Evolution of Three-Finger Toxin Genes in Neotropical Colubrine Snakes (Colubridae)

 , , and
, , and
Abstract
:1. Introduction
2. Results and Discussion
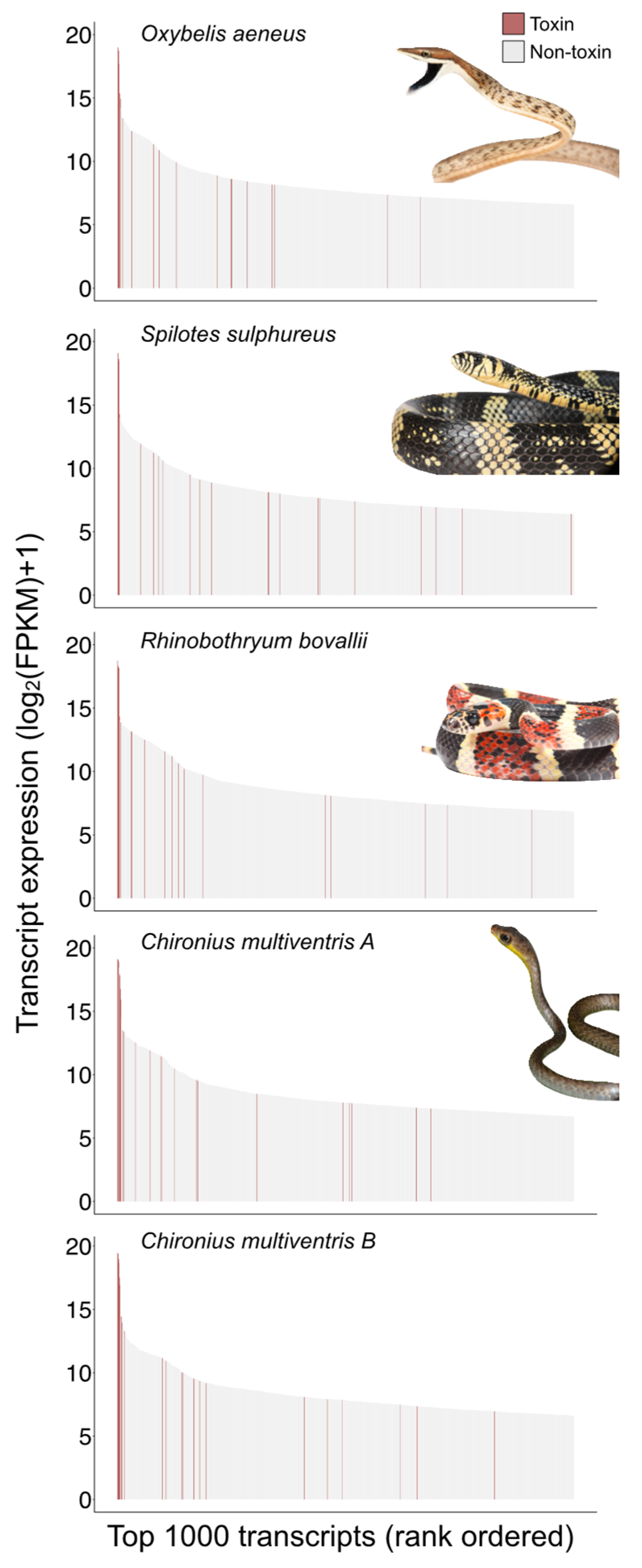
2.1. Colubrine Venom Transcriptomes Are Dominated by 3FTxs
2.2. Parallel Evolution of Heterodimeric Sequences
2.3. Colubrine 3FTxs Are under Positive Selection
3. Conclusions
4. Materials and Methods
4.1. Specimen Selection
4.2. MicroCT Scanning
4.3. RNA Extraction and Transcriptome Assembly
4.4. Gene Identification and Abundance Estimate
4.5. Gene Tree Construction
4.6. Protein Prediction
4.7. Selection Tests
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mackessy, S.P.; Saviola, A.J. Understanding Biological Roles of Venoms among the Caenophidia: The Importance of Rear-Fanged Snakes. Integr. Comp. Biol. 2016, 56, 1004–1021. [Google Scholar] [CrossRef] [PubMed]
- Saviola, A.J.; Peichoto, M.E.; Mackessy, S.P. Rear-Fanged Snake Venoms: An Untapped Source of Novel Compounds and Potential Drug Leads. Toxins Rev. 2014, 33, 185–201. [Google Scholar] [CrossRef]
- Pyron, R.A.; Burbrink, F.T.; Wiens, J.J. A Phylogeny and Revised Classification of Squamata, Including 4161 Species of Lizards and Snakes. BMC Evol. Biol. 2013, 13, 1. [Google Scholar] [CrossRef] [PubMed]
- Westeen, E.P.; Durso, A.M.; Grundler, M.C.; Rabosky, D.L.; Davis Rabosky, A.R. What Makes a Fang? Phylogenetic and Ecological Controls on Tooth Evolution in Rear-Fanged Snakes. BMC Evol. Biol. 2020, 20, 80. [Google Scholar] [CrossRef]
- Avella, I.; Wüster, W.; Luiselli, L.; Martínez-Freiría, F. Toxic Habits: An Analysis of General Trends and Biases in Snake Venom Research. Toxins 2022, 14, 884. [Google Scholar] [CrossRef] [PubMed]
- Junqueira-de-Azevedo, I.L.M.; Campos, P.F.; Ching, A.T.C.; Mackessy, S.P. Colubrid Venom Composition: An -Omics Perspective. Toxins 2016, 8, 230. [Google Scholar] [CrossRef]
- Cerda, P.A.; Crowe-Riddell, J.M.; Gonçalves, D.J.P.; Larson, D.A.; Duda, T.F.; Davis Rabosky, A.R. Divergent Specialization of Simple Venom Gene Profiles among Rear-Fanged Snake Genera (Helicops and Leptodeira, Dipsadinae, Colubridae). Toxins 2022, 14, 489. [Google Scholar] [CrossRef]
- McGivern, J.J.; Wray, K.P.; Margres, M.J.; Couch, M.E.; Mackessy, S.P.; Rokyta, D.R. RNA-Seq and High-Definition Mass Spectrometry Reveal the Complex and Divergent Venoms of Two Rear-Fanged Colubrid Snakes. BMC Genom. 2014, 15, 1061. [Google Scholar] [CrossRef]
- Ching, A.T.C.; Paes Leme, A.F.; Zelanis, A.; Rocha, M.M.T.; Furtado, M.D.F.D.; Silva, D.A.; Trugilho, M.R.O.; da Rocha, S.L.G.; Perales, J.; Ho, P.L.; et al. Venomics Profiling of Thamnodynastes Strigatus Unveils Matrix Metalloproteinases and Other Novel Proteins Recruited to the Toxin Arsenal of Rear-Fanged Snakes. J. Proteome Res. 2012, 11, 1152–1162. [Google Scholar] [CrossRef]
