
Sulforaphane Inhibits Foam Cell Formation and Atherosclerosis via Mechanisms Involving the Modulation of Macrophage Cholesterol Transport and the Related Phenotype
 and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Antibodies and Reagents
2.2. Animals and Treatments
2.3. Quantification of Atherosclerotic Lesions
2.4. Serum Lipid Profile Analysis
2.5. Immunofluorescence
2.6. Dihydroethidium (DHE) Staining
2.7. Mouse Peritoneal Macrophage Isolation and Culture
2.8. THP-1 Cell Differentiation and Culture
2.9. Cell Viability Assay
2.10. Foam Cell Formation Assay
2.11. Dil-oxLDL Uptake Assay
2.12. Cholesterol Efflux Assay
2.13. Detection of Reactive Oxygen Species (ROS)
2.14. Mitochondrial Membrane Potential (MMP) Assay
2.15. Western Blotting
2.16. Statistical Analysis
3. Results
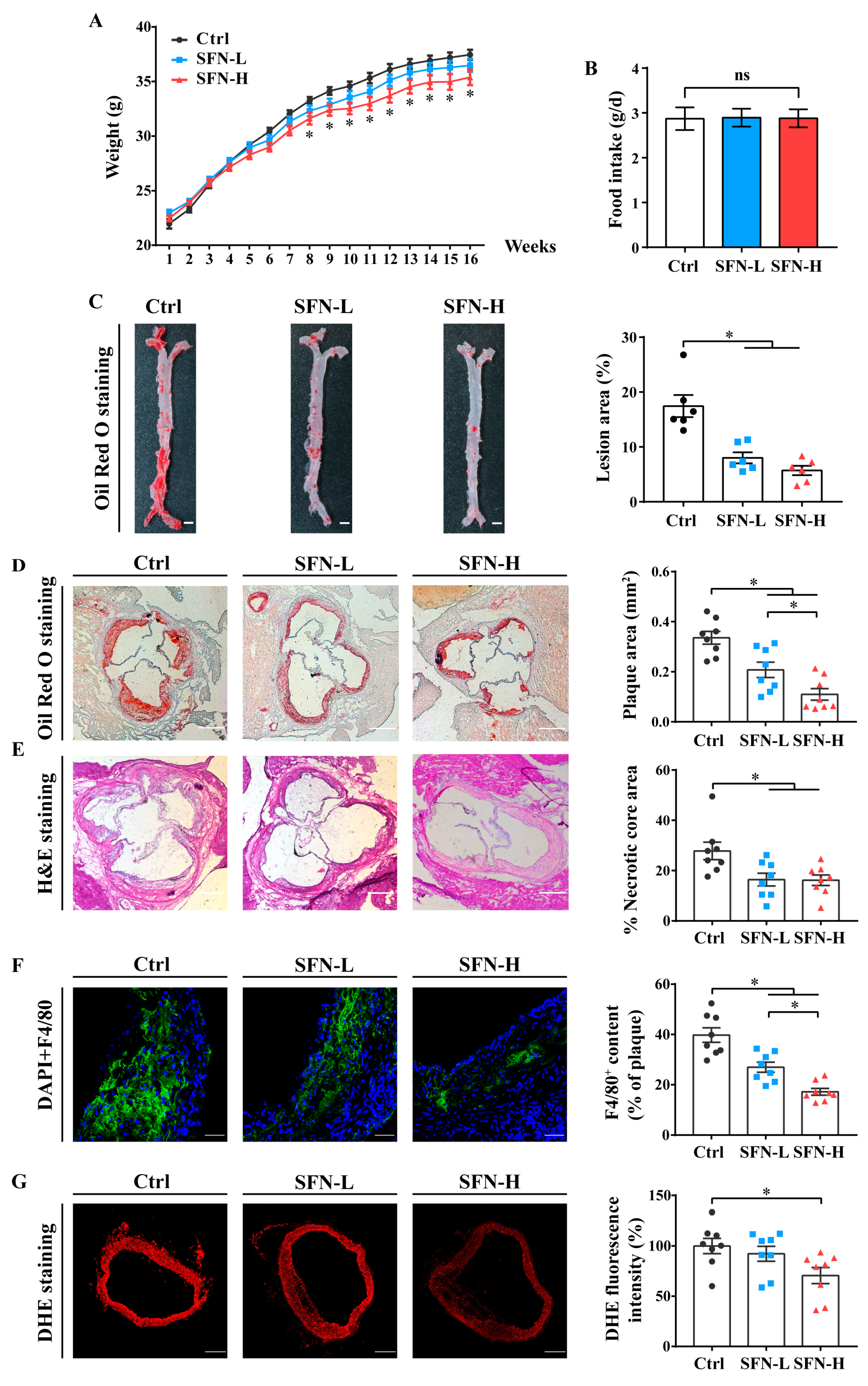
3.1. SFN Attenuates Atherosclerosis Progression and Improves the Serum Lipid Profile in ApoE−/− Mice
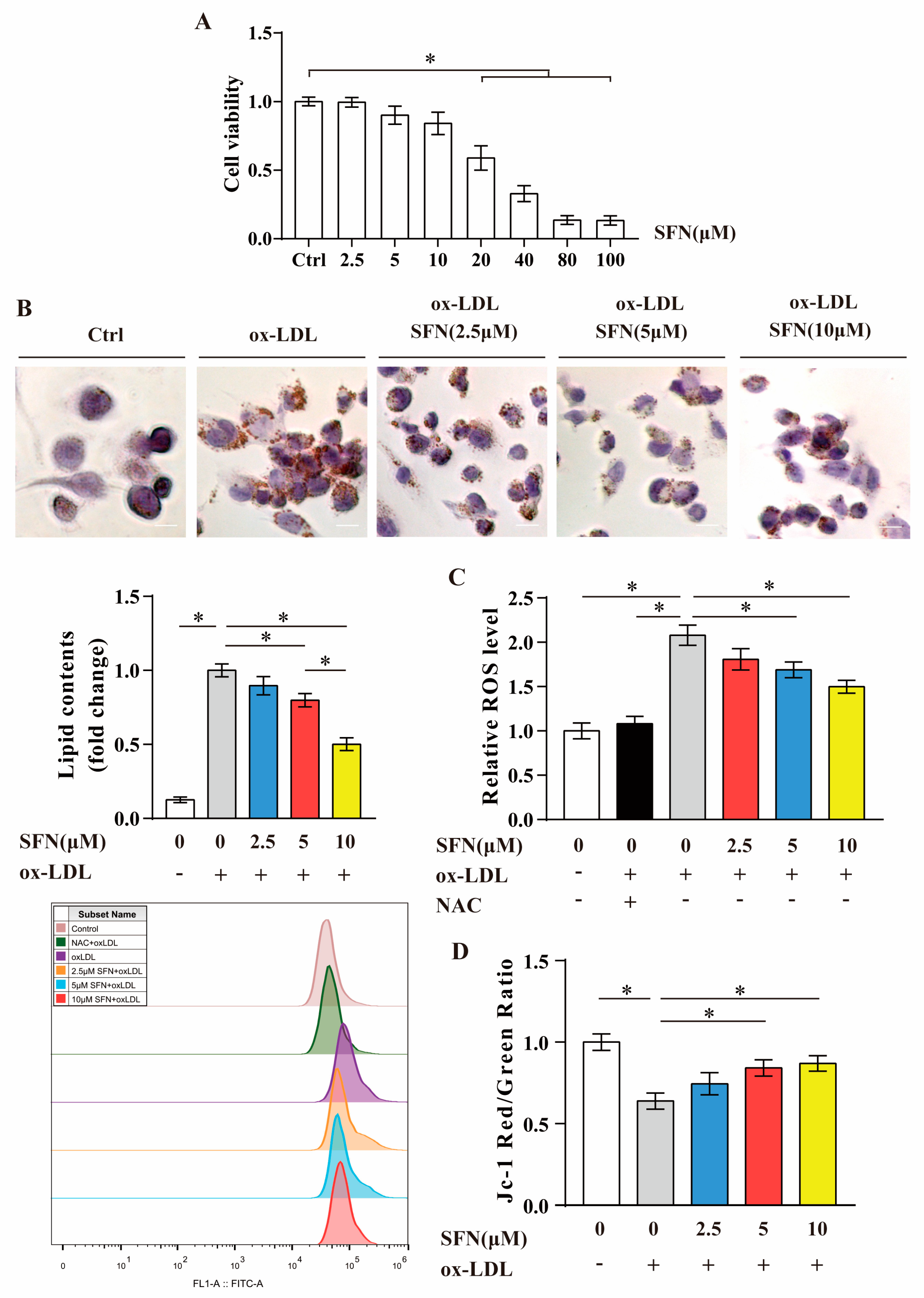
3.2. SFN Inhibits ox-LDL-Induced Foam Cell Formation, ROS Generation and Mitochondrial Dysfunction in THP-1-Derived Macrophages
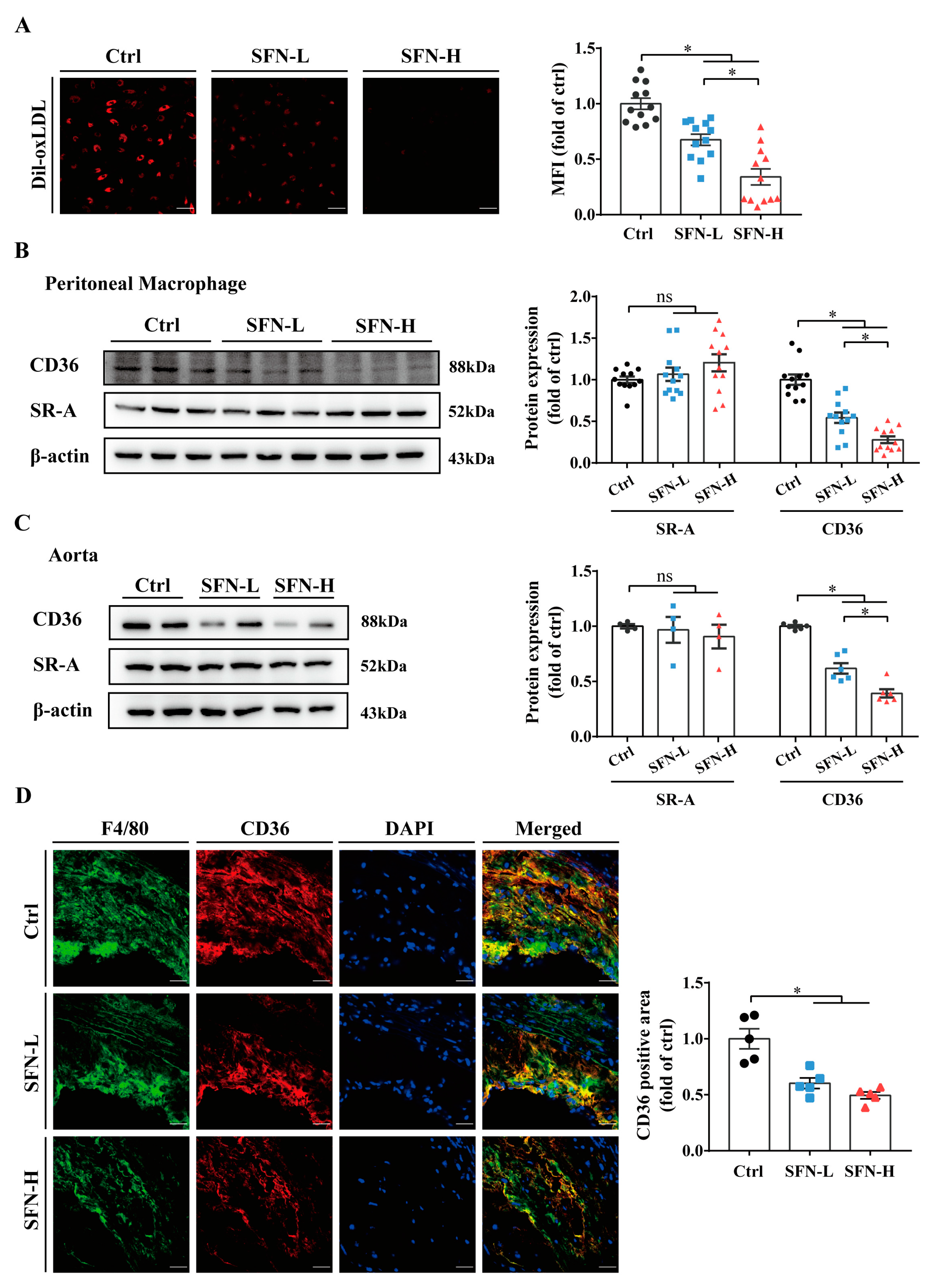
3.3. SFN Inhibits Macrophages’ Cholesterol Uptake and CD36 Expression in ApoE−/− Mice
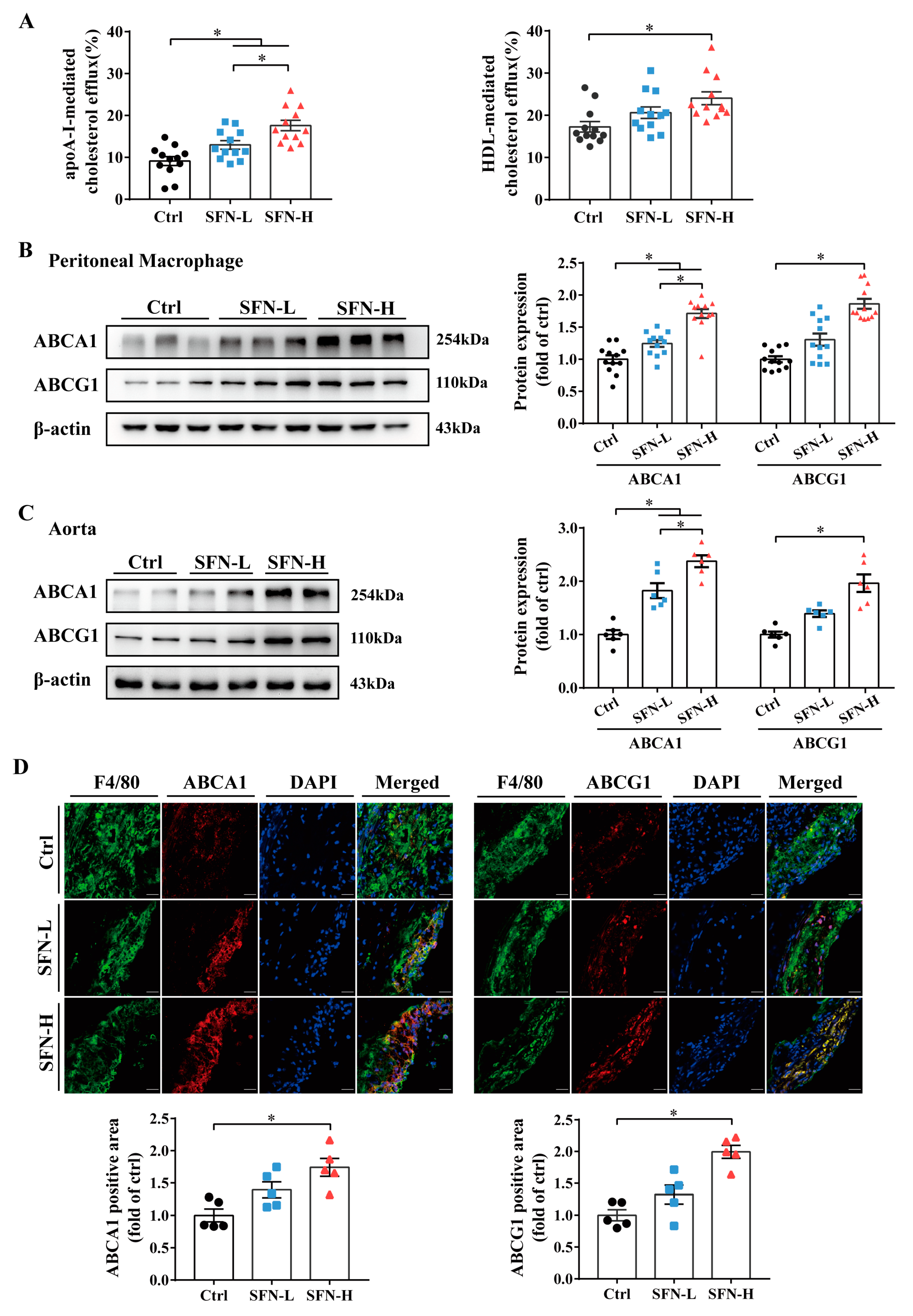
3.4. SFN Promotes Macrophages’ Cholesterol Efflux and ABCA1/G1 Expression in ApoE−/− Mice
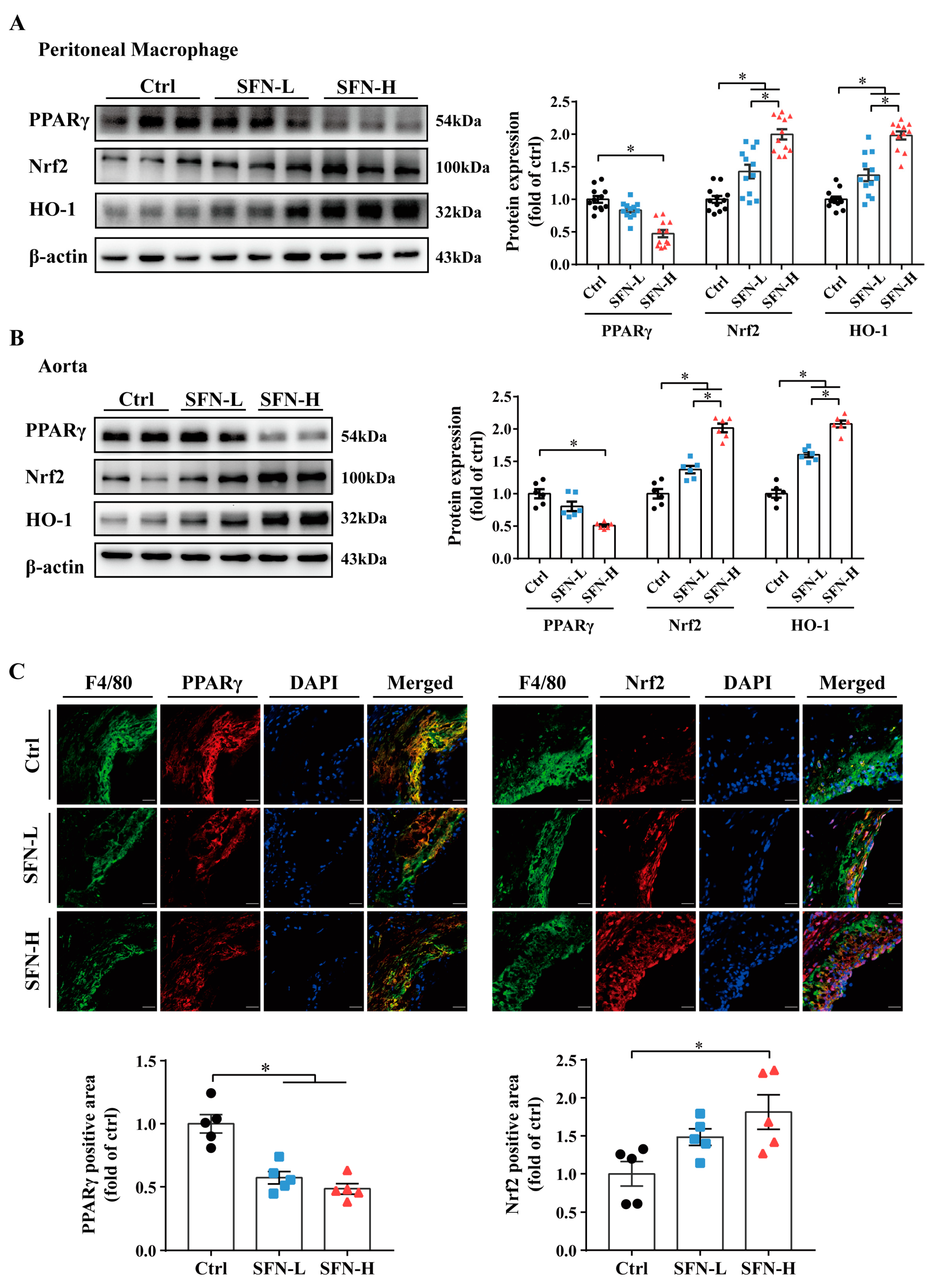
3.5. Downregulation of PPARγ and Upregulation of Nrf2/HO-1 Expression by SFN in ApoE−/− Mice
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Moore, K.J.; Sheedy, F.J.; Fisher, E.A. Macrophages in atherosclerosis: A dynamic balance. Nat. Rev. Immunol. 2013, 13, 709–721. [Google Scholar] [CrossRef] [PubMed]
- Lusis, A.J. Atherosclerosis. Nature 2000, 407, 233–241. [Google Scholar] [CrossRef] [PubMed]
- Moore, K.J.; Tabas, I. Macrophages in the pathogenesis of atherosclerosis. Cell 2011, 145, 341–355. [Google Scholar] [CrossRef] [PubMed]
- Wynn, T.A.; Chawla, A.; Pollard, J.W. Macrophage biology in development, homeostasis and disease. Nature 2013, 496, 445–455. [Google Scholar] [CrossRef]
- Oliveira, H.C.F.; Vercesi, A.E. Mitochondrial bioenergetics and redox dysfunctions in hypercholesterolemia and atherosclerosis. Mol. Aspects Med. 2020, 71, 100840. [Google Scholar] [CrossRef]
- Brune, B.; Dehne, N.; Grossmann, N.; Jung, M.; Namgaladze, D.; Schmid, T.; von Knethen, A.; Weigert, A. Redox control of inflammation in macrophages. Antioxid. Redox Signal. 2013, 19, 595–637. [Google Scholar] [CrossRef]
- Forstermann, U.; Xia, N.; Li, H. Roles of Vascular Oxidative Stress and Nitric Oxide in the Pathogenesis of Atherosclerosis. Circ. Res. 2017, 120, 713–735. [Google Scholar] [CrossRef]
- Wang, D.; Yang, Y.; Lei, Y.; Tzvetkov, N.T.; Liu, X.; Yeung, A.W.K.; Xu, S.; Atanasov, A.G. Targeting Foam Cell Formation in Atherosclerosis: Therapeutic Potential of Natural Products. Pharmacol. Rev. 2019, 71, 596–670. [Google Scholar] [CrossRef]
- Yang, M.; Silverstein, R.L. CD36 signaling in vascular redox stress. Free Radic. Biol. Med. 2019, 136, 159–171. [Google Scholar] [CrossRef]
- Makinen, P.I.; Lappalainen, J.P.; Heinonen, S.E.; Leppanen, P.; Lahteenvuo, M.T.; Aarnio, J.V.; Heikkila, J.; Turunen, M.P.; Yla-Herttuala, S. Silencing of either SR-A or CD36 reduces atherosclerosis in hyperlipidaemic mice and reveals reciprocal upregulation of these receptors. Cardiovasc. Res. 2010, 88, 530–538. [Google Scholar] [CrossRef]
