
Endoplasmic Reticulum Stress and Its Impact on Adipogenesis: Molecular Mechanisms Implicated

Abstract
:1. Introduction
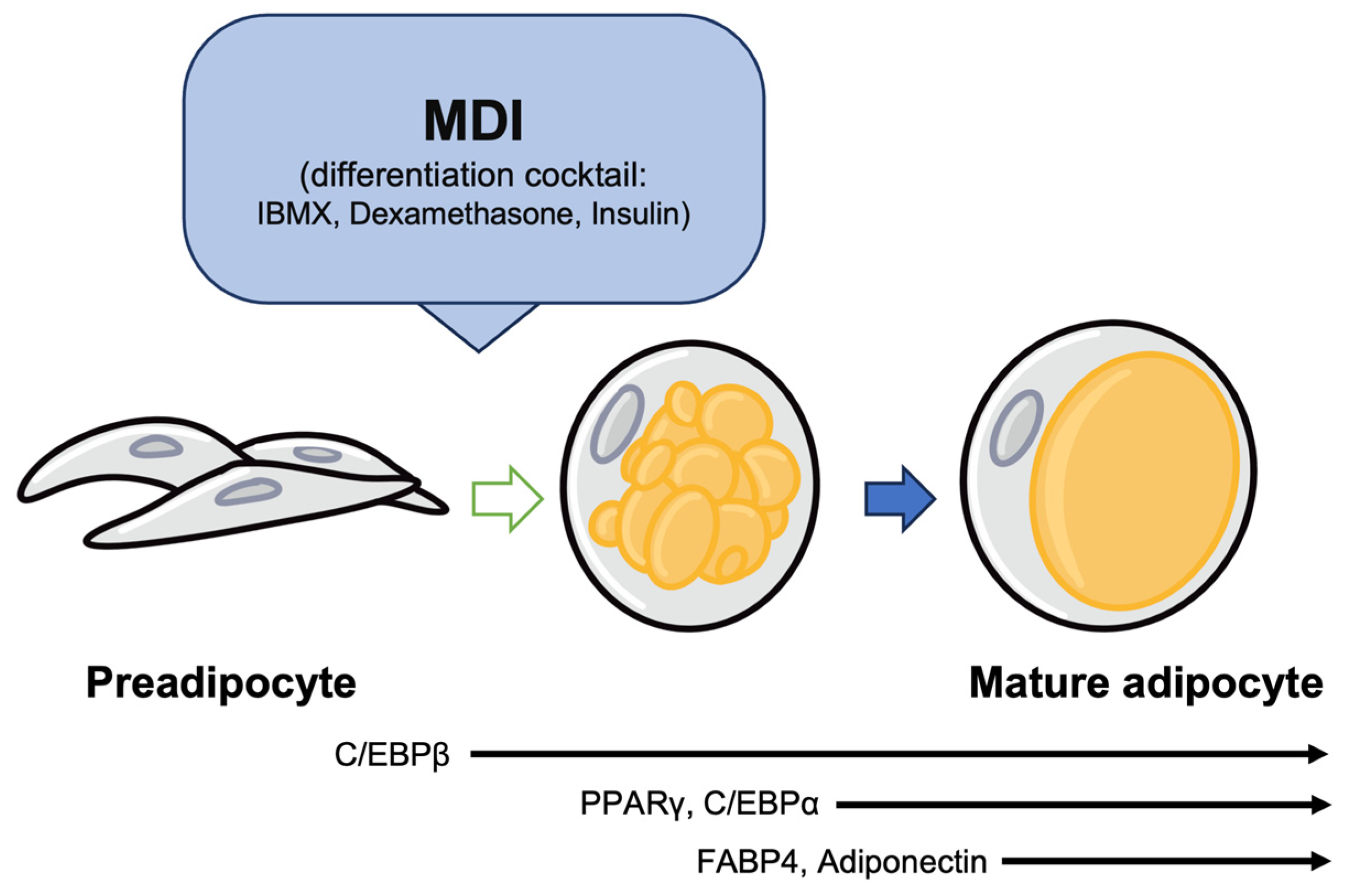
2. The Process of Adipogenesis
3. UPR Signaling Pathways (IRE1-XBP1, PERK, ATF6) in Obesity
3.1. The IRE1-XBP1 Pathway and Lipid Metabolism
3.2. The PERK Pathway and Insulin Sensitivity
3.3. The ATF6 Pathway and Inflammation
4. ER Stress and Adipogenesis
4.1. Effects of ER Stress on Transcription Factor Involed in Adipogenesis (PPARγ, C/EBPs)
4.2. Relationship between CHOP and a Transcription Factor Involved in Adipogenesis
4.3. Crosstalk between ER Stress and Other Signaling Pathways in Adipogenesis
4.3.1. ER Stress and UPR in Adipogenesis
4.3.2. ER Stress and Wingless/Integrated (Wnt) Signaling in Adipogenesis
4.3.3. ER Stress and mTOR Signaling in Adipogenesis
4.3.4. ER Stress and Insulin Signaling in Adipogenesis
4.3.5. ER Stress and Nuclear Receptors in Adipogenesis
5. Cyclophilin Family in Adipogenesis
5.1. CypA
5.2. CypB
6. The Cellular Consequences of Excessive ER Stress in the Adipose Tissue
6.1. Altered Lipid Metabolism and Dynamics of the Lipid Droplets
6.2. Adipokine Dysregulation and Metabolic Inflammation
6.3. Correlation between Aged Adipose Tissue and ER Stress
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| IRE1 | inositol-requiring enzyme1 |
| XBP1 | X-box binding protein 1 |
| PERK | protein kinase RNA-like ER kinase |
| PPARγ | proliferator-activated receptor γ |
| C/EBPα | CCAAT-enhancer-binding protein α |
| C/EBPβ | CCAAT-enhancer-binding protein β |
| TAG | triacylglycerol |
| CHOP | C/EBP homologous protein |
| NF-κB | nuclear factor-kappa B |
| AKT | protein kinase B |
| TNF-α | tumor necrosis factor-alpha |
| JNK | c-Jun N-terminal kinase |
| IL-6 | interleukin-6 |
| IL-8 | interleukin-8 |
| FABP4 | fatty acid-binding protein 4 |
| GLUT4 | glucose transporter type 4 |
| IL-1β | interleukin-1β |
| mTOR | mammalian target of rapamycin |
| mTORC1 | mTOR Complex 1 |
| AMPK | AMP-activated protein kinase |
| TSC1 | tuberous sclerosis complex 1 |
| TSC2 | tuberous sclerosis complex 2 |
| SREBP | sterol regulatory element-binding protein |
| IKK | inhibitor of nuclear factor kappa-B kinase |
| FFA | free fatty acids |
| IRS-1 | insulin receptor substrate-1 |
| IRS-2 | insulin receptor substrate-2 |
| LXRs | liver X receptors |
| HDL | high-density lipoprotein |
| ABCA1 | ATP-binding cassette A1 |
| ABCG1 | ATP-binding cassette G1 |
| CETP | cholesteryl ester transfer protein |
| Lpcat3 | lysophosphatidylcholine acyltransferase 3 |
| PLs | phospholipids |
| CypA | cyclophilin A |
| CypB | cyclophilin B |
| CsA | cyclosporine A |
References
- Park, J.H.; Moon, J.H.; Kim, H.J.; Kong, M.H.; Oh, Y.H. Sedentary Lifestyle: Overview of Updated Evidence of Potential Health Risks. Korean J. Fam. Med. 2020, 41, 365–373. [Google Scholar] [CrossRef] [PubMed]
- Ajoolabady, A.; Liu, S.; Klionsky, D.J.; Lip, G.Y.H.; Tuomilehto, J.; Kavalakatt, S.; Pereira, D.M.; Samali, A.; Ren, J. ER stress in obesity pathogenesis and management. Trends Pharmacol. Sci. 2022, 43, 97–109. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, E. Endoplasmic Reticulum Stress and Obesity. Adv. Exp. Med. Biol. 2017, 960, 261–276. [Google Scholar] [PubMed]
- Kawasaki, N.; Asada, R.; Saito, A.; Kanemoto, S.; Imaizumi, K. Obesity-induced endoplasmic reticulum stress causes chronic inflammation in adipose tissue. Sci. Rep. 2012, 2, 799. [Google Scholar] [CrossRef] [PubMed]
- Moncan, M.; Mnich, K.; Blomme, A.; Almanza, A.; Samali, A.; Gorman, A.M. Regulation of lipid metabolism by the unfolded protein response. J. Cell Mol. Med. 2021, 25, 1359–1370. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Maity, S. ER Stress-Sensor Proteins and ER-Mitochondrial Crosstalk-Signaling Beyond (ER) Stress Response. Biomolecules 2021, 11, 173. [Google Scholar] [CrossRef]
- Liang, W.L.; Cai, M.R.; Zhang, M.Q.; Cui, S.; Zhang, T.R.; Cheng, W.H.; Wu, Y.H.; Ou, W.J.; Jia, Z.H.; Zhang, S.F. Chinese Herbal Medicine Alleviates Myocardial Ischemia/Reperfusion Injury by Regulating Endoplasmic Reticulum Stress. Evid. Based Complement. Altern. Med. 2021, 2021, 4963346. [Google Scholar] [CrossRef]
- Yanagitani, K.; Kimata, Y.; Kadokura, H.; Kohno, K. Translational pausing ensures membrane targeting and cytoplasmic splicing of XBP1u mRNA. Science 2011, 331, 586–589. [Google Scholar] [CrossRef] [PubMed]