- Brahma, R.K.; McCleary, R.J.R.; Kini, R.M.; Doley, R. Venom Gland Transcriptomics for Identifying, Cataloging, and Characterizing Venom Proteins in Snakes. Toxicon 2015, 93, 1–10. [Google Scholar] [CrossRef]
- Xie, B.; Dashevsky, D.; Rokyta, D.; Ghezellou, P.; Fathinia, B.; Shi, Q.; Richardson, M.K.; Fry, B.G. Dynamic Genetic Differentiation Drives the Widespread Structural and Functional Convergent Evolution of Snake Venom Proteinaceous Toxins. BMC Biol. 2022, 20, 4. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, E.P.; Rautsaw, R.M.; Mason, A.J.; Strickland, J.L.; Parkinson, C.L. Duvernoy’s Gland Transcriptomics of the Plains Black-Headed Snake, Tantilla Nigriceps (Squamata, Colubridae): Unearthing the Venom of Small Rear-Fanged Snakes. Toxins 2021, 13, 336. [Google Scholar] [CrossRef] [PubMed]
- Modahl, C.M.; Mrinalini; Frietze, S.; Mackessy, S.P. Adaptive Evolution of Distinct Prey-Specific Toxin Genes in Rear-Fanged Snake Venom. Proc. R. Soc. B: Biol. Sci. 2018, 285, 20181003. [Google Scholar] [CrossRef] [PubMed]
- Pawlak, J.; Mackessy, S.P.; Sixberry, N.M.; Stura, E.A.; Le Du, M.H.; Ménez, R.; Foo, C.S.; Ménez, A.; Nirthanan, S.; Kini, R.M. Irditoxin, a Novel Covalently Linked Heterodimeric Three-Finger Toxin with High Taxon-Specific Neurotoxicity. FASEB J. 2009, 23, 534–545. [Google Scholar] [CrossRef]
- Pawlak, J.; Mackessy, S.P.; Fry, B.G.; Bhatia, M.; Mourier, G.; Fruchart-Gaillard, C.; Servent, D.; Ménez, R.; Stura, E.; Ménez, A.; et al. Denmotoxin, a Three-Finger Toxin from the Colubrid Snake Boiga Dendrophila (Mangrove catsnake) with Bird-Specific Activity. J. Biol. Chem. 2006, 281, 29030–29041. [Google Scholar] [CrossRef]
- Modahl, C.M.; Brahma, R.K.; Koh, C.Y.; Shioi, N.; Kini, R.M. Omics Technologies for Profiling Toxin Diversity and Evolution in Snake Venom: Impacts on the Discovery of Therapeutic and Diagnostic Agents. Annu. Rev. Anim. Biosci. 2020, 8, 91–116. [Google Scholar] [CrossRef]
- Modahl, C.M.; Mackessy, S.P. Venoms of Rear-Fanged Snakes: New Proteins and Novel Activities. Front. Ecol. Evol. 2019, 7, 1–18. [Google Scholar] [CrossRef]
- Modahl, C.M.; Mackessy, S.P. Full-Length Venom Protein CDNA Sequences from Venom-Derived MRNA: Exploring Compositional Variation and Adaptive Multigene Evolution. PLoS Negl. Trop. Dis. 2016, 10, e0004587. [Google Scholar] [CrossRef]
- Chang, C.C.; Lee, C.Y. Isolation of neurotoxins from the venom of bungarus multicinctus and their modes of neuromuscular blocking action. Arch. Int. Pharmacodyn. Ther. 1963, 144, 241–257. [Google Scholar] [CrossRef]