- Febbraio, M.; Hajjar, D.P.; Silverstein, R.L. CD36: A class B scavenger receptor involved in angiogenesis, atherosclerosis, inflammation, and lipid metabolism. J. Clin. Investig. 2001, 108, 785–791. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.M. CD36, a scavenger receptor implicated in atherosclerosis. Exp. Mol. Med. 2014, 46, e99. [Google Scholar] [CrossRef] [PubMed]
- Kuchibhotla, S.; Vanegas, D.; Kennedy, D.J.; Guy, E.; Nimako, G.; Morton, R.E.; Febbraio, M. Absence of CD36 protects against atherosclerosis in ApoE knock-out mice with no additional protection provided by absence of scavenger receptor A I/II. Cardiovasc. Res. 2008, 78, 185–196. [Google Scholar] [CrossRef] [PubMed]
- Febbraio, M.; Podrez, E.A.; Smith, J.D.; Hajjar, D.P.; Hazen, S.L.; Hoff, H.F.; Sharma, K.; Silverstein, R.L. Targeted disruption of the class B scavenger receptor CD36 protects against atherosclerotic lesion development in mice. J. Clin. Investig. 2000, 105, 1049–1056. [Google Scholar] [CrossRef]
- Liu, Y.; Zhong, Y.; Chen, H.; Wang, D.; Wang, M.; Ou, J.S.; Xia, M. Retinol-Binding Protein-Dependent Cholesterol Uptake Regulates Macrophage Foam Cell Formation and Promotes Atherosclerosis. Circulation 2017, 135, 1339–1354. [Google Scholar] [CrossRef]
- Febbraio, M.; Guy, E.; Silverstein, R.L. Stem cell transplantation reveals that absence of macrophage CD36 is protective against atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 2333–2338. [Google Scholar] [CrossRef]
- Handberg, A.; Levin, K.; Hojlund, K.; Beck-Nielsen, H. Identification of the oxidized low-density lipoprotein scavenger receptor CD36 in plasma: A novel marker of insulin resistance. Circulation 2006, 114, 1169–1176. [Google Scholar] [CrossRef]
- Castano, D.; Rattanasopa, C.; Monteiro-Cardoso, V.F.; Corliano, M.; Liu, Y.; Zhong, S.; Rusu, M.; Liehn, E.A.; Singaraja, R.R. Lipid efflux mechanisms, relation to disease and potential therapeutic aspects. Adv. Drug Deliv. Rev. 2020, 159, 54–93. [Google Scholar] [CrossRef]
- Rosenson, R.S.; Brewer, H.B., Jr.; Davidson, W.S.; Fayad, Z.A.; Fuster, V.; Goldstein, J.; Hellerstein, M.; Jiang, X.C.; Phillips, M.C.; Rader, D.J.; et al. Cholesterol efflux and atheroprotection: Advancing the concept of reverse cholesterol transport. Circulation 2012, 125, 1905–1919. [Google Scholar] [CrossRef]
- Uto-Kondo, H.; Ayaori, M.; Ogura, M.; Nakaya, K.; Ito, M.; Suzuki, A.; Takiguchi, S.; Yakushiji, E.; Terao, Y.; Ozasa, H.; et al. Coffee consumption enhances high-density lipoprotein-mediated cholesterol efflux in macrophages. Circ. Res. 2010, 106, 779–787. [Google Scholar] [CrossRef]
- Yvan-Charvet, L.; Ranalletta, M.; Wang, N.; Han, S.; Terasaka, N.; Li, R.; Welch, C.; Tall, A.R. Combined deficiency of ABCA1 and ABCG1 promotes foam cell accumulation and accelerates atherosclerosis in mice. J. Clin. Investig. 2007, 117, 3900–3908. [Google Scholar] [CrossRef] [PubMed]
- Westerterp, M.; Murphy, A.J.; Wang, M.; Pagler, T.A.; Vengrenyuk, Y.; Kappus, M.S.; Gorman, D.J.; Nagareddy, P.R.; Zhu, X.; Abramowicz, S.; et al. Deficiency of ATP-binding cassette transporters A1 and G1 in macrophages increases inflammation and accelerates atherosclerosis in mice. Circ. Res. 2013, 112, 1456–1465. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Xia, M.; Yan, X.; Li, D.; Wang, L.; Xu, Y.; Jin, T.; Ling, W. Gut microbiota metabolism of anthocyanin promotes reverse cholesterol transport in mice via repressing miRNA-10b. Circ. Res. 2012, 111, 967–981. [Google Scholar] [CrossRef] [PubMed]
- Marcil, M.; Brooks-Wilson, A.; Clee, S.M.; Roomp, K.; Zhang, L.H.; Yu, L.; Collins, J.A.; van Dam, M.; Molhuizen, H.O.; Loubster, O.; et al. Mutations in the ABC1 gene in familial HDL deficiency with defective cholesterol efflux. Lancet 1999, 354, 1341–1346. [Google Scholar] [CrossRef] [PubMed]