- Gardner, B.M.; Walter, P. Unfolded proteins are Ire1-activating ligands that directly induce the unfolded protein response. Science 2011, 333, 1891–1894. [Google Scholar] [CrossRef]
- Casagrande, R.; Stern, P.; Diehn, M.; Shamu, C.; Osario, M.; Zuniga, M.; Brown, P.O.; Ploegh, H. Degradation of proteins from the ER of S. cerevisiae requires an intact unfolded protein response pathway. Mol. Cell 2000, 5, 729–735. [Google Scholar] [CrossRef]
- Friedlander, R.; Jarosch, E.; Urban, J.; Volkwein, C.; Sommer, T. A regulatory link between ER-associated protein degradation and the unfolded-protein response. Nat. Cell Biol. 2000, 2, 379–384. [Google Scholar] [CrossRef]
- Travers, K.J.; Patil, C.K.; Wodicka, L.; Lockhart, D.J.; Weissman, J.S.; Walter, P. Functional and genomic analyses reveal an essential coordination between the unfolded protein response and ER-associated degradation. Cell 2000, 101, 249–258. [Google Scholar] [CrossRef]
- Ko, J.; Kim, J.Y.; Chae, M.K.; Lee, E.J.; Yoon, J.S. PERK mediates oxidative stress and adipogenesis in Graves’ orbitopathy pathogenesis. J. Mol. Endocrinol. 2021, 66, 313–323. [Google Scholar] [CrossRef]
- Jiao, P.; Ma, J.; Feng, B.; Zhang, H.; Diehl, J.A.; Chin, Y.E.; Yan, W.; Xu, H. FFA-induced adipocyte inflammation and insulin resistance: Involvement of ER stress and IKKbeta pathways. Obesity 2011, 19, 483–491. [Google Scholar] [CrossRef]
- Fernandes-da-Silva, A.; Miranda, C.S.; Santana-Oliveira, D.A.; Oliveira-Cordeiro, B.; Rangel-Azevedo, C.; Silva-Veiga, F.M.; Martins, F.F.; Souza-Mello, V. Endoplasmic reticulum stress as the basis of obesity and metabolic diseases: Focus on adipose tissue, liver, and pancreas. Eur. J. Nutr. 2021, 60, 2949–2960. [Google Scholar] [CrossRef]
- Zatterale, F.; Longo, M.; Naderi, J.; Raciti, G.A.; Desiderio, A.; Miele, C.; Beguinot, F. Chronic Adipose Tissue Inflammation Linking Obesity to Insulin Resistance and Type 2 Diabetes. Front. Physiol. 2019, 10, 1607. [Google Scholar] [CrossRef] [PubMed]
- Moreno, M.J.; Martinez, J.A. Adipose tissue: A storage and secretory organ. An. Del Sist. Sanit. De Navar. 2002, 25 (Suppl. S1), 29–39. [Google Scholar]
- Arrese, E.L.; Soulages, J.L. Insect fat body: Energy, metabolism, and regulation. Annu. Rev. Entomol. 2010, 55, 207–225. [Google Scholar] [CrossRef] [PubMed]
- Fain, J.N. Release of interleukins and other inflammatory cytokines by human adipose tissue is enhanced in obesity and primarily due to the nonfat cells. Vitam. Horm. 2006, 74, 443–477. [Google Scholar]
- Han, J.S.; Jeon, Y.G.; Oh, M.; Lee, G.; Nahmgoong, H.; Han, S.M.; Choi, J.; Kim, Y.Y.; Shin, K.C.; Kim, J.; et al. Adipocyte HIF2α functions as a thermostat via PKA Cα regulation in beige adipocytes. Nat. Commun. 2022, 13, 3268. [Google Scholar] [CrossRef]
- Park, A.; Kim, K.-e.; Park, I.; Lee, S.H.; Park, K.-Y.; Jung, M.; Li, X.; Sleiman, M.B.; Lee, S.J.; Kim, D.-S.; et al. Mitochondrial matrix protein LETMD1 maintains thermogenic capacity of brown adipose tissue in male mice. Nat. Commun. 2023, 14, 3746. [Google Scholar] [CrossRef] [PubMed]
- Rosen, E.D.; Spiegelman, B.M. What we talk about when we talk about fat. Cell 2014, 156, 20–44. [Google Scholar] [CrossRef] [PubMed]
- Shuster, A.; Patlas, M.; Pinthus, J.H.; Mourtzakis, M. The clinical importance of visceral adiposity: A critical review of methods for visceral adipose tissue analysis. Br. J. Radiol. 2012, 85, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Coelho, M.; Oliveira, T.; Fernandes, R. Biochemistry of adipose tissue: An endocrine organ. Arch. Med. Sci. 2013, 9, 191–200. [Google Scholar] [CrossRef]
- Deng, Y.; Scherer, P.E. Adipokines as novel biomarkers and regulators of the metabolic syndrome. Ann. N. Y. Acad. Sci. 2010, 1212, E1–E19. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, A.F.; Rocha, D.Q.; McIntyre, R.S.; Mesquita, L.M.; Kohler, C.A.; Hyphantis, T.N.; Sales, P.M.; Machado-Vieira, R.; Berk, M. Adipokines as emerging depression biomarkers: A systematic review and meta-analysis. J. Psychiatr. Res. 2014, 59, 28–37. [Google Scholar] [CrossRef]
- Landecho, M.F.; Tuero, C.; Valenti, V.; Bilbao, I.; de la Higuera, M.; Fruhbeck, G. Relevance of Leptin and Other Adipokines in Obesity-Associated Cardiovascular Risk. Nutrients 2019, 11, 2664. [Google Scholar] [CrossRef]
- Greenberg, A.S.; Obin, M.S. Obesity and the role of adipose tissue in inflammation and metabolism. Am. J. Clin. Nutr. 2006, 83, 461S–465S. [Google Scholar] [CrossRef]
