; Bhakdi, S. Quantitation of monomeric and oligomeric forms of membrane-bound staphylococcal alpha-toxin by enzyme-linked immunosorbent assay with a neutralizing monoclonal antibody. Infect. Immun. 1987, 55, 2933–2939. [Google Scholar]
- Reichwein, J.; Hugo, F.; Roth, M.; Sinner, A.; Bhakdi, S. Quantitative analysis of the binding and oligomerization of staphylococcal alpha-toxin in target erythrocyte membranes. Infect. Immun. 1987, 55, 2940–2944. [Google Scholar]
- Bhakdi, S.; Fussle, R.; Tranum-Jensen, J. Staphylococcal alpha-toxin: Oligomerization of hydrophilic monomers to form amphiphilic hexamers induced through contact with deoxycholate detergent micelles. Proc. Natl. Acad. Sci. USA 1981, 78, 5475–5479. [Google Scholar] [CrossRef]
- Recsei, P.; Kreiswirth, B.; O’Reilly, M.; Schlievert, P.; Gruss, A.; Novick, R.P. Regulation of exoprotein gene expression in Staphylococcus aureus by agar. Mol. Gen. Genet. 1986, 202, 58–61. [Google Scholar] [CrossRef]
- Peng, H.L.; Novick, R.P.; Kreiswirth, B.; Kornblum, J.; Schlievert, P. Cloning, characterization, and sequencing of an accessory gene regulator (agr) in Staphylococcus aureus. J. Bacteriol. 1988, 170, 4365–4372. [Google Scholar]
- Novick, R.P.; Ross, H.F.; Projan, S.J.; Kornblum, J.; Kreiswirth, B.; Moghazeh, S. Synthesis of staphylococcal virulence factors is controlled by a regulatory RNA molecule. EMBO J. 1993, 12, 3967–3975. [Google Scholar]
- Lina, G.; Jarraud, S.; Ji, G.; Greenland, T.; Pedraza, A.; Etienne, J.; Novick, R.P.; Vandenesch, F. Transmembrane topology and histidine protein kinase activity of agrC, the agr signal receptor in Staphylococcus aureus. Mol. Microbiol. 1998, 28, 655–662. [Google Scholar] [CrossRef]
- Lyon, G.J.; Wright, J.S.; Muir, T.W.; Novick, R.P. Key determinants of receptor activation in the agr autoinducing peptides of Staphylococcus aureus. Biochemistry 2002, 41, 10095–10104. [Google Scholar] [CrossRef]
- Koenig, R.L.; Ray, J.L.; Maleki, S.J.; Smeltzer, M.S.; Hurlburt, B.K. Staphylococcus aureus agrA binding to the RNAIII-agr regulatory region. J. Bacteriol. 2004, 186, 7549–7555. [Google Scholar] [CrossRef]
- McNiven, A.C.; Arbuthnott, J.P. Cell-associated alpha-toxin from Staphylococcus aureus. J. Med. Microbiol. 1972, 5, 123–127. [Google Scholar] [CrossRef]
- Xiong, Y.Q.; Willard, J.; Yeaman, M.R.; Cheung, A.L.; Bayer, A.S. Regulation of Staphylococcus aureus alpha-toxin gene (hla) expression by agr, sarA, and sae in vitro and in experimental infective endocarditis. J. Infect. Dis. 2006, 194, 1267–1275. [Google Scholar] [CrossRef]
- Reyes, D.; Andrey, D.O.; Monod, A.; Kelley, W.L.; Zhang, G.; Cheung, A.L. Coordinated regulation by agrA, sarA, and sarR to control agr expression in Staphylococcus aureus. J. Bacteriol. 2011, 193, 6020–6031. [Google Scholar] [CrossRef]
- Cheung, A.L.; Chien, Y.T.; Bayer, A.S. Hyperproduction of alpha-hemolysin in a sigB mutant is associated with elevated sarA expression in Staphylococcus aureus. Infect. Immun. 1999, 67, 1331–1337. [Google Scholar]
- Cheung, A.L.; Eberhardt, K.J.; Chung, E.; Yeaman, M.R.; Sullam, P.M.; Ramos, M.; Bayer, A.S. Diminished virulence of a sar-/agr-mutant of Staphylococcus aureus in the rabbit model of endocarditis. J. Clin. Invest. 1994, 94, 1815–1822. [Google Scholar] [CrossRef]