- Mody, P.; Joshi, P.H.; Khera, A.; Ayers, C.R.; Rohatgi, A. Beyond Coronary Calcification, Family History, and C-Reactive Protein: Cholesterol Efflux Capacity and Cardiovascular Risk Prediction. J. Am. Coll. Cardiol. 2016, 67, 2480–2487. [Google Scholar] [CrossRef]
- Khera, A.V.; Cuchel, M.; de la Llera-Moya, M.; Rodrigues, A.; Burke, M.F.; Jafri, K.; French, B.C.; Phillips, J.A.; Mucksavage, M.L.; Wilensky, R.L.; et al. Cholesterol efflux capacity, high-density lipoprotein function, and atherosclerosis. N. Engl. J. Med. 2011, 364, 127–135. [Google Scholar] [CrossRef]
- Rohatgi, A.; Khera, A.; Berry, J.D.; Givens, E.G.; Ayers, C.R.; Wedin, K.E.; Neeland, I.J.; Yuhanna, I.S.; Rader, D.R.; de Lemos, J.A.; et al. HDL cholesterol efflux capacity and incident cardiovascular events. N. Engl. J. Med. 2014, 371, 2383–2393. [Google Scholar] [CrossRef]
- Li, H.; Xia, Y.; Liu, H.Y.; Guo, H.; He, X.Q.; Liu, Y.; Wu, D.T.; Mai, Y.H.; Li, H.B.; Zou, L.; et al. Nutritional values, beneficial effects, and food applications of broccoli (Brassica oleracea var. italica Plenck). Trends Food Sci. Technol. 2022, 119, 288–308. [Google Scholar] [CrossRef]
- Shehatou, G.S.; Suddek, G.M. Sulforaphane attenuates the development of atherosclerosis and improves endothelial dysfunction in hypercholesterolemic rabbits. Exp. Biol. Med. 2016, 241, 426–436. [Google Scholar] [CrossRef]
- Matsui, T.; Nakamura, N.; Ojima, A.; Nishino, Y.; Yamagishi, S.I. Sulforaphane reduces advanced glycation end products (AGEs)-induced inflammation in endothelial cells and rat aorta. Nutr. Metab. Cardiovasc. Dis. 2016, 26, 797–807. [Google Scholar] [CrossRef]
- Nallasamy, P.; Si, H.; Babu, P.V.; Pan, D.; Fu, Y.; Brooke, E.A.; Shah, H.; Zhen, W.; Zhu, H.; Liu, D.; et al. Sulforaphane reduces vascular inflammation in mice and prevents TNF-alpha-induced monocyte adhesion to primary endothelial cells through interfering with the NF-kappaB pathway. J. Nutr. Biochem. 2014, 25, 824–833. [Google Scholar] [CrossRef] [PubMed]
- Shawky, N.M.; Pichavaram, P.; Shehatou, G.S.; Suddek, G.M.; Gameil, N.M.; Jun, J.Y.; Segar, L. Sulforaphane improves dysregulated metabolic profile and inhibits leptin-induced VSMC proliferation: Implications toward suppression of neointima formation after arterial injury in western diet-fed obese mice. J. Nutr. Biochem. 2016, 32, 73–84. [Google Scholar] [CrossRef] [PubMed]
- Yoo, S.H.; Lim, Y.; Kim, S.J.; Yoo, K.D.; Yoo, H.S.; Hong, J.T.; Lee, M.Y.; Yun, Y.P. Sulforaphane inhibits PDGF-induced proliferation of rat aortic vascular smooth muscle cell by up-regulation of p53 leading to G1/S cell cycle arrest. Vascul. Pharmacol. 2013, 59, 44–51. [Google Scholar] [CrossRef] [PubMed]
- Shawky, N.M.; Segar, L. Sulforaphane inhibits platelet-derived growth factor-induced vascular smooth muscle cell proliferation by targeting mTOR/p70S6kinase signaling independent of Nrf2 activation. Pharmacol. Res. 2017, 119, 251–264. [Google Scholar] [CrossRef]
- Kwon, J.S.; Joung, H.; Kim, Y.S.; Shim, Y.S.; Ahn, Y.; Jeong, M.H.; Kee, H.J. Sulforaphane inhibits restenosis by suppressing inflammation and the proliferation of vascular smooth muscle cells. Atherosclerosis 2012, 225, 41–49. [Google Scholar] [CrossRef]
- Gillespie, S.; Holloway, P.M.; Becker, F.; Rauzi, F.; Vital, S.A.; Taylor, K.A.; Stokes, K.Y.; Emerson, M.; Gavins, F.N.E. The isothiocyanate sulforaphane modulates platelet function and protects against cerebral thrombotic dysfunction. Br. J. Pharmacol. 2018, 175, 3333–3346. [Google Scholar] [CrossRef]