- Bluher, M. Adipose tissue dysfunction contributes to obesity related metabolic diseases. Best Pract. Res. Clin. Endocrinol. Metab. 2013, 27, 163–177. [Google Scholar] [CrossRef]
- Jung, U.J.; Choi, M.S. Obesity and its metabolic complications: The role of adipokines and the relationship between obesity, inflammation, insulin resistance, dyslipidemia and nonalcoholic fatty liver disease. Int. J. Mol. Sci. 2014, 15, 6184–6223. [Google Scholar] [CrossRef] [PubMed]
- Mitrovic, B.; Gluvic, Z.M.; Obradovic, M.; Radunovic, M.; Rizzo, M.; Banach, M.; Isenovic, E.R. Non-alcoholic fatty liver disease, metabolic syndrome, and type 2 diabetes mellitus: Where do we stand today? Arch. Med. Sci. 2023, 19, 884–894. [Google Scholar] [CrossRef]
- Zhou, X.-D.; Cai, J.; Targher, G.; Byrne, C.D.; Shapiro, M.D.; Sung, K.-C.; Somers, V.K.; Chahal, C.A.A.; George, J.; Chen, L.-L.; et al. Metabolic dysfunction-associated fatty liver disease and implications for cardiovascular risk and disease prevention. Cardiovasc. Diabetol. 2022, 21, 270. [Google Scholar] [CrossRef] [PubMed]
- Marquez, M.P.; Alencastro, F.; Madrigal, A.; Jimenez, J.L.; Blanco, G.; Gureghian, A.; Keagy, L.; Lee, C.; Liu, R.; Tan, L.; et al. The Role of Cellular Proliferation in Adipogenic Differentiation of Human Adipose Tissue-Derived Mesenchymal Stem Cells. Stem. Cells Dev. 2017, 26, 1578–1595. [Google Scholar] [CrossRef]
- Lefterova, M.I.; Lazar, M.A. New developments in adipogenesis. Trends Endocrinol. Metab. 2009, 20, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Chawla, A.; Schwarz, E.J.; Dimaculangan, D.D.; Lazar, M.A. Peroxisome proliferator-activated receptor (PPAR) gamma: Adipose-predominant expression and induction early in adipocyte differentiation. Endocrinology 1994, 135, 798–800. [Google Scholar] [CrossRef] [PubMed]
- Farmer, S.R. Transcriptional control of adipocyte formation. Cell Metab. 2006, 4, 263–273. [Google Scholar] [CrossRef] [PubMed]
- McKnight, S.L.; Lane, M.D.; Gluecksohn-Waelsch, S. Is CCAAT/enhancer-binding protein a central regulator of energy metabolism? Genes Dev. 1989, 3, 2021–2024. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Bucher, N.L.; Farmer, S.R. Induction of peroxisome proliferator-activated receptor gamma during the conversion of 3T3 fibroblasts into adipocytes is mediated by C/EBPbeta, C/EBPdelta, and glucocorticoids. Mol. Cell Biol. 1996, 16, 4128–4136. [Google Scholar] [CrossRef]
- Robert, A.W.; Marcon, B.H.; Dallagiovanna, B.; Shigunov, P. Adipogenesis, Osteogenesis, and Chondrogenesis of Human Mesenchymal Stem/Stromal Cells: A Comparative Transcriptome Approach. Front. Cell Dev. Biol. 2020, 8, 561. [Google Scholar] [CrossRef]
- Ahmadian, M.; Duncan, R.E.; Jaworski, K.; Sarkadi-Nagy, E.; Sul, H.S. Triacylglycerol metabolism in adipose tissue. Future Lipidol. 2007, 2, 229–237. [Google Scholar] [CrossRef]
- Nakamura, T.; Shiojima, S.; Hirai, Y.; Iwama, T.; Tsuruzoe, N.; Hirasawa, A.; Katsuma, S.; Tsujimoto, G. Temporal gene expression changes during adipogenesis in human mesenchymal stem cells. Biochem. Biophys. Res. Commun. 2003, 303, 306–312. [Google Scholar] [CrossRef]
- Pittenger, M.F.; Mackay, A.M.; Beck, S.C.; Jaiswal, R.K.; Douglas, R.; Mosca, J.D.; Moorman, M.A.; Simonetti, D.W.; Craig, S.; Marshak, D.R. Multilineage potential of adult human mesenchymal stem cells. Science 1999, 284, 143–147. [Google Scholar] [CrossRef]
- Wan, Q.; Calhoun, C.; Zahr, T.; Qiang, L. Uncoupling Lipid Synthesis from Adipocyte Development. Biomedicines 2023, 11, 1132. [Google Scholar] [CrossRef] [PubMed]
- Read, A.; Schroder, M. The Unfolded Protein Response: An Overview. Biology 2021, 10, 384. [Google Scholar] [CrossRef] [PubMed]
- Sicari, D.; Delaunay-Moisan, A.; Combettes, L.; Chevet, E.; Igbaria, A. A guide to assessing endoplasmic reticulum homeostasis and stress in mammalian systems. FEBS J. 2020, 287, 27–42. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, H.; Matsui, T.; Yamamoto, A.; Okada, T.; Mori, K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell 2001, 107, 881–891. [Google Scholar] [CrossRef] [PubMed]
- Park, S.M.; Kang, T.I.; So, J.S. Roles of XBP1s in Transcriptional Regulation of Target Genes. Biomedicines 2021, 9, 791. [Google Scholar] [CrossRef]
