
Impact of High Salt-Intake on a Natural Gut Ecosystem in Wildling Mice
 , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals and Diet
2.2. DNA Extraction
2.3. 16S rRNA Gene Amplification and Sequencing
2.4. Processing and Statistical Analysis of 16S rRNA Gene Sequencing Data
3. Results
3.1. HSD Affects Diversity and Composition of CLM and Wildling Gut Microbiota
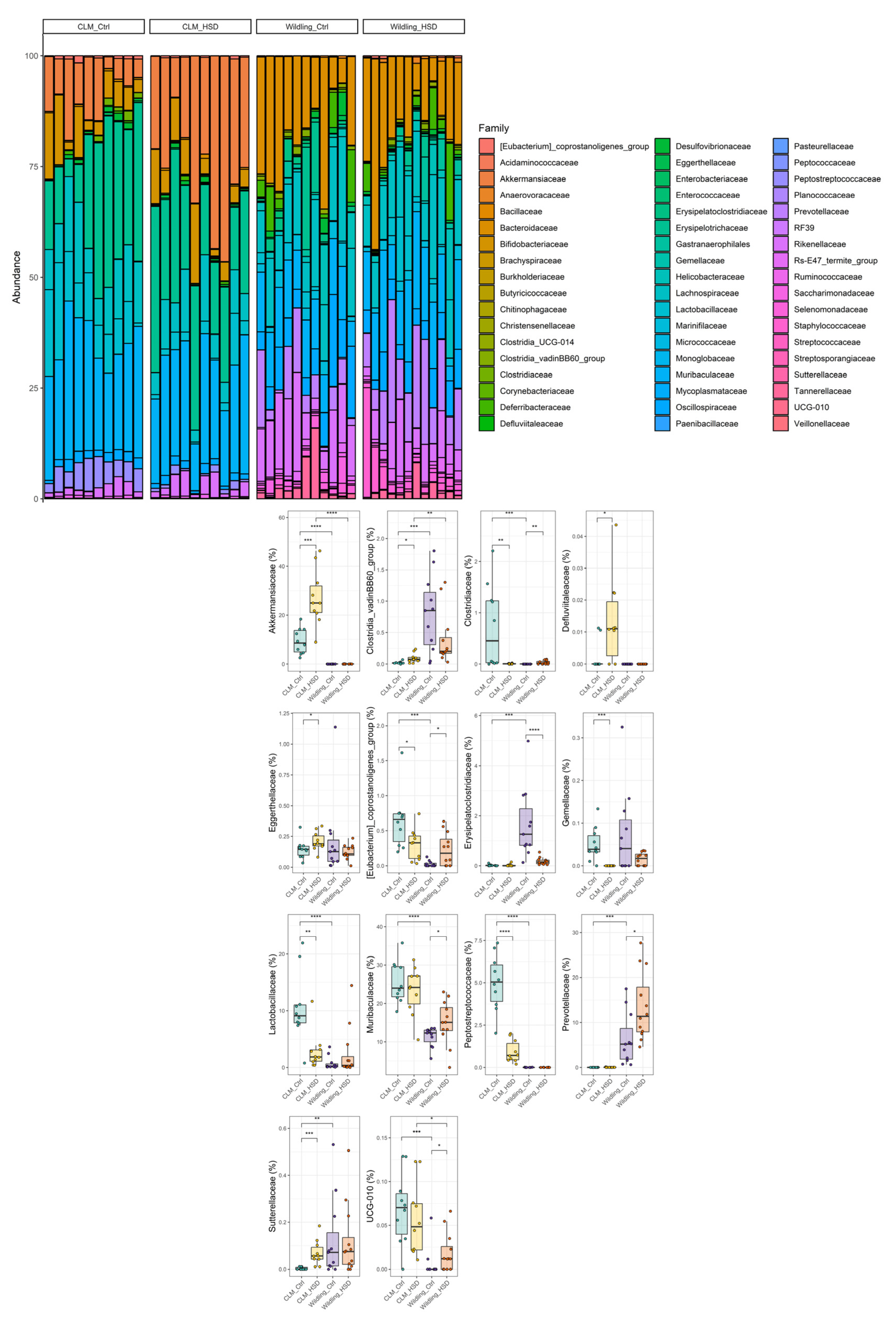
3.2. Gut Microbial Composition of Wildling Mice Is More Resistant to HSD than CLM
3.3. HSD Affects Predictive Microbial Functions in CLM but Not in Wildling Mice
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Candela, M.; Biagi, E.; Turroni, S.; Maccaferri, S.; Figini, P.; Brigidi, P. Dynamic efficiency of the human intestinal microbiota. Crit. Rev. Microbiol. 2015, 41, 165–171. [Google Scholar] [CrossRef] [PubMed]
- Candela, M.; Biagi, E.; Maccaferri, S.; Turroni, S.; Brigidi, P. Intestinal microbiota is a plastic factor responding to environmental changes. Trends Microbiol. 2012, 20, 385–391. [Google Scholar] [CrossRef] [PubMed]
- Boets, E.; Gomand, S.V.; Deroover, L.; Preston, T.; Vermeulen, K.; De Preter, V.; Hamer, H.M.; Van den Mooter, G.; De Vuyst, L.; Courtin, C.M. Systemic availability and metabolism of colonic-derived short-chain fatty acids in healthy subjects: A stable isotope study. J. Physiol. 2017, 595, 541–555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, J.; McKenzie, C.; Potamitis, M.; Thorburn, A.N.; Mackay, C.R.; Macia, L. The role of short-chain fatty acids in health and disease. Adv. Immunol. 2014, 121, 91–119. [Google Scholar] [PubMed]
- Kumar, J.; Rani, K.; Datt, C. Molecular link between dietary fibre, gut microbiota and health. Mol. Biol. Rep. 2020, 47, 6229–6237. [Google Scholar] [CrossRef] [PubMed]
- Bilotta, A.J.; Cong, Y. Gut microbiota metabolite regulation of host defenses at mucosal surfaces: Implication in precision medicine. Precis. Clin. Med. 2019, 2, 110–119. [Google Scholar] [CrossRef]
- Rooks, M.G.; Garrett, W.S. Gut microbiota, metabolites and host immunity. Nat. Rev. Immunol. 2016, 16, 341–352. [Google Scholar] [CrossRef]
- Rodríguez, J.M.; Murphy, K.; Stanton, C.; Ross, R.P.; Kober, O.I.; Juge, N.; Avershina, E.; Rudi, K.; Narbad, A.; Jenmalm, M.C. The composition of the gut microbiota throughout life, with an emphasis on early life. Microb. Ecol. Health Dis. 2015, 26, 26050. [Google Scholar] [CrossRef]
- Arrieta, M.-C.; Stiemsma, L.T.; Amenyogbe, N.; Brown, E.M.; Finlay, B. The intestinal microbiome in early life: Health and disease. Front. Immunol. 2014, 5, 427. [Google Scholar] [CrossRef] [Green Version]
- Chung, W.S.F.; Walker, A.W.; Louis, P.; Parkhill, J.; Vermeiren, J.; Bosscher, D.; Duncan, S.H.; Flint, H.J. Modulation of the human gut microbiota by dietary fibres occurs at the species level. BMC Biol. 2016, 14, 3. [Google Scholar] [CrossRef] [Green Version]