- Cuadrado, A.; Rojo, A.I.; Wells, G.; Hayes, J.D.; Cousin, S.P.; Rumsey, W.L.; Attucks, O.C.; Franklin, S.; Levonen, A.L.; Kensler, T.W.; et al. Therapeutic targeting of the NRF2 and KEAP1 partnership in chronic diseases. Nat. Rev. Drug Discov. 2019, 18, 295–317. [Google Scholar] [CrossRef]
- Mimura, J.; Itoh, K. Role of Nrf2 in the pathogenesis of atherosclerosis. Free Radic. Biol. Med. 2015, 88, 221–232. [Google Scholar] [CrossRef]
- Collins, A.R.; Gupte, A.A.; Ji, R.; Ramirez, M.R.; Minze, L.J.; Liu, J.Z.; Arredondo, M.; Ren, Y.; Deng, T.; Wang, J.; et al. Myeloid deletion of nuclear factor erythroid 2-related factor 2 increases atherosclerosis and liver injury. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 2839–2846. [Google Scholar] [CrossRef]
- Barajas, B.; Che, N.; Yin, F.; Rowshanrad, A.; Orozco, L.D.; Gong, K.W.; Wang, X.; Castellani, L.W.; Reue, K.; Lusis, A.J.; et al. NF-E2-related factor 2 promotes atherosclerosis by effects on plasma lipoproteins and cholesterol transport that overshadow antioxidant protection. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 58–66. [Google Scholar] [CrossRef]
- Ruotsalainen, A.K.; Lappalainen, J.P.; Heiskanen, E.; Merentie, M.; Sihvola, V.; Napankangas, J.; Lottonen-Raikaslehto, L.; Kansanen, E.; Adinolfi, S.; Kaarniranta, K.; et al. Nuclear factor E2-related factor 2 deficiency impairs atherosclerotic lesion development but promotes features of plaque instability in hypercholesterolaemic mice. Cardiovasc. Res. 2019, 115, 243–254. [Google Scholar] [CrossRef] [PubMed]
- Ruotsalainen, A.K.; Inkala, M.; Partanen, M.E.; Lappalainen, J.P.; Kansanen, E.; Makinen, P.I.; Heinonen, S.E.; Laitinen, H.M.; Heikkila, J.; Vatanen, T.; et al. The absence of macrophage Nrf2 promotes early atherogenesis. Cardiovasc. Res. 2013, 98, 107–115. [Google Scholar] [CrossRef] [PubMed]
- Ooi, B.K.; Goh, B.H.; Yap, W.H. Oxidative Stress in Cardiovascular Diseases: Involvement of Nrf2 Antioxidant Redox Signaling in Macrophage Foam Cells Formation. Int. J. Mol. Sci. 2017, 18, 2336. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez-Cuevas, J.; Galicia-Moreno, M.; Monroy-Ramirez, H.C.; Sandoval-Rodriguez, A.; Garcia-Banuelos, J.; Santos, A.; Armendariz-Borunda, J. The Role of NRF2 in Obesity-Associated Cardiovascular Risk Factors. Antioxidants 2022, 11, 235. [Google Scholar] [CrossRef]
- Ishii, T.; Itoh, K.; Ruiz, E.; Leake, D.S.; Unoki, H.; Yamamoto, M.; Mann, G.E. Role of Nrf2 in the regulation of CD36 and stress protein expression in murine macrophages: Activation by oxidatively modified LDL and 4-hydroxynonenal. Circ. Res. 2004, 94, 609–616. [Google Scholar] [CrossRef]
- Hong, M.; Li, J.; Li, S.; Almutairi, M.M. Resveratrol Derivative, Trans-3, 5, 4’-Trimethoxystilbene, Prevents the Developing of Atherosclerotic Lesions and Attenuates Cholesterol Accumulation in Macrophage Foam Cells. Mol. Nutr. Food Res. 2020, 64, e1901115. [Google Scholar] [CrossRef]
- Lu, Q.; Tang, S.L.; Liu, X.Y.; Zhao, G.J.; Ouyang, X.P.; Lv, Y.C.; He, P.P.; Yao, F.; Chen, W.J.; Tang, Y.Y.; et al. Tertiary-butylhydroquinone upregulates expression of ATP-binding cassette transporter A1 via nuclear factor E2-related factor 2/heme oxygenase-1 signaling in THP-1 macrophage-derived foam cells. Circ. J. 2013, 77, 2399–2408. [Google Scholar] [CrossRef]
- Chen, L.W.; Tsai, M.C.; Chern, C.Y.; Tsao, T.P.; Lin, F.Y.; Chen, S.J.; Tsui, P.F.; Liu, Y.W.; Lu, H.J.; Wu, W.L.; et al. A chalcone derivative, 1m-6, exhibits atheroprotective effects by increasing cholesterol efflux and reducing inflammation-induced endothelial dysfunction. Br. J. Pharmacol. 2020, 177, 5375–5392. [Google Scholar] [CrossRef]