- Gariballa, N.; Ali, B.R. Endoplasmic Reticulum Associated Protein Degradation (ERAD) in the Pathology of Diseases Related to TGFbeta Signaling Pathway: Future Therapeutic Perspectives. Front. Mol. Biosci. 2020, 7, 575608. [Google Scholar] [CrossRef]
- Bhattacharya, A.; Qi, L. ER-associated degradation in health and disease—From substrate to organism. J. Cell Sci. 2019, 132, jcs232850. [Google Scholar] [CrossRef]
- Hwang, J.; Qi, L. Quality Control in the Endoplasmic Reticulum: Crosstalk between ERAD and UPR pathways. Trends Biochem. Sci. 2018, 43, 593–605. [Google Scholar] [CrossRef]
- DuRose, J.B.; Scheuner, D.; Kaufman, R.J.; Rothblum, L.I.; Niwa, M. Phosphorylation of eukaryotic translation initiation factor 2alpha coordinates rRNA transcription and translation inhibition during endoplasmic reticulum stress. Mol. Cell Biol. 2009, 29, 4295–4307. [Google Scholar] [CrossRef] [PubMed]
- Rozpedek, W.; Pytel, D.; Mucha, B.; Leszczynska, H.; Diehl, J.A.; Majsterek, I. The Role of the PERK/eIF2alpha/ATF4/CHOP Signaling Pathway in Tumor Progression During Endoplasmic Reticulum Stress. Curr. Mol. Med. 2016, 16, 533–544. [Google Scholar] [CrossRef]
- Oyadomari, S.; Mori, M. Roles of CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ. 2004, 11, 381–389. [Google Scholar] [CrossRef] [PubMed]
- Kroeger, H.; Grimsey, N.; Paxman, R.; Chiang, W.C.; Plate, L.; Jones, Y.; Shaw, P.X.; Trejo, J.; Tsang, S.H.; Powers, E.; et al. The unfolded protein response regulator ATF6 promotes mesodermal differentiation. Sci. Signal. 2018, 11, eaan5785. [Google Scholar] [CrossRef] [PubMed]
- Hetz, C.; Chevet, E.; Oakes, S.A. Proteostasis control by the unfolded protein response. Nat. Cell Biol. 2015, 17, 829–838. [Google Scholar] [CrossRef] [PubMed]
- Szegezdi, E.; Logue, S.E.; Gorman, A.M.; Samali, A. Mediators of endoplasmic reticulum stress-induced apoptosis. EMBO Rep. 2006, 7, 880–885. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, S.P.; Kumar, K.U.; Kaufman, R.J. Phosphorylation of eukaryotic translation initiation factor 2 mediates apoptosis in response to activation of the double-stranded RNA-dependent protein kinase. J. Biol. Chem. 1998, 273, 2416–2423. [Google Scholar] [CrossRef]
- Ibrahim, I.M.; Abdelmalek, D.H.; Elfiky, A.A. GRP78: A cell’s response to stress. Life Sci. 2019, 226, 156–163. [Google Scholar] [CrossRef]
- Marzec, M.; Eletto, D.; Argon, Y. GRP94: An HSP90-like protein specialized for protein folding and quality control in the endoplasmic reticulum. Biochim. Biophys. Acta 2012, 1823, 774–787. [Google Scholar] [CrossRef]
- Grek, C.; Townsend, D.M. Protein Disulfide Isomerase Superfamily in Disease and the Regulation of Apoptosis. Cell Pathol. 2014, 1, 4–17. [Google Scholar] [CrossRef]
- Luo, B.; Lee, A.S. The critical roles of endoplasmic reticulum chaperones and unfolded protein response in tumorigenesis and anticancer therapies. Oncogene 2013, 32, 805–818. [Google Scholar] [CrossRef] [PubMed]
- Hecht, J.T.; Hayes, E.; Snuggs, M.; Decker, G.; Montufar-Solis, D.; Doege, K.; Mwalle, F.; Poole, R.; Stevens, J.; Duke, P.J. Calreticulin, PDI, Grp94 and BiP chaperone proteins are associated with retained COMP in pseudoachondroplasia chondrocytes. Matrix Biol. 2001, 20, 251–262. [Google Scholar] [CrossRef]
- Luan, W.; Li, F.; Zhang, J.; Wang, B.; Xiang, J. Cloning and expression of glucose regulated protein 78 (GRP78) in Fenneropenaeus chinensis. Mol. Biol. Rep. 2009, 36, 289–298. [Google Scholar] [CrossRef] [PubMed]
- Roufayel, R.; Kadry, S. Molecular Chaperone HSP70 and Key Regulators of Apoptosis—A Review. Curr. Mol. Med. 2019, 19, 315–325. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Wey, S.; Zhang, Y.; Ye, R.; Lee, A.S. Role of the unfolded protein response regulator GRP78/BiP in development, cancer, and neurological disorders. Antioxid. Redox Signal. 2009, 11, 2307–2316. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.S. The ER chaperone and signaling regulator GRP78/BiP as a monitor of endoplasmic reticulum stress. Methods 2005, 35, 373–381. [Google Scholar] [CrossRef] [PubMed]
- Christianson, J.C.; Shaler, T.A.; Tyler, R.E.; Kopito, R.R. OS-9 and GRP94 deliver mutant alpha1-antitrypsin to the Hrd1-SEL1L ubiquitin ligase complex for ERAD. Nat. Cell Biol. 2008, 10, 272–282. [Google Scholar] [CrossRef]
- Lippincott-Schwartz, J.; Bonifacino, J.S.; Yuan, L.C.; Klausner, R.D. Degradation from the endoplasmic reticulum: Disposing of newly synthesized proteins. Cell 1988, 54, 209–220. [Google Scholar] [CrossRef]