- Danneskiold-Samsøe, N.B.; Barros, H.D.d.F.Q.; Santos, R.; Bicas, J.L.; Cazarin, C.B.B.; Madsen, L.; Kristiansen, K.; Pastore, G.M.; Brix, S.; Júnior, M.R.M. Interplay between food and gut microbiota in health and disease. Food Res. Int. 2019, 115, 23–31. [Google Scholar] [CrossRef] [Green Version]
- Scott, K.P.; Duncan, S.H.; Flint, H.J. Dietary fibre and the gut microbiota. Nutr. Bull. 2008, 33, 201–211. [Google Scholar] [CrossRef]
- Donohoe, D.R.; Garge, N.; Zhang, X.; Sun, W.; O’Connell, T.M.; Bunger, M.K.; Bultman, S.J. The microbiome and butyrate regulate energy metabolism and autophagy in the mammalian colon. Cell Metab. 2011, 13, 517–526. [Google Scholar] [CrossRef] [Green Version]
- Gomaa, E.Z. Human gut microbiota/microbiome in health and diseases: A review. Antonie Van Leeuwenhoek 2020, 113, 2019–2040. [Google Scholar] [CrossRef]
- Requena, T.; Martínez-Cuesta, M.C.; Peláez, C. Diet and microbiota linked in health and disease. Food Funct. 2018, 9, 688–704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ericsson, A.C.; Franklin, C.L. The gut microbiome of laboratory mice: Considerations and best practices for translational research. Mamm. Genome 2021, 32, 239–250. [Google Scholar] [CrossRef] [PubMed]
- Beresford-Jones, B.S.; Forster, S.C.; Stares, M.D.; Notley, G.; Viciani, E.; Browne, H.P.; Boehmler, D.J.; Soderholm, A.T.; Kumar, N.; Vervier, K. The Mouse Gastrointestinal Bacteria Catalogue enables translation between the mouse and human gut microbiotas via functional mapping. Cell Host Microbe 2022, 30, 124–138.e8. [Google Scholar] [CrossRef]
- Fava, F.; Rizzetto, L.; Tuohy, K. Gut microbiota and health: Connecting actors across the metabolic system. Proc. Nutr. Soc. 2019, 78, 177–188. [Google Scholar] [CrossRef]
- David, L.A.; Materna, A.C.; Friedman, J.; Campos-Baptista, M.I.; Blackburn, M.C.; Perrotta, A.; Erdman, S.E.; Alm, E.J. Host lifestyle affects human microbiota on daily timescales. Genome Biol. 2014, 15, R89. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, M.; Nakayama, J. Development of the gut microbiota in infancy and its impact on health in later life. Allergol. Int. 2017, 66, 515–522. [Google Scholar] [CrossRef]
- David, L.A.; Maurice, C.F.; Carmody, R.N.; Gootenberg, D.B.; Button, J.E.; Wolfe, B.E.; Ling, A.V.; Devlin, A.S.; Varma, Y.; Fischbach, M.A. Diet rapidly and reproducibly alters the human gut microbiome. Nature 2014, 505, 559–563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- García-Montero, C.; Fraile-Martínez, O.; Gómez-Lahoz, A.M.; Pekarek, L.; Castellanos, A.J.; Noguerales-Fraguas, F.; Coca, S.; Guijarro, L.G.; García-Honduvilla, N.; Asúnsolo, A. Nutritional components in Western diet versus Mediterranean diet at the gut microbiota–immune system interplay. Implications for health and disease. Nutrients 2021, 13, 699. [Google Scholar] [CrossRef] [PubMed]