- Jiang, J.; Mo, Z.C.; Yin, K.; Zhao, G.J.; Lv, Y.C.; Ouyang, X.P.; Jiang, Z.S.; Fu, Y.; Tang, C.K. Epigallocatechin-3-gallate prevents TNF-alpha-induced NF-kappaB activation thereby upregulating ABCA1 via the Nrf2/Keap1 pathway in macrophage foam cells. Int. J. Mol. Med. 2012, 29, 946–956. [Google Scholar] [CrossRef]
- Yang, Y.; Li, X.; Peng, L.; An, L.; Sun, N.; Hu, X.; Zhou, P.; Xu, Y.; Li, P.; Chen, J. Tanshindiol C inhibits oxidized low-density lipoprotein induced macrophage foam cell formation via a peroxiredoxin 1 dependent pathway. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 882–890. [Google Scholar] [CrossRef]
- Zhang, Y.K.; Yeager, R.L.; Tanaka, Y.; Klaassen, C.D. Enhanced expression of Nrf2 in mice attenuates the fatty liver produced by a methionine- and choline-deficient diet. Toxicol. Appl. Pharmacol. 2010, 245, 326–334. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.D.; Chapman, E. The role of natural products in revealing NRF2 function. Nat. Prod. Rep. 2020, 37, 797–826. [Google Scholar] [CrossRef]
- Montaigne, D.; Butruille, L.; Staels, B. PPAR control of metabolism and cardiovascular functions. Nat. Rev. Cardiol. 2021, 18, 809–823. [Google Scholar] [CrossRef] [PubMed]
- Gross, B.; Pawlak, M.; Lefebvre, P.; Staels, B. PPARs in obesity-induced T2DM, dyslipidaemia and NAFLD. Nat. Rev. Endocrinol. 2017, 13, 36–49. [Google Scholar] [CrossRef]
- Oppi, S.; Nusser-Stein, S.; Blyszczuk, P.; Wang, X.; Jomard, A.; Marzolla, V.; Yang, K.; Velagapudi, S.; Ward, L.J.; Yuan, X.M.; et al. Macrophage NCOR1 protects from atherosclerosis by repressing a pro-atherogenic PPARgamma signature. Eur. Heart J. 2020, 41, 995–1005. [Google Scholar] [CrossRef]
- Han, L.; Shen, W.J.; Bittner, S.; Kraemer, F.B.; Azhar, S. PPARs: Regulators of metabolism and as therapeutic targets in cardiovascular disease. Part II: PPAR-beta/delta and PPAR-gamma. Future Cardiol. 2017, 13, 279–296. [Google Scholar] [CrossRef] [PubMed]
- Kvandova, M.; Majzunova, M.; Dovinova, I. The role of PPARgamma in cardiovascular diseases. Physiol. Res. 2016, 65, S343–S363. [Google Scholar] [CrossRef]
- Mirza, A.Z.; Althagafi, I.I.; Shamshad, H. Role of PPAR receptor in different diseases and their ligands: Physiological importance and clinical implications. Eur. J. Med. Chem. 2019, 166, 502–513. [Google Scholar] [CrossRef] [PubMed]
- Bugge, A.; Holst, D. PPAR agonists,—Could tissue targeting pave the way? Biochimie 2017, 136, 100–104. [Google Scholar] [CrossRef]
- Patel, B.; Mann, G.E.; Chapple, S.J. Concerted redox modulation by sulforaphane alleviates diabetes and cardiometabolic syndrome. Free Radic. Biol. Med. 2018, 122, 150–160. [Google Scholar] [CrossRef]
- Men, X.; Han, X.; Lee, S.J.; Park, K.T.; Han, J.K.; Choi, S.I.; Lee, O.H. Anti-adipogenic Effects of Sulforaphane-rich Ingredient with Broccoli Sprout and Mustard Seed in 3T3-L1 Preadipocytes. Planta Med. 2022, 89, 526–538. [Google Scholar] [CrossRef] [PubMed]
- Men, X.; Han, X.; Lee, S.J.; Oh, G.; Park, K.T.; Han, J.K.; Choi, S.I.; Lee, O.H. Anti-Obesogenic Effects of Sulforaphane-Rich Broccoli (Brassica oleracea var. italica) Sprouts and Myrosinase-Rich Mustard (Sinapis alba L.) Seeds In Vitro and In Vivo. Nutrients 2022, 14, 3814. [Google Scholar] [CrossRef] [PubMed]
- Choi, K.M.; Lee, Y.S.; Kim, W.; Kim, S.J.; Shin, K.O.; Yu, J.Y.; Lee, M.K.; Lee, Y.M.; Hong, J.T.; Yun, Y.P.; et al. Sulforaphane attenuates obesity by inhibiting adipogenesis and activating the AMPK pathway in obese mice. J. Nutr. Biochem. 2014, 25, 201–207. [Google Scholar] [CrossRef] [PubMed]