- Ushioda, R.; Hoseki, J.; Araki, K.; Jansen, G.; Thomas, D.Y.; Nagata, K. ERdj5 is required as a disulfide reductase for degradation of misfolded proteins in the ER. Science 2008, 321, 569–572. [Google Scholar] [CrossRef] [PubMed]
- Zhang, I.X.; Raghavan, M.; Satin, L.S. The Endoplasmic Reticulum and Calcium Homeostasis in Pancreatic Beta Cells. Endocrinology 2020, 161, bqz028. [Google Scholar] [CrossRef]
- Gorkhali, R.; Tian, L.; Dong, B.; Bagchi, P.; Deng, X.; Pawar, S.; Duong, D.; Fang, N.; Seyfried, N.; Yang, J. Extracellular calcium alters calcium-sensing receptor network integrating intracellular calcium-signaling and related key pathway. Sci. Rep. 2021, 11, 20576. [Google Scholar] [CrossRef] [PubMed]
- Reddy, R.K.; Mao, C.; Baumeister, P.; Austin, R.C.; Kaufman, R.J.; Lee, A.S. Endoplasmic reticulum chaperone protein GRP78 protects cells from apoptosis induced by topoisomerase inhibitors: Role of ATP binding site in suppression of caspase-7 activation. J. Biol. Chem. 2003, 278, 20915–20924. [Google Scholar] [CrossRef] [PubMed]
- Rao, R.V.; Peel, A.; Logvinova, A.; del Rio, G.; Hermel, E.; Yokota, T.; Goldsmith, P.C.; Ellerby, L.M.; Ellerby, H.M.; Bredesen, D.E. Coupling endoplasmic reticulum stress to the cell death program: Role of the ER chaperone GRP78. FEBS Lett. 2002, 514, 122–128. [Google Scholar] [CrossRef] [PubMed]
- Salganik, M.; Sergeyev, V.G.; Shinde, V.; Meyers, C.A.; Gorbatyuk, M.S.; Lin, J.H.; Zolotukhin, S.; Gorbatyuk, O.S. The loss of glucose-regulated protein 78 (GRP78) during normal aging or from siRNA knockdown augments human alpha-synuclein (alpha-syn) toxicity to rat nigral neurons. Neurobiol. Aging 2015, 36, 2213–2223. [Google Scholar] [CrossRef]
- Khanna, D.; Khanna, S.; Khanna, P.; Kahar, P.; Patel, B.M. Obesity: A Chronic Low-Grade Inflammation and Its Markers. Cureus 2022, 14, e22711. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Autieri, M.V.; Scalia, R. Adipose tissue inflammation and metabolic dysfunction in obesity. Am. J. Physiol. Cell Physiol. 2021, 320, C375–C391. [Google Scholar] [CrossRef]
- Alwarawrah, Y.; Kiernan, K.; MacIver, N.J. Changes in Nutritional Status Impact Immune Cell Metabolism and Function. Front. Immunol. 2018, 9, 1055. [Google Scholar] [CrossRef] [PubMed]
- Li, X. Endoplasmic reticulum stress regulates inflammation in adipocyte of obese rats via toll-like receptors 4 signaling. Iran J. Basic Med. Sci. 2018, 21, 502–507. [Google Scholar] [PubMed]
- Tripathi, Y.B.; Pandey, V. Obesity and endoplasmic reticulum (ER) stresses. Front. Immunol. 2012, 3, 240. [Google Scholar] [CrossRef]
- Adams, C.J.; Kopp, M.C.; Larburu, N.; Nowak, P.R.; Ali, M.M.U. Structure and Molecular Mechanism of ER Stress Signaling by the Unfolded Protein Response Signal Activator IRE1. Front. Mol. Biosci. 2019, 6, 11. [Google Scholar] [CrossRef]
- Zhao, P.; Huang, P.; Xu, T.; Xiang, X.; Sun, Y.; Liu, J.; Yan, C.; Wang, L.; Gao, J.; Cui, S.; et al. Fat body Ire1 regulates lipid homeostasis through the Xbp1s-FoxO axis in Drosophila. iScience 2021, 24, 102819. [Google Scholar] [CrossRef] [PubMed]
- Sha, H.; He, Y.; Yang, L.; Qi, L. Stressed out about obesity: IRE1alpha-XBP1 in metabolic disorders. Trends Endocrinol. Metab. 2011, 22, 374–381. [Google Scholar] [CrossRef] [PubMed]
- Madhavan, A.; Kok, B.P.; Rius, B.; Grandjean, J.M.D.; Alabi, A.; Albert, V.; Sukiasyan, A.; Powers, E.T.; Galmozzi, A.; Saez, E.; et al. Pharmacologic IRE1/XBP1s activation promotes systemic adaptive remodeling in obesity. Nat. Commun. 2022, 13, 608. [Google Scholar] [CrossRef] [PubMed]
- Chalmers, F.; van Lith, M.; Sweeney, B.; Cain, K.; Bulleid, N.J. Inhibition of IRE1alpha-mediated XBP1 mRNA cleavage by XBP1 reveals a novel regulatory process during the unfolded protein response. Wellcome Open Res. 2017, 2, 36. [Google Scholar] [CrossRef]
- Liu, S.; Ding, H.; Li, Y.; Zhang, X. Molecular Mechanism Underlying Role of the XBP1s in Cardiovascular Diseases. J. Cardiovasc. Dev. Dis. 2022, 9, 459. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.H.; Scapa, E.F.; Cohen, D.E.; Glimcher, L.H. Regulation of hepatic lipogenesis by the transcription factor XBP1. Science 2008, 320, 1492–1496. [Google Scholar] [CrossRef]
- Peng, J.; Qin, C.; Ramatchandirin, B.; Pearah, A.; Guo, S.; Hussain, M.; Yu, L.; Wondisford, F.E.; He, L. Activation of the canonical ER stress IRE1-XBP1 pathway by insulin regulates glucose and lipid metabolism. J. Biol. Chem. 2022, 298, 102283. [Google Scholar] [CrossRef]