- Soverini, M.; Rampelli, S.; Turroni, S.; Schnorr, S.L.; Quercia, S.; Castagnetti, A.; Biagi, E.; Brigidi, P.; Candela, M. Variations in the post-weaning human gut metagenome profile as result of Bifidobacterium acquisition in the western microbiome. Front. Microbiol. 2016, 7, 1058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manzel, A.; Muller, D.N.; Hafler, D.A.; Erdman, S.E.; Linker, R.A.; Kleinewietfeld, M. Role of “Western diet” in inflammatory autoimmune diseases. Curr. Allergy Asthma Rep. 2014, 14, 404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kleinewietfeld, M.; Manzel, A.; Titze, J.; Kvakan, H.; Yosef, N.; Linker, R.A.; Muller, D.N.; Hafler, D.A. Sodium chloride drives autoimmune disease by the induction of pathogenic TH17 cells. Nature 2013, 496, 518–522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haase, S.; Wilck, N.; Kleinewietfeld, M.; Müller, D.N.; Linker, R.A. Sodium chloride triggers Th17 mediated autoimmunity. J. Neuroimmunol. 2019, 329, 9–13. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, A.L.; Kitz, A.; Wu, C.; Lowther, D.E.; Rodriguez, D.M.; Vudattu, N.; Deng, S.; Herold, K.C.; Kuchroo, V.K.; Kleinewietfeld, M. Sodium chloride inhibits the suppressive function of FOXP3+ regulatory T cells. J. Clin. Investig. 2015, 125, 4212–4222. [Google Scholar] [CrossRef] [Green Version]
- Hamad, I.; Cardilli, A.; Corte-Real, B.F.; Dyczko, A.; Vangronsveld, J.; Kleinewietfeld, M. High-Salt Diet Induces Depletion of Lactic Acid-Producing Bacteria in Murine Gut. Nutrients 2022, 14, 1171. [Google Scholar] [CrossRef]
- Wilck, N.; Matus, M.G.; Kearney, S.M.; Olesen, S.W.; Forslund, K.; Bartolomaeus, H.; Haase, S.; Mähler, A.; Balogh, A.; Markó, L. Salt-responsive gut commensal modulates TH 17 axis and disease. Nature 2017, 551, 585–589. [Google Scholar] [CrossRef]
- Wei, Y.; Lu, C.; Chen, J.; Cui, G.; Wang, L.; Yu, T.; Yang, Y.; Wu, W.; Ding, Y.; Li, L. High salt diet stimulates gut Th17 response and exacerbates TNBS-induced colitis in mice. Oncotarget 2017, 8, 70. [Google Scholar] [CrossRef] [Green Version]
- He, F.J.; Li, J.; MacGregor, G.A. Effect of longer-term modest salt reduction on blood pressure. Cochrane Database Syst. Rev. 2013, 346, f1325. [Google Scholar] [CrossRef] [PubMed]
- Hu, L.; Zhu, S.; Peng, X.; Li, K.; Peng, W.; Zhong, Y.; Kang, C.; Cao, X.; Liu, Z.; Zhao, B. High salt elicits brain inflammation and cognitive dysfunction, accompanied by alternations in the gut microbiota and decreased SCFA production. J. Alzheimer’s Dis. 2020, 77, 629–640. [Google Scholar] [CrossRef]
- Tubbs, A.L.; Liu, B.; Rogers, T.D.; Sartor, R.B.; Miao, E.A. Dietary salt exacerbates experimental colitis. J. Immunol. 2017, 199, 1051–1059. [Google Scholar] [CrossRef] [Green Version]
- Muller, D.N.; Wilck, N.; Haase, S.; Kleinewietfeld, M.; Linker, R.A. Sodium in the microenvironment regulates immune responses and tissue homeostasis. Nat. Rev. Immunol. 2019, 19, 243–254. [Google Scholar] [CrossRef] [PubMed]