- Tian, S.; Lei, P.; Teng, C.; Sun, Y.; Song, X.; Li, B.; Shan, Y. Targeting PLIN2/PLIN5-PPARgamma: Sulforaphane Disturbs the Maturation of Lipid Droplets. Mol. Nutr. Food Res. 2019, 63, e1900183. [Google Scholar] [CrossRef]
- Du, K.; Fan, Y.; Li, D. Sulforaphane ameliorates lipid profile in rodents: An updated systematic review and meta-analysis. Sci. Rep. 2021, 11, 7804. [Google Scholar] [CrossRef]
- Zhao, J.F.; Shyue, S.K.; Kou, Y.R.; Lu, T.M.; Lee, T.S. Transient Receptor Potential Ankyrin 1 Channel Involved in Atherosclerosis and Macrophage-Foam Cell Formation. Int. J. Biol. Sci. 2016, 12, 812–823. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Li, Y.; Wang, W.; Han, X.; Han, J.; Chen, M.; Zhang, J.; Wang, C.; Li, S.; Luo, J.; et al. Nuclear Factor Erythroid 2 Related Factor 2 Activator JC-5411 Inhibits Atherosclerosis Through Suppression of Inflammation and Regulation of Lipid Metabolism. Front. Pharmacol. 2020, 11, 532568. [Google Scholar] [CrossRef]
- Wu, L.; Noyan Ashraf, M.H.; Facci, M.; Wang, R.; Paterson, P.G.; Ferrie, A.; Juurlink, B.H. Dietary approach to attenuate oxidative stress, hypertension, and inflammation in the cardiovascular system. Proc. Natl. Acad. Sci. USA 2004, 101, 7094–7099. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Jiang, M.; Chen, Y.; Zhang, S.; Zhang, W.; Yang, X.; Li, X.; Li, Y.; Duan, S.; Han, J.; et al. Inhibition of Macrophage CD36 Expression and Cellular Oxidized Low Density Lipoprotein (oxLDL) Accumulation by Tamoxifen: A PEROXISOME PROLIFERATOR-ACTIVATED RECEPTOR (PPAR)gamma-DEPENDENT MECHANISM. J. Biol. Chem. 2016, 291, 16977–16989. [Google Scholar] [CrossRef]
- Han, J.; Zhou, X.; Yokoyama, T.; Hajjar, D.P.; Gotto, A.M., Jr.; Nicholson, A.C. Pitavastatin downregulates expression of the macrophage type B scavenger receptor, CD36. Circulation 2004, 109, 790–796. [Google Scholar] [CrossRef]
- Chen, Y.; Yang, M.; Huang, W.; Chen, W.; Zhao, Y.; Schulte, M.L.; Volberding, P.; Gerbec, Z.; Zimmermann, M.T.; Zeighami, A.; et al. Mitochondrial Metabolic Reprogramming by CD36 Signaling Drives Macrophage Inflammatory Responses. Circ. Res. 2019, 125, 1087–1102. [Google Scholar] [CrossRef] [PubMed]
- Im, Y.S.; Gwon, M.H.; Yun, J.M. Protective effects of phenethyl isothiocyanate on foam cell formation by combined treatment of oxidized low-density lipoprotein and lipopolysaccharide in THP-1 macrophage. Food Sci. Nutr. 2021, 9, 3269–3279. [Google Scholar] [CrossRef] [PubMed]
- Gwon, M.H.; Im, Y.S.; Seo, A.R.; Kim, K.Y.; Moon, H.R.; Yun, J.M. Phenethyl Isothiocyanate Protects against High Fat/Cholesterol Diet-Induced Obesity and Atherosclerosis in C57BL/6 Mice. Nutrients 2020, 12, 3657. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.T.; Li, C.H.; Dai, T.T.; Tao, F.L.; Wang, M.W.; Wang, C.Y.; Han, Z.L.; Sun, N.X.; Zhao, Y.N.; Wang, D.L. Effects of allyl isothiocyanate on the expression, function, and its mechanism of ABCA1 and ABCG1 in pulmonary of COPD rats. Int. Immunopharmacol. 2021, 101, 108373. [Google Scholar] [CrossRef]
- Aranda-Rivera, A.K.; Cruz-Gregorio, A.; Aparicio-Trejo, O.E.; Tapia, E.; Sanchez-Lozada, L.G.; Garcia-Arroyo, F.E.; Amador-Martinez, I.; Orozco-Ibarra, M.; Fernandez-Valverde, F.; Pedraza-Chaverri, J. Sulforaphane Protects against Unilateral Ureteral Obstruction-Induced Renal Damage in Rats by Alleviating Mitochondrial and Lipid Metabolism Impairment. Antioxidants 2022, 11, 1854. [Google Scholar] [CrossRef]
- Zhao, X.; Sun, G.; Ting, S.M.; Song, S.; Zhang, J.; Edwards, N.J.; Aronowski, J. Cleaning up after ICH: The role of Nrf2 in modulating microglia function and hematoma clearance. J. Neurochem. 2015, 133, 144–152. [Google Scholar] [CrossRef]