- Valeva, A.; Hellmann, N.; Walev, I.; Strand, D.; Plate, M.; Boukhallouk, F.; Brack, A.; Hanada, K.; Decker, H.; Bhakdi, S. Evidence that clustered phosphocholine head groups serve as sites for binding and assembly of an oligomeric protein pore. J. Biol. Chem. 2006, 281, 26014–26021. [Google Scholar] [CrossRef]
- Menestrina, G. Ionic channels formed by Staphylococcus aureus alpha-toxin: Voltage-dependent inhibition by divalent and trivalent cations. J. Membr. Biol. 1986, 90, 177–190. [Google Scholar] [CrossRef]
- Watanabe, M.; Tomita, T.; Yasuda, T. Membrane-damaging action of staphylococcal alpha-toxin on phospholipid-cholesterol liposomes. Biochim. Biophys. Acta 1987, 898, 257–265. [Google Scholar] [CrossRef]
- Belmonte, G.; Cescatti, L.; Ferrari, B.; Nicolussi, T.; Ropele, M.; Menestrina, G. Pore formation by Staphylococcus aureus alpha-toxin in lipid bilayers. Dependence upon temperature and toxin concentration. Eur. Biophys. J. 1987, 14, 349–358. [Google Scholar]
- Ikigai, H.; Nakae, T. Assembly of the alpha-toxin-hexamer of Staphylococcus aureus in the liposome membrane. J. Biol. Chem. 1987, 262, 2156–2160. [Google Scholar]
- Ikigai, H.; Nakae, T. Interaction of the alpha-toxin of Staphylococcus aureus with the liposome membrane. J. Biol. Chem. 1987, 262, 2150–2155. [Google Scholar]
- Forti, S.; Menestrina, G. Staphylococcal alpha-toxin increases the permeability of lipid vesicles by cholesterol- and ph-dependent assembly of oligomeric channels. Eur. J. Biochem. 1989, 181, 767–773. [Google Scholar] [CrossRef]
- Schwiering, M.; Brack, A.; Stork, R.; Hellmann, N. Lipid and phase specificity of alpha-toxin from S. aureus. Biochim. Biophys. Acta 2013, 1828, 1962–1972. [Google Scholar] [CrossRef]
- Galdiero, S.; Gouaux, E. High resolution crystallographic studies of alpha-hemolysin-phospholipid complexes define heptamer-lipid head group interactions: Implication for understanding protein-lipid interactions. Protein Sci. 2004, 13, 1503–1511. [Google Scholar] [CrossRef]
- Tweten, R.K. Cholesterol-dependent cytolysins, a family of versatile pore-forming toxins. Infect. Immun. 2005, 73, 6199–6209. [Google Scholar] [CrossRef]
- Thay, B.; Wai, S.N.; Oscarsson, J. Staphylococcus aureus alpha-toxin-dependent induction of host cell death by membrane-derived vesicles. PLoS One 2013, 8, e54661. [Google Scholar] [CrossRef]
- Lee, E.Y.; Choi, D.Y.; Kim, D.K.; Kim, J.W.; Park, J.O.; Kim, S.; Kim, S.H.; Desiderio, D.M.; Kim, Y.K.; Kim, K.P.; et al. Gram-positive bacteria produce membrane vesicles: Proteomics-based characterization of Staphylococcus aureus-derived membrane vesicles. Proteomics 2009, 9, 5425–5436. [Google Scholar] [CrossRef]
- Wilke, G.A.; Bubeck Wardenburg, J. Role of a disintegrin and metalloprotease 10 in Staphylococcus aureus alpha-hemolysin-mediated cellular injury. Proc. Natl. Acad. Sci. USA 2010, 107, 13473–13478. [Google Scholar] [CrossRef]
- Gouaux, E.; Hobaugh, M.; Song, L. Alpha-hemolysin, gamma-hemolysin, and leukocidin from Staphylococcus aureus: Distant in sequence but similar in structure. Protein Sci. 1997, 6, 2631–2635. [Google Scholar] [CrossRef]
- Valeva, A.; Palmer, M.; Bhakdi, S. Staphylococcal α-toxin:Formation of the heptameric pore is partially cooperative and proceeds through multiple intermediate stages? Biochemistry 1997, 36, 13298–13304. [Google Scholar] [CrossRef]
- Valeva, A.; Pongs, J.; Bhakdi, S.; Palmer, M. Staphylococcal alpha-toxin: The role of the n-terminus in formation of the heptameric pore—A fluorescence study. Biochim. Biophys. Acta 1997, 1325, 281–286. [Google Scholar] [CrossRef]