- Burr, A.H.; Bhattacharjee, A.; Hand, T.W. Nutritional modulation of the microbiome and immune response. J. Immunol. 2020, 205, 1479–1487. [Google Scholar] [CrossRef] [PubMed]
- Roca-Saavedra, P.; Mendez-Vilabrille, V.; Miranda, J.M.; Nebot, C.; Cardelle-Cobas, A.; Franco, C.M.; Cepeda, A. Food additives, contaminants and other minor components: Effects on human gut microbiota—A review. J. Physiol. Biochem. 2018, 74, 69–83. [Google Scholar] [CrossRef]
- Côrte-Real, B.F.; Hamad, I.; Hornero, R.A.; Geisberger, S.; Roels, J.; Van Zeebroeck, L.; Dyczko, A.; van Gisbergen, M.W.; Kurniawan, H.; Wagner, A. Sodium perturbs mitochondrial respiration and induces dysfunctional Tregs. Cell Metab. 2023, 35, 299–315.e298. [Google Scholar] [CrossRef] [PubMed]
- Zagato, E.; Pozzi, C.; Bertocchi, A.; Schioppa, T.; Saccheri, F.; Guglietta, S.; Fosso, B.; Melocchi, L.; Nizzoli, G.; Troisi, J. Endogenous murine microbiota member Faecalibaculum rodentium and its human homologue protect from intestinal tumour growth. Nat. Microbiol. 2020, 5, 511–524. [Google Scholar] [CrossRef] [PubMed]
- Mao, G.; Li, S.; Orfila, C.; Shen, X.; Zhou, S.; Linhardt, R.J.; Ye, X.; Chen, S. Depolymerized RG-I-enriched pectin from citrus segment membranes modulates gut microbiota, increases SCFA production, and promotes the growth of Bifidobacterium spp., Lactobacillus spp. and Faecalibaculum spp. Food Funct. 2019, 10, 7828–7843. [Google Scholar] [CrossRef]
- Miranda, P.M.; De Palma, G.; Serkis, V.; Lu, J.; Louis-Auguste, M.P.; McCarville, J.L.; Verdu, E.F.; Collins, S.M.; Bercik, P. High salt diet exacerbates colitis in mice by decreasing Lactobacillus levels and butyrate production. Microbiome 2018, 6, 57. [Google Scholar] [CrossRef]
- Chen, L.; He, F.J.; Dong, Y.; Huang, Y.; Wang, C.; Harshfield, G.A.; Zhu, H. Modest sodium reduction increases circulating short-chain fatty acids in untreated hypertensives: A randomized, double-blind, placebo-controlled trial. Hypertension 2020, 76, 73–79. [Google Scholar] [CrossRef] [PubMed]
- Lukovac, S.; Belzer, C.; Pellis, L.; Keijser, B.J.; de Vos, W.M.; Montijn, R.C.; Roeselers, G. Differential modulation by Akkermansia muciniphila and Faecalibacterium prausnitzii of host peripheral lipid metabolism and histone acetylation in mouse gut organoids. MBio 2014, 5, e01438-14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dao, M.C.; Everard, A.; Aron-Wisnewsky, J.; Sokolovska, N.; Prifti, E.; Verger, E.O.; Kayser, B.D.; Levenez, F.; Chilloux, J.; Hoyles, L. Akkermansia muciniphila and improved metabolic health during a dietary intervention in obesity: Relationship with gut microbiome richness and ecology. Gut 2016, 65, 426–436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Llewellyn, S.R.; Britton, G.J.; Contijoch, E.J.; Vennaro, O.H.; Mortha, A.; Colombel, J.-F.; Grinspan, A.; Clemente, J.C.; Merad, M.; Faith, J.J. Interactions between diet and the intestinal microbiota alter intestinal permeability and colitis severity in mice. Gastroenterology 2018, 154, 1037–1046.e1032. [Google Scholar] [CrossRef]