- Utkin, Y.N. Last Decade Update for Three-Finger Toxins: Newly Emerging Structures and Biological Activities. World J. Biol. Chem. 2019, 10, 17–27. [Google Scholar] [CrossRef]
- Kini, R.M.; Doley, R. Structure, Function and Evolution of Three-Finger Toxins: Mini Proteins with Multiple Targets. Toxicon 2010, 56, 855–867. [Google Scholar] [CrossRef] [PubMed]
- Dashevsky, D.; Fry, B.G. Ancient Diversification of Three-Finger Toxins in Micrurus Coral Snakes. J. Mol. Evol. 2018, 86, 58–67. [Google Scholar] [CrossRef] [PubMed]
- Fry, B.G.; Wüster, W.; Kini, R.M.; Brusic, V.; Khan, A.; Venkataraman, D.; Rooney, A.P. Molecular Evolution and Phylogeny of Elapid Snake Venom Three-Finger Toxins. J. Mol. Evol. 2003, 57, 110–129. [Google Scholar] [CrossRef]
- Dashevsky, D.; Rokyta, D.; Frank, N.; Nouwens, A.; Fry, B.G. Electric Blue: Molecular Evolution of Three-Finger Toxins in the Long-Glanded Coral Snake Species Calliophis Bivirgatus. Toxins 2021, 13, 124. [Google Scholar] [CrossRef] [PubMed]
- Dashevsky, D.; Debono, J.; Rokyta, D.; Nouwens, A.; Josh, P.; Fry, B.G. Three-Finger Toxin Diversification in the Venoms of Cat-Eye Snakes (Colubridae: Boiga). J. Mol. Evol. 2018, 86, 531–545. [Google Scholar] [CrossRef] [PubMed]
- Fry, B.G.; Lumsden, N.G.; Wüster, W.; Wickramaratna, J.C.; Hodgson, W.C.; Manjunatha Kini, R. Isolation of a Neurotoxin (α-Colubritoxin) from a Nonvenomous Colubrid: Evidence for Early Origin of Venom in Snakes. J. Mol. Evol. 2003, 57, 446–452. [Google Scholar] [CrossRef]
- Heyborne, W.H.; Mackessy, S.P. Venoms of New World Vinesnakes (Oxybelis aeneus and O. Fulgidus). Toxicon 2021, 190, 22–30. [Google Scholar] [CrossRef]
- Calvete, J.J.; Bonilla, F.; Granados-Martínez, S.; Sanz, L.; Lomonte, B.; Sasa, M. Venomics of the Duvernoy’s Gland Secretion of the False Coral Snake Rhinobothryum Bovallii (Andersson, 1916) and Assessment of Venom Lethality towards Synapsid and Diapsid Animal Models. J. Proteom. 2020, 225, 103882. [Google Scholar] [CrossRef]
- Modahl, C.M.; Saviola, A.J.; Mackessy, S.P. Integration of Transcriptomic and Proteomic Approaches for Snake Venom Profiling. Expert. Rev. Proteom. 2021, 18, 827–834. [Google Scholar] [CrossRef]
- Schramer, T.D.; Rautsaw, R.M.; Bayona-Serrano, J.D.; Nystrom, G.S.; West, T.R.; Ortiz-Medina, J.A.; Sabido-Alpuche, B.; Meneses-Millán, M.; Borja, M.; Junqueira-de-Azevedo, I.L.M.; et al. An Integrative View of the Toxic Potential of Conophis Lineatus (Dipsadidae: Xenodontinae), a Medically Relevant Rear-Fanged Snake. Toxicon 2022, 205, 38–52. [Google Scholar] [CrossRef]