- Wang, Q.; Zhou, H.; Bu, Q.; Wei, S.; Li, L.; Zhou, J.; Zhou, S.; Su, W.; Liu, M.; Liu, Z.; et al. Role of XBP1 in regulating the progression of non-alcoholic steatohepatitis. J. Hepatol. 2022, 77, 312–325. [Google Scholar] [CrossRef] [PubMed]
- Markussen, L.K.; Rondini, E.A.; Johansen, O.S.; Madsen, J.G.S.; Sustarsic, E.G.; Marcher, A.B.; Hansen, J.B.; Gerhart-Hines, Z.; Granneman, J.G.; Mandrup, S. Lipolysis regulates major transcriptional programs in brown adipocytes. Nat. Commun. 2022, 13, 3956. [Google Scholar] [CrossRef]
- Wang, S.; Chen, Z.; Lam, V.; Han, J.; Hassler, J.; Finck, B.N.; Davidson, N.O.; Kaufman, R.J. IRE1alpha-XBP1s induces PDI expression to increase MTP activity for hepatic VLDL assembly and lipid homeostasis. Cell Metab. 2012, 16, 473–486. [Google Scholar] [CrossRef]
- Morishita, Y.; Kellogg, A.P.; Larkin, D.; Chen, W.; Vadrevu, S.; Satin, L.; Liu, M.; Arvan, P. Cell death-associated lipid droplet protein CIDE-A is a noncanonical marker of endoplasmic reticulum stress. JCI Insight 2021, 6, e143980. [Google Scholar] [CrossRef]
- Smedley, G.D.; Walker, K.E.; Yuan, S.H. The Role of PERK in Understanding Development of Neurodegenerative Diseases. Int. J. Mol. Sci. 2021, 22, 8146. [Google Scholar] [CrossRef]
- Oakes, S.A. Endoplasmic Reticulum Stress Signaling in Cancer Cells. Am. J. Pathol. 2020, 190, 934–946. [Google Scholar] [CrossRef]
- Hetz, C.; Zhang, K.; Kaufman, R.J. Mechanisms, regulation and functions of the unfolded protein response. Nat. Rev. Mol. Cell Biol. 2020, 21, 421–438. [Google Scholar] [CrossRef] [PubMed]
- Cui, W.; Li, J.; Ron, D.; Sha, B. The structure of the PERK kinase domain suggests the mechanism for its activation. Acta Crystallogr. D Biol. Crystallogr. 2011, 67 Pt 5, 423–428. [Google Scholar] [CrossRef]
- Pitale, P.M.; Gorbatyuk, O.; Gorbatyuk, M. Neurodegeneration: Keeping ATF4 on a Tight Leash. Front. Cell Neurosci. 2017, 11, 410. [Google Scholar] [CrossRef] [PubMed]
- Prentki, M.; Nolan, C.J. Islet beta cell failure in type 2 diabetes. J. Clin. Invest. 2006, 116, 1802–1812. [Google Scholar] [CrossRef] [PubMed]
- Boland, B.B.; Rhodes, C.J.; Grimsby, J.S. The dynamic plasticity of insulin production in beta-cells. Mol. Metab. 2017, 6, 958–973. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; McGrath, B.; Cavener, D.R. PERK (EIF2AK3) regulates proinsulin trafficking and quality control in the secretory pathway. Diabetes 2010, 59, 1937–1947. [Google Scholar] [CrossRef] [PubMed]
- Scheuner, D.; Kaufman, R.J. The unfolded protein response: A pathway that links insulin demand with beta-cell failure and diabetes. Endocr. Rev. 2008, 29, 317–333. [Google Scholar] [CrossRef]
- Liu, M.; Huang, Y.; Xu, X.; Li, X.; Alam, M.; Arunagiri, A.; Haataja, L.; Ding, L.; Wang, S.; Itkin-Ansari, P.; et al. Normal and defective pathways in biogenesis and maintenance of the insulin storage pool. J. Clin. Invest. 2021, 131, e142240. [Google Scholar] [CrossRef] [PubMed]
- Rohli, K.E.; Boyer, C.K.; Blom, S.E.; Stephens, S.B. Nutrient Regulation of Pancreatic Islet beta-Cell Secretory Capacity and Insulin Production. Biomolecules 2022, 12, 335. [Google Scholar] [CrossRef] [PubMed]
- Rabhi, N.; Salas, E.; Froguel, P.; Annicotte, J.S. Role of the unfolded protein response in beta cell compensation and failure during diabetes. J. Diabetes Res. 2014, 2014, 795171. [Google Scholar] [CrossRef] [PubMed]
- Kitakaze, K.; Oyadomari, M.; Zhang, J.; Hamada, Y.; Takenouchi, Y.; Tsuboi, K.; Inagaki, M.; Tachikawa, M.; Fujitani, Y.; Okamoto, Y.; et al. ATF4-mediated transcriptional regulation protects against beta-cell loss during endoplasmic reticulum stress in a mouse model. Mol. Metab. 2021, 54, 101338. [Google Scholar] [CrossRef] [PubMed]
- Berry, C.; Lal, M.; Binukumar, B.K. Crosstalk Between the Unfolded Protein Response, MicroRNAs, and Insulin Signaling Pathways: In Search of Biomarkers for the Diagnosis and Treatment of Type 2 Diabetes. Front. Endocrinol. 2018, 9, 210. [Google Scholar] [CrossRef]
- de la Cadena, S.G.; Hernandez-Fonseca, K.; Camacho-Arroyo, I.; Massieu, L. Glucose deprivation induces reticulum stress by the PERK pathway and caspase-7- and calpain-mediated caspase-12 activation. Apoptosis 2014, 19, 414–427. [Google Scholar] [CrossRef]
- Lee, J.H.; Lee, J. Endoplasmic Reticulum (ER) Stress and Its Role in Pancreatic beta-Cell Dysfunction and Senescence in Type 2 Diabetes. Int. J. Mol. Sci. 2022, 23, 4843. [Google Scholar] [CrossRef]