- Kawate, T.; Gouaux, E. Arresting and releasing staphylococcal α-hemolysin at intermediate stages of pore formation by engineered disulfide bonds. Protein Sci. 2003, 12, 997–1006. [Google Scholar] [CrossRef]
- Jursch, R.; Hildebrand, A.; Hobom, G.; Tranum-Jensen, J.; Ward, R.; Kehoe, M.; Bhakdi, S. Histidine residues near the N-terminus of staphylococcal alpha-toxin as reporters of regions that are critical for oligomerization and pore formation. Infect. Immun. 1994, 62, 2249–2256. [Google Scholar]
- Menzies, B.E.; Kernodle, D.S. Site-directed mutagenesis of the alpha-toxin gene of Staphylococcus aureus: Role of histidines in toxin activity in vitro and in a murine model. Infect. Immun. 1994, 62, 1843–1847. [Google Scholar]
- Walker, B.; Bayley, H. Key residues for membrane binding, oligomerization, and pore forming activity of staphylococcal alpha-hemolysin identified by cysteine scanning mutagenesis and targeted chemical modification. J. Biol. Chem. 1995, 270, 23065–23071. [Google Scholar]
- Walker, B.; Bayley, H. Restoration of pore-forming activity in staphylococcal alpha-hemolysin by targeted covalent modification. Protein Eng. 1995, 8, 491–495. [Google Scholar]
- Jayasinghe, L.; Miles, G.; Bayley, H. Role of the amino latch of staphylococcal alpha-hemolysin in pore formation: A co-operative interaction between the N-terminus and position 217. J. Biol. Chem. 2006, 281, 2195–2204. [Google Scholar] [CrossRef]
- Meesters, C.; Brack, A.; Hellmann, N.; Decker, H. Structural characterization of the alpha-hemolysin monomer from Staphylococcus aureus. Proteins 2009, 75, 118–126. [Google Scholar] [CrossRef]
- Foletti, D.; Strop, P.; Shaughnessy, L.; Hasa-Moreno, A.; Casas, M.G.; Russell, M.; Bee, C.; Wu, S.; Pham, A.; Zeng, Z.; et al. Mechanism of action and in vivo efficacy of a human-derived antibody against Staphylococcus aureus alpha-hemolysin. J. Mol. Biol. 2013, 425, 1641–1654. [Google Scholar] [CrossRef]
- Bhakdi, S.; Jursch, R.; Broker, M.; Ronneberger, H.; Hungerer, K.D. Functionally inactive S. aureus alpha-toxin containing a single amino acid substitution: Potential usefulness as a vaccine. Behring Inst. Mitteilungen 1994, 95, 80–84. [Google Scholar]
- Ragle, B.E.; Bubeck Wardenburg, J. Anti-alpha-hemolysin monoclonal antibodies mediate protection against Staphylococcus aureus pneumonia. Infect. Immun. 2009, 77, 2712–2718. [Google Scholar] [CrossRef]
- Tkaczyk, C.; Hua, L.; Varkey, R.; Shi, Y.; Dettinger, L.; Woods, R.; Barnes, A.; MacGill, R.S.; Wilson, S.; Chowdhury, P.; et al. Identification of anti-alpha toxin monoclonal antibodies that reduce the severity of Staphylococcus aureus dermonecrosis and exhibit a correlation between affinity and potency. Clin. Vaccine Immunol. 2012, 19, 377–385. [Google Scholar] [CrossRef]
- Adhikari, R.P.; Ajao, A.O.; Aman, M.J.; Karauzum, H.; Sarwar, J.; Lydecker, A.D.; Johnson, J.K.; Nguyen, C.; Chen, W.H.; Roghmann, M.C. Lower antibody levels to Staphylococcus aureus exotoxins are associated with sepsis in hospitalized adults with invasive S. aureus infections. J. Infect. Dis. 2012, 206, 915–923. [Google Scholar] [CrossRef]
- Roundtree, P.M.; Beard, M.A. Further observations on infections with phage type 80 staphylococci in australia. Med. J. Aust. 1958, 2, 789–795. [Google Scholar]
- Gillespie, W.A.; Alder, V.G. Control of an outbreak of staphylococcal infection in a hospital. Lancet 1957, 272, 632–634. [Google Scholar] [CrossRef]