- Casewell, N.R.; Jackson, T.N.W.; Laustsen, A.H.; Sunagar, K. Causes and Consequences of Snake Venom Variation. Trends Pharmacol. Sci. 2020, 41, 570–581. [Google Scholar] [CrossRef] [PubMed]
- Facente, J.; McGivern, J.J.; Seavy, M.; Wray, K.P.; Rokyta, D.R. Contrasting Modes and Tempos of Venom Expression Evolution in Two Snake Species. Genetics 2015, 199, 165–176. [Google Scholar] [CrossRef]
- Zhang, Z.Y.; Lv, Y.; Wu, W.; Yan, C.; Tang, C.Y.; Peng, C.; Li, J.T. The Structural and Functional Divergence of a Neglected Three-Finger Toxin Subfamily in Lethal Elapids. Cell Rep. 2022, 40, 111079. [Google Scholar] [CrossRef] [PubMed]
- Sunagar, K.; Jackson, T.N.W.; Undheim, E.A.B.; Ali, S.A.; Antunes, A.; Fry, B.G. Three-Fingered RAVERs: Rapid Accumulation of Variations in Exposed Residues of Snake Venom Toxins. Toxins 2013, 5, 2172–2208. [Google Scholar] [CrossRef]
- Wong, E.S.W.; Belov, K. Venom Evolution through Gene Duplications. Gene 2012, 496, 1–7. [Google Scholar] [CrossRef]
- Hargreaves, A.D.; Swain, M.T.; Hegarty, M.J.; Logan, D.W.; Mulley, J.F. Restriction and Recruitment-Gene Duplication and the Origin and Evolution of Snake Venom Toxins. Genome Biol. Evol. 2014, 6, 2088–2095. [Google Scholar] [CrossRef]
- Lynch, V.J. Inventing an Arsenal: Adaptive Evolution and Neofunctionalization of Snake Venom Phospholipase A2 Genes. BMC Evol. Biol. 2007, 7, 2. [Google Scholar] [CrossRef]
- Brust, A.; Sunagar, K.; Undheim, E.A.B.; Vetter, I.; Yang, D.C.; Casewell, N.R.; Jackson, T.N.W.; Koludarov, I.; Alewood, P.F.; Hodgson, W.C.; et al. Differential Evolution and Neofunctionalization of Snake Venom Metalloprotease Domains. Mol. Cell Proteom. 2013, 12, 651–663. [Google Scholar] [CrossRef]
- Pla, D.; Petras, D.; Saviola, A.J.; Modahl, C.M.; Sanz, L.; Pérez, A.; Juárez, E.; Frietze, S.; Dorrestein, P.C.; Mackessy, S.P.; et al. Transcriptomics-Guided Bottom-up and Top-down Venomics of Neonate and Adult Specimens of the Arboreal Rear-Fanged Brown Treesnake, Boiga Irregularis, from Guam. J. Proteom. 2018, 174, 71–84. [Google Scholar] [CrossRef]
- Roberto, I.J.; Souza, A.R. Review of Prey Items Recorded for Snakes of the Genus Chironius (Squamata, Colubridae), Including the First Record of Osteocephalus as Prey. Herpetol. Notes 2020, 13, 1–5. [Google Scholar]
- Callahan, S.; Crowe-Riddell, J.M.; Nagesan, R.S.; Gray, J.A.; Davis Rabosky, A.R. A Guide for Optimal Iodine Staining and High-Throughput DiceCT Scanning in Snakes. Ecol. Evol. 2021, 11, 11587–11603. [Google Scholar] [CrossRef] [PubMed]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data [Online]. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc (accessed on 20 August 2021).