- Berer, K.; Gerdes, L.A.; Cekanaviciute, E.; Jia, X.; Xiao, L.; Xia, Z.; Liu, C.; Klotz, L.; Stauffer, U.; Baranzini, S.E. Gut microbiota from multiple sclerosis patients enables spontaneous autoimmune encephalomyelitis in mice. Proc. Natl. Acad. Sci. USA 2017, 114, 10719–10724. [Google Scholar] [CrossRef] [Green Version]
- Sanchez, J.M.S.; DePaula-Silva, A.B.; Libbey, J.E.; Fujinami, R.S. Role of diet in regulating the gut microbiota and multiple sclerosis. Clin. Immunol. 2022, 235, 108379. [Google Scholar] [CrossRef]
- Mak, I.W.; Evaniew, N.; Ghert, M. Lost in translation: Animal models and clinical trials in cancer treatment. Am. J. Transl. Res. 2014, 6, 114. [Google Scholar]
- Payne, K.J.; Crooks, G.M. Immune-cell lineage commitment: Translation from mice to humans. Immunity 2007, 26, 674–677. [Google Scholar] [CrossRef] [Green Version]
- Seok, J.; Warren, H.S.; Cuenca, A.G.; Mindrinos, M.N.; Baker, H.V.; Xu, W.; Richards, D.R.; McDonald-Smith, G.P.; Gao, H.; Hennessy, L. Genomic responses in mouse models poorly mimic human inflammatory diseases. Proc. Natl. Acad. Sci. USA 2013, 110, 3507–3512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, T.L.A.; Vieira-Silva, S.; Liston, A.; Raes, J. How informative is the mouse for human gut microbiota research? Dis. Model. Mech. 2015, 8, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Rosshart, S.P.; Vassallo, B.G.; Angeletti, D.; Hutchinson, D.S.; Morgan, A.P.; Takeda, K.; Hickman, H.D.; McCulloch, J.A.; Badger, J.H.; Ajami, N.J. Wild mouse gut microbiota promotes host fitness and improves disease resistance. Cell 2017, 171, 1015–1028.e1013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, T.A.; Phifer-Rixey, M.; Mack, K.L.; Sheehan, M.J.; Lin, D.; Bi, K.; Nachman, M.W. Host genetic determinants of the gut microbiota of wild mice. Mol. Ecol. 2019, 28, 3197–3207. [Google Scholar] [CrossRef] [PubMed]
- Maurice, C.F.; CL Knowles, S.; Ladau, J.; Pollard, K.S.; Fenton, A.; Pedersen, A.B.; Turnbaugh, P.J. Marked seasonal variation in the wild mouse gut microbiota. ISME J. 2015, 9, 2423–2434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosshart, S.P.; Herz, J.; Vassallo, B.G.; Hunter, A.; Wall, M.K.; Badger, J.H.; McCulloch, J.A.; Anastasakis, D.G.; Sarshad, A.A.; Leonardi, I. Laboratory mice born to wild mice have natural microbiota and model human immune responses. Science 2019, 365, eaaw4361. [Google Scholar] [CrossRef] [PubMed]
- Hild, B.; Dreier, M.S.; Oh, J.H.; McCulloch, J.A.; Badger, J.H.; Guo, J.; Thefaine, C.E.; Umarova, R.; Hall, K.D.; Gavrilova, O. Neonatal exposure to a wild-derived microbiome protects mice against diet-induced obesity. Nat. Metab. 2021, 3, 1042–1057. [Google Scholar] [CrossRef] [PubMed]