- Sharma, R.B.; Landa-Galvan, H.V.; Alonso, L.C. Living Dangerously: Protective and Harmful ER Stress Responses in Pancreatic beta-Cells. Diabetes 2021, 70, 2431–2443. [Google Scholar] [CrossRef]
- Kong, F.J.; Wu, J.H.; Sun, S.Y.; Zhou, J.Q. The endoplasmic reticulum stress/autophagy pathway is involved in cholesterol-induced pancreatic beta-cell injury. Sci. Rep. 2017, 7, 44746. [Google Scholar] [CrossRef] [PubMed]
- Tran, K.; Li, Y.; Duan, H.; Arora, D.; Lim, H.Y.; Wang, W. Identification of small molecules that protect pancreatic beta cells against endoplasmic reticulum stress-induced cell death. ACS Chem. Biol. 2014, 9, 2796–2806. [Google Scholar] [CrossRef]
- Turishcheva, E.; Vildanova, M.; Onishchenko, G.; Smirnova, E. The Role of Endoplasmic Reticulum Stress in Differentiation of Cells of Mesenchymal Origin. Biochemistry 2022, 87, 916–931. [Google Scholar] [CrossRef] [PubMed]
- Bobrovnikova-Marjon, E.; Pytel, D.; Riese, M.J.; Vaites, L.P.; Singh, N.; Koretzky, G.A.; Witze, E.S.; Diehl, J.A. PERK utilizes intrinsic lipid kinase activity to generate phosphatidic acid, mediate Akt activation, and promote adipocyte differentiation. Mol. Cell Biol. 2012, 32, 2268–2278. [Google Scholar] [CrossRef] [PubMed]
- Cohen, D.M.; Won, K.J.; Nguyen, N.; Lazar, M.A.; Chen, C.S.; Steger, D.J. ATF4 licenses C/EBPbeta activity in human mesenchymal stem cells primed for adipogenesis. Elife 2015, 4, e06821. [Google Scholar] [CrossRef]
- Yuzefovych, L.V.; Musiyenko, S.I.; Wilson, G.L.; Rachek, L.I. Mitochondrial DNA damage and dysfunction, and oxidative stress are associated with endoplasmic reticulum stress, protein degradation and apoptosis in high fat diet-induced insulin resistance mice. PLoS ONE 2013, 8, e54059. [Google Scholar] [CrossRef] [PubMed]
- Kapetanaki, S.; Kumawat, A.K.; Persson, K.; Demirel, I. The Fibrotic Effects of TMAO on Human Renal Fibroblasts Is Mediated by NLRP3, Caspase-1 and the PERK/Akt/mTOR Pathway. Int. J. Mol. Sci. 2021, 22, 11864. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Hai, C. Redox modulation of adipocyte differentiation: Hypothesis of “Redox Chain” and novel insights into intervention of adipogenesis and obesity. Free Radic. Biol. Med. 2015, 89, 99–125. [Google Scholar] [CrossRef]
- Lowe, C.E.; Dennis, R.J.; Obi, U.; O’Rahilly, S.; Rochford, J.J. Investigating the involvement of the ATF6alpha pathway of the unfolded protein response in adipogenesis. Int. J. Obes. 2012, 36, 1248–1251. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Kaufman, R.J. Signaling the unfolded protein response from the endoplasmic reticulum. J. Biol. Chem. 2004, 279, 25935–25938. [Google Scholar] [CrossRef]
- van Laar, T.; van der Eb, A.J.; Terleth, C. Mif1: A missing link between the unfolded protein response pathway and ER-associated protein degradation? Curr. Protein Pept. Sci. 2001, 2, 169–190. [Google Scholar] [CrossRef]
- Yoshida, H.; Matsui, T.; Hosokawa, N.; Kaufman, R.J.; Nagata, K.; Mori, K. A time-dependent phase shift in the mammalian unfolded protein response. Dev. Cell 2003, 4, 265–271. [Google Scholar] [CrossRef]
- Qu, J.; Zou, T.; Lin, Z. The Roles of the Ubiquitin-Proteasome System in the Endoplasmic Reticulum Stress Pathway. Int. J. Mol. Sci. 2021, 22, 1526. [Google Scholar] [CrossRef]
- Chen, X.; Shen, J.; Prywes, R. The luminal domain of ATF6 senses endoplasmic reticulum (ER) stress and causes translocation of ATF6 from the ER to the Golgi. J. Biol. Chem. 2002, 277, 13045–13052. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Prywes, R. Dependence of site-2 protease cleavage of ATF6 on prior site-1 protease digestion is determined by the size of the luminal domain of ATF6. J. Biol. Chem. 2004, 279, 43046–43051. [Google Scholar] [CrossRef]
- Thuerauf, D.J.; Morrison, L.E.; Hoover, H.; Glembotski, C.C. Coordination of ATF6-mediated transcription and ATF6 degradation by a domain that is shared with the viral transcription factor, VP16. J. Biol. Chem. 2002, 277, 20734–20739. [Google Scholar] [CrossRef] [PubMed]
- Stengel, S.T.; Fazio, A.; Lipinski, S.; Jahn, M.T.; Aden, K.; Ito, G.; Wottawa, F.; Kuiper, J.W.P.; Coleman, O.I.; Tran, F.; et al. Activating Transcription Factor 6 Mediates Inflammatory Signals in Intestinal Epithelial Cells Upon Endoplasmic Reticulum Stress. Gastroenterology 2020, 159, 1357–1374.e10. [Google Scholar] [CrossRef]