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A Flexible Trimmer for Illumina Sequence Data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Haas, B.J.; Papanicolaou, A.; Yassour, M.; Grabherr, M.; Blood, P.D.; Bowden, J.; Couger, M.B.; Eccles, D.; Li, B.; Lieber, M.; et al. De Novo Transcript Sequence Reconstruction from RNA-Seq Using the Trinity Platform for Reference Generation and Analysis. Nat. Protoc. 2013, 8, 1494–1512. [Google Scholar] [CrossRef] [PubMed]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-Length Transcriptome Assembly from RNA-Seq Data without a Reference Genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [PubMed]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and Applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for Clustering the next-Generation Sequencing Data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef]
- Li, B.; Dewey, C.N. RSEM: Accurate Transcript Quantification from RNA-Seq Data with or without a Reference Genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast Gapped-Read Alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Bayona-Serrano, J.D.; Viala, V.L.; Rautsaw, R.M.; Schramer, T.D.; Barros-Carvalho, G.A.; Nishiyama, M.Y.; Freitas-De-Sousa, L.A.; Moura-Da-Silva, A.M.; Parkinson, C.L.; Grazziotin, F.G.; et al. Replacement and Parallel Simplification of Nonhomologous Proteinases Maintain Venom Phenotypes in Rear-Fanged Snakes. Mol. Biol. Evol. 2020, 37, 3563–3575. [Google Scholar] [CrossRef]
- Margres, M.J.; Aronow, K.; Loyacano, J.; Rokyta, D.R. The Venom-Gland Transcriptome of the Eastern Coral Snake (Micrurus fulvius) Reveals High Venom Complexity in the Intragenomic Evolution of Venoms. BMC Genom. 2013, 14, 531. [Google Scholar] [CrossRef]
- Rokyta, D.R.; Wray, K.P.; Margres, M.J. The Genesis of an Exceptionally Lethal Venom in the Timber Rattlesnake (Crotalus horridus) Revealed through Comparative Venom-Gland Transcriptomics. BMC Genom. 2013, 14, 394. [Google Scholar] [CrossRef] [PubMed]
- Rokyta, D.R.; Lemmon, A.R.; Margres, M.J.; Aronow, K. The Venom-Gland Transcriptome of the Eastern Diamondback Rattlesnake (Crotalus adamanteus). BMC Genom. 2012, 13, 312. [Google Scholar] [CrossRef] [PubMed]
- Vonk, F.J.; Casewell, N.R.; Henkel, C.V.; Heimberg, A.M.; Jansen, H.J.; McCleary, R.J.R.; Kerkkamp, H.M.E.; Vos, R.A.; Guerreiro, I.; Calvete, J.J.; et al. The King Cobra Genome Reveals Dynamic Gene Evolution and Adaptation in the Snake Venom System. Proc. Natl. Acad. Sci. USA 2013, 110, 20651–20656. [Google Scholar] [CrossRef]
- R Core Team R: A Language and Environment for Statistical Computing. Available online: https://www.r-project.org/ (accessed on 25 August 2021).