- Caporaso, J.G.; Lauber, C.L.; Walters, W.A.; Berg-Lyons, D.; Lozupone, C.A.; Turnbaugh, P.J.; Fierer, N.; Knight, R. Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc. Natl. Acad. Sci. USA 2011, 108, 4516–4522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [Green Version]
- Lozupone, C.; Lladser, M.E.; Knights, D.; Stombaugh, J.; Knight, R. UniFrac: An effective distance metric for microbial community comparison. ISME J. 2011, 5, 169–172. [Google Scholar] [CrossRef] [Green Version]
- Oksanen, J.; Simpson, G.; Blanchet, F.; Kindt, R.; Legendre, P.; Minchin, P.; O’Hara, R.; Solymos, P.; Stevens, M.; Szoecs, E.; et al. Vegan: Community Ecology Package. Version 2.6-4. 11 October 2022. Available online: https://CRAN.R-project.org/package=vegan (accessed on 26 November 2022).
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011, 12, R60. [Google Scholar] [CrossRef] [Green Version]
- Douglas, G.M.; Maffei, V.J.; Zaneveld, J.R.; Yurgel, S.N.; Brown, J.R.; Taylor, C.M.; Huttenhower, C.; Langille, M.G. PICRUSt2 for prediction of metagenome functions. Nat. Biotechnol. 2020, 38, 685–688. [Google Scholar] [CrossRef]
- Neal, B.; Wu, Y.; Feng, X.; Zhang, R.; Zhang, Y.; Shi, J.; Zhang, J.; Tian, M.; Huang, L.; Li, Z. Effect of salt substitution on cardiovascular events and death. N. Engl. J. Med. 2021, 385, 1067–1077. [Google Scholar] [CrossRef]
- Arroyo Hornero, R.; Hamad, I.; Côrte-Real, B.; Kleinewietfeld, M. The impact of dietary components on regulatory T cells and disease. Front. Immunol. 2020, 11, 253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, G.D.; Chen, J.; Hoffmann, C.; Bittinger, K.; Chen, Y.Y.; Keilbaugh, S.A.; Bewtra, M.; Knights, D.; Walters, W.A.; Knight, R.; et al. Linking long-term dietary patterns with gut microbial enterotypes. Science 2011, 334, 105–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ju, T.; Kong, J.Y.; Stothard, P.; Willing, B.P. Defining the role of Parasutterella, a previously uncharacterized member of the core gut microbiota. ISME J. 2019, 13, 1520–1534. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.J.; Wu, H.; Wu, S.D.; Lu, N.; Wang, Y.T.; Liu, H.N.; Dong, L.; Liu, T.T.; Shen, X.Z. Parasutterella, in association with irritable bowel syndrome and intestinal chronic inflammation. J. Gastroenterol. Hepatol. 2018, 33, 1844–1852. [Google Scholar] [CrossRef] [PubMed]
- Clavel, T.; Duck, W.; Charrier, C.; Wenning, M.; Elson, C.; Haller, D. Enterorhabdus caecimuris sp. nov., a member of the family Coriobacteriaceae isolated from a mouse model of spontaneous colitis, and emended description of the genus Enterorhabdus Clavel et al. 2009. Int. J. Syst. Evol. Microbiol. 2010, 60, 1527–1531. [Google Scholar] [CrossRef] [Green Version]
- Machiels, K.; Joossens, M.; Sabino, J.; De Preter, V.; Arijs, I.; Eeckhaut, V.; Ballet, V.; Claes, K.; Van Immerseel, F.; Verbeke, K. A decrease of the butyrate-producing species Roseburia hominis and Faecalibacterium prausnitzii defines dysbiosis in patients with ulcerative colitis. Gut 2014, 63, 1275–1283. [Google Scholar] [CrossRef]