- Aubouy, A.; Olagnier, D.; Bertin, G.; Ezinmegnon, S.; Majorel, C.; Mimar, S.; Massougbodji, A.; Deloron, P.; Pipy, B.; Coste, A. Nrf2-driven CD36 and HO-1 gene expression in circulating monocytes correlates with favourable clinical outcome in pregnancy-associated malaria. Malar. J. 2015, 14, 358. [Google Scholar] [CrossRef]
- McGovern, T.; Farahnak, S.; Chen, M.; Larsson, K.; Martin, J.G.; Adner, M. Organic dust, causing both oxidative stress and Nrf2 activation, is phagocytized by bronchial epithelial cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2019, 317, L305–L316. [Google Scholar] [CrossRef]
- Bahiraii, S.; Brenner, M.; Yan, F.; Weckwerth, W.; Heiss, E.H. Sulforaphane diminishes moonlighting of pyruvate kinase M2 and interleukin 1beta expression in M1 (LPS) macrophages. Front. Immunol. 2022, 13, 935692. [Google Scholar] [CrossRef]
- Pal, S.; Konkimalla, V.B. Sulforaphane regulates phenotypic and functional switching of both induced and spontaneously differentiating human monocytes. Int. Immunopharmacol. 2016, 35, 85–98. [Google Scholar] [CrossRef]
- Olagnier, D.; Lavergne, R.A.; Meunier, E.; Lefevre, L.; Dardenne, C.; Aubouy, A.; Benoit-Vical, F.; Ryffel, B.; Coste, A.; Berry, A.; et al. Nrf2, a PPARgamma alternative pathway to promote CD36 expression on inflammatory macrophages: Implication for malaria. PLoS Pathog. 2011, 7, e1002254. [Google Scholar] [CrossRef] [PubMed]
- Bewley, M.A.; Budd, R.C.; Ryan, E.; Cole, J.; Collini, P.; Marshall, J.; Kolsum, U.; Beech, G.; Emes, R.D.; Tcherniaeva, I.; et al. Opsonic Phagocytosis in Chronic Obstructive Pulmonary Disease Is Enhanced by Nrf2 Agonists. Am. J. Respir. Crit. Care Med. 2018, 198, 739–750. [Google Scholar] [CrossRef] [PubMed]
- Harvey, C.J.; Thimmulappa, R.K.; Sethi, S.; Kong, X.; Yarmus, L.; Brown, R.H.; Feller-Kopman, D.; Wise, R.; Biswal, S. Targeting Nrf2 signaling improves bacterial clearance by alveolar macrophages in patients with COPD and in a mouse model. Sci. Transl. Med. 2011, 3, 78ra32. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Gibbons, J.G.; DeLoid, G.M.; Bedugnis, A.S.; Thimmulappa, R.K.; Biswal, S.; Kobzik, L. Immunomodulators targeting MARCO expression improve resistance to postinfluenza bacterial pneumonia. Am. J. Physiol. Lung Cell. Mol. Physiol. 2017, 313, L138–L153. [Google Scholar] [CrossRef] [PubMed]
- Alonso-Pineiro, J.A.; Gonzalez-Rovira, A.; Sanchez-Gomar, I.; Moreno, J.A.; Duran-Ruiz, M.C. Nrf2 and Heme Oxygenase-1 Involvement in Atherosclerosis Related Oxidative Stress. Antioxidants 2021, 10, 1463. [Google Scholar] [CrossRef] [PubMed]
- Ungvari, Z.; Tarantini, S.; Nyul-Toth, A.; Kiss, T.; Yabluchanskiy, A.; Csipo, T.; Balasubramanian, P.; Lipecz, A.; Benyo, Z.; Csiszar, A. Nrf2 dysfunction and impaired cellular resilience to oxidative stressors in the aged vasculature: From increased cellular senescence to the pathogenesis of age-related vascular diseases. Geroscience 2019, 41, 727–738. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Davies, K.J.A.; Forman, H.J. Oxidative stress response and Nrf2 signaling in aging. Free Radic. Biol. Med. 2015, 88, 314–336. [Google Scholar] [CrossRef]
- Zakkar, M.; Van der Heiden, K.; Luong, L.A.; Chaudhury, H.; Cuhlmann, S.; Hamdulay, S.S.; Krams, R.; Edirisinghe, I.; Rahman, I.; Carlsen, H.; et al. Activation of Nrf2 in endothelial cells protects arteries from exhibiting a proinflammatory state. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 1851–1857. [Google Scholar] [CrossRef]