- Edgar, R.C. MUSCLE: Multiple Sequence Alignment with High Accuracy and High Throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An Integrated and Extendable Desktop Software Platform for the Organization and Analysis of Sequence Data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef]
- Lanfear, R.; Calcott, B.; Ho, S.Y.W.; Guindon, S. Partitionfinder: Combined Selection of Partitioning Schemes and Substitution Models for Phylogenetic Analyses. Mol. Biol. Evol. 2012, 29, 1695–1701. [Google Scholar] [CrossRef]
- Ronquist, F.; Huelsenbeck, J.P. MrBayes 3: Bayesian Phylogenetic Inference under Mixed Models. Bioinformatics 2003, 19, 1572–1574. [Google Scholar] [CrossRef]
- Yang, J.; Zhang, Y. I-TASSER Server: New Development for Protein Structure and Function Predictions. Nucleic Acids Res. 2015, 43, W174–W181. [Google Scholar] [CrossRef]
- Zhou, X.; Zheng, W.; Li, Y.; Pearce, R.; Zhang, C.; Bell, E.W.; Zhang, G.; Zhang, Y. I-TASSER-MTD: A Deep-Learning-Based Platform for Multi-Domain Protein Structure and Function Prediction. Nat. Protoc. 2022, 17, 2326–2353. [Google Scholar] [CrossRef]
- Zheng, W.; Zhang, C.; Li, Y.; Pearce, R.; Bell, E.W.; Zhang, Y. Folding Non-Homologous Proteins by Coupling Deep-Learning Contact Maps with I-TASSER Assembly Simulations. Cell Rep. Methods 2021, 1, 100014. [Google Scholar] [CrossRef]
- Gao, F.; Chen, C.; Arab, D.A.; Du, Z.; He, Y.; Ho, S.Y.W. EasyCodeML: A Visual Tool for Analysis of Selection Using CodeML. Ecol. Evol. 2019, 9, 3891–3898. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z. PAML 4: Phylogenetic Analysis by Maximum Likelihood. Mol. Biol. Evol. 2007, 24, 1586–1591. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Nielsen, R.; Goldman, N.; Pedersen, A.-M.K. Codon-Substitution Models for Heterogeneous Selection Pressure at Amino Acid Sites. Genetics 2000, 155, 431–449. [Google Scholar] [CrossRef]
- Kosakovsky Pond, S.L.; Poon, A.F.Y.; Velazquez, R.; Weaver, S.; Hepler, N.L.; Murrell, B.; Shank, S.D.; Magalis, B.R.; Bouvier, D.; Nekrutenko, A.; et al. HyPhy 2.5—A Customizable Platform for Evolutionary Hypothesis Testing Using Phylogenies. Mol. Biol. Evol. 2020, 37, 295–299. [Google Scholar] [CrossRef] [PubMed]
- Murrell, B.; Wertheim, J.O.; Moola, S.; Weighill, T.; Scheffler, K.; Kosakovsky Pond, S.L. Detecting Individual Sites Subject to Episodic Diversifying Selection. PLoS Genet. 2012, 8, e1002764. [Google Scholar] [CrossRef]