- Hassall, J.E.; Rountree, P.M. Staphylococcal septicaemia. Lancet 1959, 1, 213–217. [Google Scholar] [CrossRef]
- DeLeo, F.R.; Kennedy, A.D.; Chen, L.; Bubeck Wardenburg, J.; Kobayashi, S.D.; Mathema, B.; Braughton, K.R.; Whitney, A.R.; Villaruz, A.E.; Martens, C.A.; et al. Molecular differentiation of historic phage-type 80/81 and contemporary epidemic Staphylococcus aureus. Proc. Natl. Acad. Sci. USA 2011, 108, 18091–18096. [Google Scholar] [CrossRef]
- Montgomery, C.P.; Boyle-Vavra, S.; Adem, P.V.; Lee, J.C.; Husain, A.N.; Clasen, J.; Daum, R.S. Comparison of virulence in community-associated methicillin-resistant Staphylococcus aureus pulsotypes usa300 and usa400 in a rat model of pneumonia. J. Infect. Dis. 2008, 198, 561–570. [Google Scholar] [CrossRef]
- Montgomery, C.P.; Boyle-Vavra, S.; Daum, R.S. Importance of the global regulators agr and saeRS in the pathogenesis of CA-MRSA USA300 infection. PLoS One 2010, 5, e15177. [Google Scholar] [CrossRef]
- Bubeck Wardenburg, J.; Schneewind, O. Vaccine protection against Staphylococcus aureus pneumonia. J. Exp. Med. 2008, 205, 287–294. [Google Scholar] [CrossRef]
- Barretti, P.; Montelli, A.C.; Batalha, J.E.; Caramori, J.C.; Cunha Mde, L. The role of virulence factors in the outcome of staphylococcal peritonitis in capd patients. BMC Infect. Dis. 2009, 9, 212. [Google Scholar] [CrossRef]
- Patel, A.H.; Nowlan, P.; Weavers, E.D.; Foster, T. Virulence of protein a-deficient and alpha-toxin-deficient mutants of Staphylococcus aureus isolated by allele replacement. Infect. Immun. 1987, 55, 3103–3110. [Google Scholar]
- Menzies, B.E.; Kernodle, D.S. Passive immunization with antiserum to a nontoxic alpha-toxin mutant from Staphylococcus aureus is protective in a murine model. Infect. Immun. 1996, 64, 1839–1841. [Google Scholar]
- O’Reilly, M.; de Azavedo, J.C.; Kennedy, S.; Foster, T.J. Inactivation of the alpha-haemolysin gene of Staphylococcus aureus 8325-4 by site-directed mutagenesis and studies on the expression of its haemolysins. Microbial. Pathog. 1986, 1, 125–138. [Google Scholar] [CrossRef]
- Rauch, S.; DeDent, A.C.; Kim, H.K.; Bubeck Wardenburg, J.; Missiakas, D.M.; Schneewind, O. Abscess formation and alpha-hemolysin induced toxicity in a mouse model of Staphylococcus aureus peritoneal infection. Infect. Immun. 2012, 80, 3721–3732. [Google Scholar] [CrossRef]
- Bayer, A.S.; Ramos, M.D.; Menzies, B.E.; Yeaman, M.R.; Shen, A.J.; Cheung, A.L. Hyperproduction of alpha-toxin by Staphylococcus aureus results in paradoxically reduced virulence in experimental endocarditis: A host defense role for platelet microbicidal proteins. Infect. Immun. 1997, 65, 4652–4660. [Google Scholar]
- Bramley, A.J.; Patel, A.H.; O’Reilly, M.; Foster, R.; Foster, T.J. Roles of alpha-toxin and beta-toxin in virulence of Staphylococcus aureus for the mouse mammary gland. Infect. Immun. 1989, 57, 2489–2494. [Google Scholar]
- Jonsson, P.; Lindberg, M.; Haraldsson, I.; Wadstrom, T. Virulence of staphylococcus aureus in a mouse mastitis model: Studies of alpha hemolysin, coagulase, and protein a as possible virulence determinants with protoplast fusion and gene cloning. Infect. Immun. 1985, 49, 765–769. [Google Scholar]
- McGee, M.P.; Kreger, A.; Leake, E.S.; Harshman, S. Toxicity of staphylococcal alpha toxin for rabbit alveolar macrophages. Infect. Immun. 1983, 39, 439–444. [Google Scholar]