- Imhann, F.; Vila, A.V.; Bonder, M.J.; Fu, J.; Gevers, D.; Visschedijk, M.C.; Spekhorst, L.M.; Alberts, R.; Franke, L.; Van Dullemen, H.M. Interplay of host genetics and gut microbiota underlying the onset and clinical presentation of inflammatory bowel disease. Gut 2018, 67, 108–119. [Google Scholar] [CrossRef]
- Silveira-Nunes, G.; Durso, D.F.; Jr, L.R.A.O.; Cunha, E.H.M.; Maioli, T.U.; Vieira, A.T.; Speziali, E.; Corrêa-Oliveira, R.; Martins-Filho, O.A.; Teixeira-Carvalho, A. Hypertension is associated with intestinal microbiota dysbiosis and inflammation in a Brazilian population. Front. Pharmacol. 2020, 11, 258. [Google Scholar] [CrossRef] [Green Version]
- Sommer, F.; Bäckhed, F. The gut microbiota—Masters of host development and physiology. Nat. Rev. Microbiol. 2013, 11, 227–238. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Jia, H.; Zhou, C.; Yang, Y.; Zhao, Y.; Yang, M.; Zou, Z. Variations in gut microbiota and fecal metabolic phenotype associated with depression by 16S rRNA gene sequencing and LC/MS-based metabolomics. J. Pharm. Biomed. Anal. 2017, 138, 231–239. [Google Scholar] [CrossRef] [PubMed]
- Derrien, M.; Belzer, C.; de Vos, W.M. Akkermansia muciniphila and its role in regulating host functions. Microb. Pathog. 2017, 106, 171–181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ansaldo, E.; Slayden, L.C.; Ching, K.L.; Koch, M.A.; Wolf, N.K.; Plichta, D.R.; Brown, E.M.; Graham, D.B.; Xavier, R.J.; Moon, J.J. Akkermansia muciniphila induces intestinal adaptive immune responses during homeostasis. Science 2019, 364, 1179–1184. [Google Scholar] [CrossRef]
- Bodogai, M.; O’Connell, J.; Kim, K.; Kim, Y.; Moritoh, K.; Chen, C.; Gusev, F.; Vaughan, K.; Shulzhenko, N.; Mattison, J.A. Commensal bacteria contribute to insulin resistance in aging by activating innate B1a cells. Sci. Transl. Med. 2018, 10, eaat4271. [Google Scholar] [CrossRef]
- Everard, A.; Belzer, C.; Geurts, L.; Ouwerkerk, J.P.; Druart, C.; Bindels, L.B.; Guiot, Y.; Derrien, M.; Muccioli, G.G.; Delzenne, N.M. Cross-talk between Akkermansia muciniphila and intestinal epithelium controls diet-induced obesity. Proc. Natl. Acad. Sci. USA 2013, 110, 9066–9071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osman, M.A.; Neoh, H.-m.; Ab Mutalib, N.-S.; Chin, S.-F.; Mazlan, L.; Raja Ali, R.A.; Zakaria, A.D.; Ngiu, C.S.; Ang, M.Y.; Jamal, R. Parvimonas micra, Peptostreptococcus stomatis, Fusobacterium nucleatum and Akkermansia muciniphila as a four-bacteria biomarker panel of colorectal cancer. Sci. Rep. 2021, 11, 2925. [Google Scholar] [CrossRef]
- Baldini, F.; Hertel, J.; Sandt, E.; Thinnes, C.C.; Neuberger-Castillo, L.; Pavelka, L.; Betsou, F.; Krüger, R.; Thiele, I.; Consortium, N.-P. Parkinson’s disease-associated alterations of the gut microbiome can invoke disease-relevant metabolic changes. bioRxiv 2019, 691030. Available online: https://www.researchgate.net/publication/334202416_Parkinson's_disease-associated_alterations_of_the_gut_microbiome_can_invoke_disease-relevant_metabolic_changes (accessed on 13 January 2023). [CrossRef] [Green Version]