- Ali, M.; Bonay, M.; Vanhee, V.; Vinit, S.; Deramaudt, T.B. Comparative effectiveness of 4 natural and chemical activators of Nrf2 on inflammation, oxidative stress, macrophage polarization, and bactericidal activity in an in vitro macrophage infection model. PLoS ONE 2020, 15, e0234484. [Google Scholar] [CrossRef]
- Jiang, M.; Li, X. Activation of PPARgamma does not contribute to macrophage ABCA1 expression and ABCA1-mediated cholesterol efflux to apoAI. Biochem. Biophys. Res. Commun. 2017, 482, 849–856. [Google Scholar] [CrossRef]
- Liu, Y.G.; Yan, J.L.; Ji, Y.Q.; Nie, W.J.; Jiang, Y. Black mulberry ethanol extract attenuates atherosclerosis-related inflammatory factors and downregulates PPARgamma and CD36 genes in experimental atherosclerotic rats. Food Funct. 2020, 11, 2997–3005. [Google Scholar] [CrossRef] [PubMed]
- Morihara, N.; Ide, N.; Weiss, N. Aged garlic extract inhibits CD36 expression in human macrophages via modulation of the PPARgamma pathway. Phytother. Res. 2010, 24, 602–608. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, J.L.; Liu, Y.Y.; Li, Z.Z.; Zhuang, Q.Z.; Tang, W.Z.; Xiong, Y.; Huang, X.Z. Amentoflavone prevents ox-LDL-induced lipid accumulation by suppressing the PPARgamma/CD36 signal pathway. Toxicol. Appl. Pharmacol. 2021, 431, 115733. [Google Scholar] [CrossRef] [PubMed]
- Tang, F.T.; Cao, Y.; Wang, T.Q.; Wang, L.J.; Guo, J.; Zhou, X.S.; Xu, S.W.; Liu, W.H.; Liu, P.Q.; Huang, H.Q. Tanshinone IIA attenuates atherosclerosis in ApoE(-/-) mice through down-regulation of scavenger receptor expression. Eur. J. Pharmacol. 2011, 650, 275–284. [Google Scholar] [CrossRef]
- Sajja, R.K.; Prasad, S.; Tang, S.; Kaisar, M.A.; Cucullo, L. Blood-brain barrier disruption in diabetic mice is linked to Nrf2 signaling deficits: Role of ABCB10? Neurosci. Lett. 2017, 653, 152–158. [Google Scholar] [CrossRef]
- Hanssen, K.M.; Haber, M.; Fletcher, J.I. Targeting multidrug resistance-associated protein 1 (MRP1)-expressing cancers: Beyond pharmacological inhibition. Drug Resist. Updates 2021, 59, 100795. [Google Scholar] [CrossRef] [PubMed]
- Colin, S.; Chinetti-Gbaguidi, G.; Staels, B. Macrophage phenotypes in atherosclerosis. Immunol. Rev. 2014, 262, 153–166. [Google Scholar] [CrossRef] [PubMed]
- Chinetti-Gbaguidi, G.; Colin, S.; Staels, B. Macrophage subsets in atherosclerosis. Nat. Rev. Cardiol. 2015, 12, 10–17. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Groups | TC | TG | LDL-C | HDL-C |
|---|---|---|---|---|
| (mmol/L) | (mmol/L) | (mmol/L) | (mmol/L) | |
| Ctrl | 17.04 ± 2.52 | 2.21 ± 0.37 | 14.52 ± 2.60 | 1.47 ± 0.27 |
| SFN-L | 14.58 ± 2.06 | 1.82 ± 0.27 | 11.23 ± 1.27 1 | 1.81 ± 0.24 |
| SFN-H | 11.67 ± 1.82 1 | 1.39 ± 0.18 1 | 9.61 ± 1.09 1 | 2.19 ± 0.21 1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, S.; Zhang, Y.; Zheng, X.; Wang, Z.; Wang, P.; Zhang, M.; Shen, M.; Bao, Y.; Li, D. Sulforaphane Inhibits Foam Cell Formation and Atherosclerosis via Mechanisms Involving the Modulation of Macrophage Cholesterol Transport and the Related Phenotype. Nutrients 2023, 15, 2117. https://doi.org/10.3390/nu15092117
Liu S, Zhang Y, Zheng X, Wang Z, Wang P, Zhang M, Shen M, Bao Y, Li D. Sulforaphane Inhibits Foam Cell Formation and Atherosclerosis via Mechanisms Involving the Modulation of Macrophage Cholesterol Transport and the Related Phenotype. Nutrients. 2023; 15(9):2117. https://doi.org/10.3390/nu15092117
Chicago/Turabian StyleLiu, Shiyan, Yuan Zhang, Xiangyu Zheng, Ziling Wang, Pan Wang, Mengdi Zhang, Mengfan Shen, Yongping Bao, and Dan Li. 2023. "Sulforaphane Inhibits Foam Cell Formation and Atherosclerosis via Mechanisms Involving the Modulation of Macrophage Cholesterol Transport and the Related Phenotype" Nutrients 15, no. 9: 2117. https://doi.org/10.3390/nu15092117