- Yarovinsky, T.O.; Monick, M.M.; Husmann, M.; Hunninghake, G.W. Interferons increase cell resistance to staphylococcal alpha-toxin. Infect. Immun. 2008, 76, 571–577. [Google Scholar] [CrossRef]
- Hruz, P.; Zinkernagel, A.S.; Jenikova, G.; Botwin, G.J.; Hugot, J.P.; Karin, M.; Nizet, V.; Eckmann, L. Nod2 contributes to cutaneous defense against Staphylococcus aureus through alpha-toxin-dependent innate immune activation. Proc. Natl. Acad. Sci. USA 2009, 106, 12873–12878. [Google Scholar] [CrossRef]
- Lizak, M.; Yarovinsky, T.O. Phospholipid scramblase 1 mediates type I interferon-induced protection against staphylococcal alpha-toxin. Cell Host Microbe 2012, 11, 70–80. [Google Scholar] [CrossRef]
- Seals, D.F. The adams family of metalloproteases: Multidomain proteins with multiple functions. Genes Dev. 2003, 17, 7–30. [Google Scholar] [CrossRef]
- Edwards, D.; Handsley, M.; Pennington, C. The adam metalloproteinases. Mol. Aspects Med. 2008, 29, 258–289. [Google Scholar] [CrossRef]
- Nagano, O.; Murakami, D.; Hartmann, D.; de Strooper, B.; Saftig, P.; Iwatsubo, T.; Nakajima, M.; Shinohara, M.; Saya, H. Cell-matrix interaction via cd44 is independently regulated by different metalloproteinases activated in response to extracellular Ca(2+) influx and PKC activation. J. Cell Biol. 2004, 165, 893–902. [Google Scholar] [CrossRef]
- Allinson, T.M.; Parkin, E.T.; Turner, A.J.; Hooper, N.M. Adams family members as amyloid precursor protein alpha-secretases. J. Neurosci. Res. 2003, 74, 342–352. [Google Scholar] [CrossRef]
- Maretzky, T.; Reiss, K.; Ludwig, A.; Buchholz, J.; Scholz, F.; Proksch, E.; de Strooper, B.; Hartmann, D.; Saftig, P. Adam10 mediates E-cadherin shedding and regulates epithelial cell-cell adhesion, migration, and beta-catenin translocation. Proc. Natl. Acad. Sci. USA 2005, 102, 9182–9187. [Google Scholar] [CrossRef]
- Janes, P.W.; Saha, N.; Barton, W.A.; Kolev, M.V.; Wimmer-Kleikamp, S.H.; Nievergall, E.; Blobel, C.P.; Himanen, J.P.; Lackmann, M.; Nikolov, D.B. Adam meets eph: An adam substrate recognition module acts as a molecular switch for ephrin cleavage in trans. Cell 2005, 123, 291–304. [Google Scholar] [CrossRef]
- Gardiner, E.E.; Karunakaran, D.; Shen, Y.; Arthur, J.F.; Andrews, R.K.; Berndt, M.C. Controlled shedding of platelet glycoprotein (GP)VI and GPIb-IX-V by adam family metalloproteinases. J. Thromb. Haemost. 2007, 5, 1530–1537. [Google Scholar] [CrossRef]
- Schulte, A.; Schulz, B.; Andrzejewski, M.; Hundhausen, C.; Mletzko, S.; Achilles, J.; Reiss, K.; Paliga, K.; Weber, C.; Rosejohn, S. Sequential processing of the transmembrane chemokines cx3cl1 and cxcl16 by α- and γ-secretases. Biochem. Biophys. Res. Commun. 2007, 358, 233–240. [Google Scholar] [CrossRef]
- Schulz, B.; Pruessmeyer, J.; Maretzky, T.; Ludwig, A.; Blobel, C.P.; Saftig, P.; Reiss, K. Adam10 regulates endothelial permeability and t-cell transmigration by proteolysis of vascular endothelial cadherin. Circ. Res. 2008, 102, 1192–1201. [Google Scholar] [CrossRef]
- Gibb, D.R.; El Shikh, M.; Kang, D.J.; Rowe, W.J.; El Sayed, R.; Cichy, J.; Yagita, H.; Tew, J.G.; Dempsey, P.J.; Crawford, H.C.; et al. Adam10 is essential for notch2-dependent marginal zone b cell development and CD23 cleavage in vivo. J. Exp. Med. 2010, 207, 623–635. [Google Scholar] [CrossRef]
- Saftig, P.; Reiss, K. The “a disintegrin and metalloproteases” ADAM10 and ADAM17: Novel drug targets with therapeutic potential? Eur. J. Cell Biol. 2011, 90, 527–535. [Google Scholar] [CrossRef]