- Hertel, J.; Harms, A.C.; Heinken, A.; Baldini, F.; Thinnes, C.C.; Glaab, E.; Vasco, D.A.; Pietzner, M.; Stewart, I.D.; Wareham, N.J. Integrated analyses of microbiome and longitudinal metabolome data reveal microbial-host interactions on sulfur metabolism in Parkinson’s disease. Cell Rep. 2019, 29, 1767–1777.e8. [Google Scholar] [CrossRef] [Green Version]
- Cekanaviciute, E.; Yoo, B.B.; Runia, T.F.; Debelius, J.W.; Singh, S.; Nelson, C.A.; Kanner, R.; Bencosme, Y.; Lee, Y.K.; Hauser, S.L. Gut bacteria from multiple sclerosis patients modulate human T cells and exacerbate symptoms in mouse models. Proc. Natl. Acad. Sci. USA 2017, 114, 10713–10718. [Google Scholar] [CrossRef] [Green Version]
- Nagpal, R.; Neth, B.J.; Wang, S.; Mishra, S.P.; Craft, S.; Yadav, H. Gut mycobiome and its interaction with diet, gut bacteria and alzheimer’s disease markers in subjects with mild cognitive impairment: A pilot study. EBioMedicine 2020, 59, 102950. [Google Scholar] [CrossRef] [PubMed]
- Mims, T.S.; Abdallah, Q.A.; Stewart, J.D.; Watts, S.P.; White, C.T.; Rousselle, T.V.; Gosain, A.; Bajwa, A.; Han, J.C.; Willis, K.A. The gut mycobiome of healthy mice is shaped by the environment and correlates with metabolic outcomes in response to diet. Commun. Biol. 2021, 4, 281. [Google Scholar] [CrossRef] [PubMed]
- Van Tilburg Bernardes, E.; Pettersen, V.K.; Gutierrez, M.W.; Laforest-Lapointe, I.; Jendzjowsky, N.G.; Cavin, J.-B.; Vicentini, F.A.; Keenan, C.M.; Ramay, H.R.; Samara, J. Intestinal fungi are causally implicated in microbiome assembly and immune development in mice. Nat. Commun. 2020, 11, 2577. [Google Scholar] [CrossRef] [PubMed]
- Underhill, D.M.; Iliev, I.D. The mycobiota: Interactions between commensal fungi and the host immune system. Nat. Rev. Immunol. 2014, 14, 405–416. [Google Scholar] [CrossRef] [PubMed]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cardilli, A.; Hamad, I.; Dyczko, A.; Thijs, S.; Vangronsveld, J.; Müller, D.N.; Rosshart, S.P.; Kleinewietfeld, M. Impact of High Salt-Intake on a Natural Gut Ecosystem in Wildling Mice. Nutrients 2023, 15, 1565. https://doi.org/10.3390/nu15071565
Cardilli A, Hamad I, Dyczko A, Thijs S, Vangronsveld J, Müller DN, Rosshart SP, Kleinewietfeld M. Impact of High Salt-Intake on a Natural Gut Ecosystem in Wildling Mice. Nutrients. 2023; 15(7):1565. https://doi.org/10.3390/nu15071565
Chicago/Turabian StyleCardilli, Alessio, Ibrahim Hamad, Aleksandra Dyczko, Sofie Thijs, Jaco Vangronsveld, Dominik N. Müller, Stephan P. Rosshart, and Markus Kleinewietfeld. 2023. "Impact of High Salt-Intake on a Natural Gut Ecosystem in Wildling Mice" Nutrients 15, no. 7: 1565. https://doi.org/10.3390/nu15071565