- Murrell, B.; Moola, S.; Mabona, A.; Weighill, T.; Sheward, D.; Kosakovsky Pond, S.L.; Scheffler, K. FUBAR: A Fast, Unconstrained Bayesian AppRoximation for Inferring Selection. Mol. Biol. Evol. 2013, 30, 1196–1205. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Taxonomy | 3FTx | CRiSP | PLA2 | SVMP | SVSP | Other |
|---|---|---|---|---|---|---|
| Spilotes sulphureus | 41 | 16 | 10 | 29 | 55 | 60 |
| Rhinobothryum bovallii | 5 | 7 | 3 | 20 | 57 | 58 |
| Oxybelis aeneus | 11 | 6 | 9 | 9 | 23 | 38 |
| Chironius multiventris A | 7 | 21 | 8 | 27 | 58 | 53 |
| Chironius multiventris B | 10 | 25 | 4 | 34 | 56 | 55 |
| Model | np | Ln L | Estimates of Parameters | Model Compared | LRT p-Value | ||
|---|---|---|---|---|---|---|---|
| M0 | 62 | −2330.419 | ω0 = 1.347 | M0 vs. M1 | p < 0.001 | ||
| M3 | 66 | −2249.816 | p = 0.192 ω = 0.136 | 0.355 0.762 | 0.453 3.028 | ||
| M1 | 63 | −2280.078 | p = 0.380 ω = 0.156 | 0.620 1 | M1 vs. M2 | p < 0.001 | |
| M2 | 65 | −2250.100 | p = 0.273 ω = 0.213 | 0.294 1 | 0.433 3.192 | ||
| M7 | 63 | −2291.574 | p = 0.051 q = 0.056 | M7 vs. M8 | p < 0.001 | ||
| M8 | 65 | −2249.800 | p0 = 0.401 q = 0.524 | p1 = 0.447 ω = 3.049 | p = 0.652 | ||
| M2 | MEME | FUBAR | |
|---|---|---|---|
| Whole alignment | 25 | 30 | 21 |
| N-terminus region | 3 | 5 | 3 |
| Loop II | 8 | 6 | 3 |
| Loop V | 0 | 4 | 4 |
| Specimen | Field No. | Museum No. | SVL (mm) | Mass (g) | Capture Locality |
|---|---|---|---|---|---|
| Spilotes sulphureus | RAB562 | MUSM 37565 | 1840 | 1250 | EBVC, Peru |
| Rhinobothryum bovallii | RAB3018 | UMMZ 247120 | 1030 | 71.4 | RB, Nicaragua |
| Oxybelis aeneus | RAB3102 | UMMZ 247113 | 865 | 73.1 | Momotombo, Nicaragua |
| Chironius multiventris A | RAB3332 | UMMZ 249111 | 810 | 160 | EBLA, Peru |
| Chironius multiventris B | RAB3577 | MUSM 39803 | 739 | 85 | EBLA, Peru |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Srodawa, K.; Cerda, P.A.; Davis Rabosky, A.R.; Crowe-Riddell, J.M. Evolution of Three-Finger Toxin Genes in Neotropical Colubrine Snakes (Colubridae). Toxins 2023, 15, 523. https://doi.org/10.3390/toxins15090523
Srodawa K, Cerda PA, Davis Rabosky AR, Crowe-Riddell JM. Evolution of Three-Finger Toxin Genes in Neotropical Colubrine Snakes (Colubridae). Toxins. 2023; 15(9):523. https://doi.org/10.3390/toxins15090523
Chicago/Turabian StyleSrodawa, Kristy, Peter A. Cerda, Alison R. Davis Rabosky, and Jenna M. Crowe-Riddell. 2023. "Evolution of Three-Finger Toxin Genes in Neotropical Colubrine Snakes (Colubridae)" Toxins 15, no. 9: 523. https://doi.org/10.3390/toxins15090523