- Horiuchi, K.; Le Gall, S.; Schulte, M.; Yamaguchi, T.; Reiss, K.; Murphy, G.; Toyama, Y.; Hartmann, D.; Saftig, P.; Blobel, C.P. Substrate selectivity of epidermal growth factor-receptor ligand sheddases and their regulation by phorbol esters and calcium influx. Mol. Biol. Cell 2006, 18, 176–188. [Google Scholar] [CrossRef]
- Hartmann, D.; de Strooper, B.; Serneels, L.; Craessaerts, K.; Herreman, A.; Annaert, W.; Umans, L.; Lubke, T.; Lena Illert, A.; von Figura, K.; et al. The disintegrin/metalloprotease adam 10 is essential for notch signalling but not for alpha-secretase activity in fibroblasts. Hum. Mol. Genet. 2002, 11, 2615–2624. [Google Scholar] [CrossRef]
- Tian, L.; Wu, X.; Chi, C.; Han, M.; Xu, T.; Zhuang, Y. Adam10 is essential for proteolytic activation of notch during thymocyte development. Int. Immunol. 2008, 20, 1181–1187. [Google Scholar] [CrossRef]
- Weber, S.; Niessen, M.T.; Prox, J.; Lullmann-Rauch, R.; Schmitz, A.; Schwanbeck, R.; Blobel, C.P.; Jorissen, E.; de Strooper, B.; Niessen, C.M.; et al. The disintegrin/metalloproteinase ADAM10 is essential for epidermal integrity and notch-mediated signaling. Development 2011, 138, 495–505. [Google Scholar] [CrossRef]
- Glomski, K.; Monette, S.; Manova, K.; de Strooper, B.; Saftig, P.; Blobel, C.P. Deletion of ADAM10 in endothelial cells leads to defects in organ-specific vascular structures. Blood 2011, 118, 1163–1174. [Google Scholar] [CrossRef]
- Zhang, C.; Tian, L.; Chi, C.; Wu, X.; Yang, X.; Han, M.; Xu, T.; Zhuang, Y.; Deng, K. ADAM10 is essential for early embryonic cardiovascular development. Dev. Dyn. 2010, 239, 2594–2602. [Google Scholar] [CrossRef]
- Jorissen, E.; Prox, J.; Bernreuther, C.; Weber, S.; Schwanbeck, R.; Serneels, L.; Snellinx, A.; Craessaerts, K.; Thathiah, A.; Tesseur, I.; et al. The disintegrin/metalloproteinase ADAM10 is essential for the establishment of the brain cortex. J. Neurosci. 2010, 30, 4833–4844. [Google Scholar] [CrossRef]
- Yoda, M.; Kimura, T.; Tohmonda, T.; Uchikawa, S.; Koba, T.; Takito, J.; Morioka, H.; Matsumoto, M.; Link, D.C.; Chiba, K.; et al. Dual functions of cell-autonomous and non-cell-autonomous ADAM10 activity in granulopoiesis. Blood 2011, 118, 6939–6942. [Google Scholar] [CrossRef]
- Bhakdi, S.; Muhly, M.; Korom, S.; Hugo, F. Release of interleukin-1 beta associated with potent cytocidal action of staphylococcal alpha-toxin on human monocytes. Infect. Immun. 1989, 57, 3512–3519. [Google Scholar]
- Suttorp, N.; Seeger, W.; Dewein, E.; Bhakdi, S.; Roka, L. Staphylococcal alpha-toxin-induced PGI2 production in endothelial cells: Role of calcium. Am. J. Physiol. 1985, 248, C127–C134. [Google Scholar]
- Rose, F.; Dahlem, G.; Guthmann, B.; Grimminger, F.; Maus, U.; Hanze, J.; Duemmer, N.; Grandel, U.; Seeger, W.; Ghofrani, H.A. Mediator generation and signaling events in alveolar epithelial cells attacked by S. aureus alpha-toxin. Am. J. Physiol. Lung Cell. Mol. Physiol. 2002, 282, L207–L214. [Google Scholar]
- Suttorp, N.; Fuhrmann, M.; Tannert-Otto, S.; Grimminger, F.; Bhadki, S. Pore-forming bacterial toxins potently induce release of nitric oxide in porcine endothelial cells. J. Exp. Med. 1993, 178, 337–341. [Google Scholar] [CrossRef]
- Seeger, W.; Birkemeyer, R.G.; Ermert, L.; Suttorp, N.; Bhakdi, S.; Duncker, H.R. Staphylococcal alpha-toxin-induced vascular leakage in isolated perfused rabbit lungs. Lab. Investig. J. Tech. Methods Pathol. 1990, 63, 341–349. [Google Scholar]
- Buerke, M.; Sibelius, U.; Grandel, U.; Buerke, U.; Grimminger, F.; Seeger, W.; Meyer, J.; Darius, H. Staphylococcus aureus alpha toxin mediates polymorphonuclear leukocyte-induced vasocontraction and endothelial dysfunction. Shock 2002, 17, 30–35. [Google Scholar]
- Suttorp, N.; Hessz, T.; Seeger, W.; Wilke, A.; Koob, R.; Lutz, F.; Drenckhahn, D. Bacterial exotoxins and endothelial permeability for water and albumin in vitro. Am. J. Physiol. 1988, 255, C368–C376. [Google Scholar]
- Maretzky, T.; Scholz, F.; Koten, B.; Proksch, E.; Saftig, P.; Reiss, K. ADAM10-mediated e-cadherin release is regulated by proinflammatory cytokines and modulates keratinocyte cohesion in eczematous dermatitis. J. Invest. Dermatol. 2008, 128, 1737–1746. [Google Scholar] [CrossRef]
- Le Gall, S.M.; Bobe, P.; Reiss, K.; Horiuchi, K.; Niu, X.D.; Lundell, D.; Gibb, D.R.; Conrad, D.; Saftig, P.; Blobel, C.P. ADAMS 10 and 17 represent differentially regulated components of a general shedding machinery for membrane proteins such as transforming growth factor alpha, l-selectin, and tumor necrosis factor alpha. Mol. Biol. Cell 2009, 20, 1785–1794. [Google Scholar] [CrossRef]
- McElroy, M.C.; Harty, H.R.; Hosford, G.E.; Boylan, G.M.; Pittet, J.F.; Foster, T.J. Alpha-toxin damages the air-blood barrier of the lung in a rat model of Staphylococcus aureus-induced pneumonia. Infect. Immun. 1999, 67, 5541–5544. [Google Scholar]
- Parker, D.; Prince, A. Immunopathogenesis of Staphylococcus aureus pulmonary infection. Semin. Immunopathol. 2012, 34, 281–297. [Google Scholar] [CrossRef]
- Miller, L.S.; Cho, J.S. Immunity against Staphylococcus aureus cutaneous infections. Nat. Rev. Immunol. 2011, 11, 505–518. [Google Scholar]
- Bartlett, A.H.; Foster, T.J.; Hayashida, A.; Park, P.W. Alpha-toxin facilitates the generation of cxc chemokine gradients and stimulates neutrophil homing in Staphylococcus aureus pneumonia. J. Infect. Dis. 2008, 198, 1529–1535. [Google Scholar] [CrossRef]
- Suttorp, N.; Buerke, M.; Tannert-Otto, S. Stimulation of paf-synthesis in pulmonary artery endothelial cells by Staphylococcus aureus alpha-toxin. Thromb. Res. 1992, 67, 243–252. [Google Scholar] [CrossRef]
- Onogawa, T. Staphylococcal alpha-toxin synergistically enhances inflammation caused by bacterial components. FEMS Immunol. Med. Microbiol. 2002, 33, 15–21. [Google Scholar]
- Fink, S.L.; Cookson, B.T. Apoptosis, pyroptosis, and necrosis: Mechanistic description of dead and dying eukaryotic cells. Infect. Immun. 2005, 73, 1907–1916. [Google Scholar] [CrossRef]
- Ragle, B.E.; Karginov, V.A.; Bubeck Wardenburg, J. Prevention and treatment of Staphylococcus aureus pneumonia with a beta-cyclodextrin derivative. Antimicrob. Agents Chemother. 2010, 54, 298–304. [Google Scholar] [CrossRef]
- Miller, L.S.; Pietras, E.M.; Uricchio, L.H.; Hirano, K.; Rao, S.; Lin, H.; O’Connell, R.M.; Iwakura, Y.; Cheung, A.L.; Cheng, G.; et al. Inflammasome-mediated production of il-1beta is required for neutrophil recruitment against Staphylococcus aureus in vivo. J. Immunol. 2007, 179, 6933–6942. [Google Scholar]
- Niebuhr, M.; Mamerow, D.; Heratizadeh, A.; Satzger, I.; Werfel, T. Staphylococcal alpha-toxin induces a higher t cell proliferation and interleukin-31 in atopic dermatitis. Int. Arch. Allergy Immunol. 2011, 156, 412–415. [Google Scholar] [CrossRef]
- Cho, J.S.; Pietras, E.M.; Garcia, N.C.; Ramos, R.I.; Farzam, D.M.; Monroe, H.R.; Magorien, J.E.; Blauvelt, A.; Kolls, J.K.; Cheung, A.L.; et al. Il-17 is essential for host defense against cutaneous Staphylococcus aureus infection in mice. J. Clin. Invest. 2010, 120, 1762–1773. [Google Scholar] [CrossRef]
- Deshmukh, H.S.; Hamburger, J.B.; Ahn, S.H.; McCafferty, D.G.; Yang, S.R.; Fowler, V.G., Jr. Critical role of NOD2 in regulating the immune response to Staphylococcus aureus. Infect. Immun. 2009, 77, 1376–1382. [Google Scholar] [CrossRef]
- Van Beelen, A.J.; Zelinkova, Z.; Taanman-Kueter, E.W.; Muller, F.J.; Hommes, D.W.; Zaat, S.A.; Kapsenberg, M.L.; de Jong, E.C. Stimulation of the intracellular bacterial sensor NOD2 programs dendritic cells to promote interleukin-17 production in human memory T cells. Immunity 2007, 27, 660–669. [Google Scholar] [CrossRef]
- Valeva, A.; Walev, I.; Gerber, A.; Klein, J.; Palmer, M.; Bhakdi, S. Staphylococcal alpha-toxin: Repair of a calcium-impermeable pore in the target cell membrane. Mol. Microbiol. 2000, 36, 467–476. [Google Scholar] [CrossRef]
- Husmann, M.; Dersch, K.; Bobkiewicz, W.; Beckmann, E.; Veerachato, G.; Bhakdi, S. Differential role of p38 mitogen activated protein kinase for cellular recovery from attack by pore-forming S. aureus alpha-toxin or streptolysin o. Biochem. Biophys. Res. Commun. 2006, 344, 1128–1134. [Google Scholar]
- Husmann, M.; Beckmann, E.; Boller, K.; Kloft, N.; Tenzer, S.; Bobkiewicz, W.; Neukirch, C.; Bayley, H.; Bhakdi, S. Elimination of a bacterial pore-forming toxin by sequential endocytosis and exocytosis. FEBS Lett. 2009, 583, 337–344. [Google Scholar] [CrossRef]
- Bhakdi, S.; Mannhardt, U.; Muhly, M.; Hugo, F.; Ronneberger, H.; Hungerer, K.D. Human hyperimmune globulin protects against the cytotoxic action of staphylococcal alpha-toxin in vitro and in vivo. Infect. Immun. 1989, 57, 3214–3220. [Google Scholar]
- Blomqvist, L.; Sjogren, A. Production and characterization of monoclonal antibodies against Staphylococcus aureus alpha-toxin. Toxicon 1988, 26, 265–273. [Google Scholar] [CrossRef]
- Spaulding, A.R.; Lin, Y.C.; Merriman, J.A.; Brosnahan, A.J.; Peterson, M.L.; Schlievert, P.M. Immunity to Staphylococcus aureus secreted proteins protects rabbits from serious illnesses. Vaccine 2012, 30, 5099–5109. [Google Scholar] [CrossRef]
- Adhikari, R.P.; Karauzum, H.; Sarwar, J.; Abaandou, L.; Mahmoudieh, M.; Boroun, A.R.; Vu, H.; Nguyen, T.; Devi, V.S.; Shulenin, S.; et al. Novel structurally designed vaccine for S. aureus alpha-hemolysin: Protection against bacteremia and pneumonia. PLoS One 2012, 7, e38567. [Google Scholar]
- Pozzi, C.; Wilk, K.; Lee, J.C.; Gening, M.; Nifantiev, N.; Pier, G.B. Opsonic and protective properties of antibodies raised to conjugate vaccines targeting six Staphylococcus aureus antigens. PLoS One 2012, 7, e46648. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Berube, B.J.; Wardenburg, J.B. Staphylococcus aureus α-Toxin: Nearly a Century of Intrigue. Toxins 2013, 5, 1140-1166. https://doi.org/10.3390/toxins5061140
Berube BJ, Wardenburg JB. Staphylococcus aureus α-Toxin: Nearly a Century of Intrigue. Toxins. 2013; 5(6):1140-1166. https://doi.org/10.3390/toxins5061140
Chicago/Turabian StyleBerube, Bryan J., and Juliane Bubeck Wardenburg. 2013. "Staphylococcus aureus α-Toxin: Nearly a Century of Intrigue" Toxins 5, no. 6: 1140-1166. https://doi.org/10.3390/toxins5061140