- Chen, Z.; Liu, Y.; Yang, L.; Liu, P.; Zhang, Y.; Wang, X. MiR-149 attenuates endoplasmic reticulum stress-induced inflammation and apoptosis in nonalcoholic fatty liver disease by negatively targeting ATF6 pathway. Immunol. Lett. 2020, 222, 40–48. [Google Scholar] [CrossRef]
- Nakajima, S.; Hiramatsu, N.; Hayakawa, K.; Saito, Y.; Kato, H.; Huang, T.; Yao, J.; Paton, A.W.; Paton, J.C.; Kitamura, M. Selective abrogation of BiP/GRP78 blunts activation of NF-kappaB through the ATF6 branch of the UPR: Involvement of C/EBPbeta and mTOR-dependent dephosphorylation of Akt. Mol. Cell Biol. 2011, 31, 1710–1718. [Google Scholar] [CrossRef] [PubMed]
- Rao, J.; Yue, S.; Fu, Y.; Zhu, J.; Wang, X.; Busuttil, R.W.; Kupiec-Weglinski, J.W.; Lu, L.; Zhai, Y. ATF6 mediates a pro-inflammatory synergy between ER stress and TLR activation in the pathogenesis of liver ischemia-reperfusion injury. Am. J. Transplant. 2014, 14, 1552–1561. [Google Scholar] [CrossRef]
- Senkal, C.E.; Ponnusamy, S.; Bielawski, J.; Hannun, Y.A.; Ogretmen, B. Antiapoptotic roles of ceramide-synthase-6-generated C16-ceramide via selective regulation of the ATF6/CHOP arm of ER-stress-response pathways. FASEB J. 2010, 24, 296–308. [Google Scholar] [CrossRef] [PubMed]
- Bommiasamy, H.; Back, S.H.; Fagone, P.; Lee, K.; Meshinchi, S.; Vink, E.; Sriburi, R.; Frank, M.; Jackowski, S.; Kaufman, R.J.; et al. ATF6alpha induces XBP1-independent expansion of the endoplasmic reticulum. J. Cell Sci. 2009, 122 Pt 10, 1626–1636. [Google Scholar] [CrossRef]
- Ghemrawi, R.; Battaglia-Hsu, S.F.; Arnold, C. Endoplasmic Reticulum Stress in Metabolic Disorders. Cells 2018, 7, 63. [Google Scholar] [CrossRef] [PubMed]
- White, U.A.; Stephens, J.M. Transcriptional factors that promote formation of white adipose tissue. Mol. Cell Endocrinol. 2010, 318, 10–14. [Google Scholar] [CrossRef] [PubMed]
- Oyadomari, S.; Harding, H.P.; Zhang, Y.; Oyadomari, M.; Ron, D. Dephosphorylation of translation initiation factor 2alpha enhances glucose tolerance and attenuates hepatosteatosis in mice. Cell Metab. 2008, 7, 520–532. [Google Scholar] [CrossRef] [PubMed]
- Park, S.W.; Ozcan, U. Potential for therapeutic manipulation of the UPR in disease. Semin. Immunopathol. 2013, 35, 351–373. [Google Scholar] [CrossRef] [PubMed]
- Tang, Q.Q.; Lane, M.D. Adipogenesis: From Stem Cell to Adipocyte. Annu. Rev. Biochem. 2012, 81, 715–736. [Google Scholar] [CrossRef] [PubMed]
- Tang, Q.Q.; Otto, T.C.; Lane, M.D. CCAAT/enhancer-binding protein beta is required for mitotic clonal expansion during adipogenesis. Proc. Natl. Acad. Sci. USA 2003, 100, 850–855. [Google Scholar] [CrossRef] [PubMed]
- Rosen, E.D.; MacDougald, O.A. Adipocyte differentiation from the inside out. Nat. Rev. Mol. Cell Biol. 2006, 7, 885–896. [Google Scholar] [CrossRef]
- El-Jack, A.K.; Hamm, J.K.; Pilch, P.F.; Farmer, S.R. Reconstitution of insulin-sensitive glucose transport in fibroblasts requires expression of both PPARgamma and C/EBPalpha. J. Biol. Chem. 1999, 274, 7946–7951. [Google Scholar] [CrossRef]
- de Winter, T.J.J.; Nusse, R. Running Against the Wnt: How Wnt/beta-Catenin Suppresses Adipogenesis. Front. Cell Dev. Biol. 2021, 9, 627429. [Google Scholar] [CrossRef]
- Funcke, J.B.; Scherer, P.E. Beyond adiponectin and leptin: Adipose tissue-derived mediators of inter-organ communication. J. Lipid Res. 2019, 60, 1648–1684. [Google Scholar] [CrossRef]
- Straub, L.G.; Scherer, P.E. Metabolic Messengers: Adiponectin. Nat. Metab. 2019, 1, 334–339. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.A.; Zhang, F.; Jiang, L.; Ye, R.; An, Y.; Shao, M.; Tao, C.; Gupta, R.K.; Scherer, P.E. Peroxisome Proliferator-Activated Receptor gamma and Its Role in Adipocyte Homeostasis and Thiazolidinedione-Mediated Insulin Sensitization. Mol. Cell Biol. 2018, 38, e00677-17. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Zhang, F.; Zhang, X.; Xue, C.; Namwanje, M.; Fan, L.; Reilly, M.P.; Hu, F.; Qiang, L. Distinct functions of PPARgamma isoforms in regulating adipocyte plasticity. Biochem. Biophys. Res. Commun. 2016, 481, 132–138. [Google Scholar] [CrossRef] [PubMed]
- Mu, F.; Jing, Y.; Ning, B.; Huang, J.; Cui, T.; Guo, Y.; You, X.; Yan, X.; Li, H.; Wang, N. Peroxisome proliferator-activated receptor gamma isoforms differentially regulate preadipocyte proliferation, apoptosis, and differentiation in chickens. Poult. Sci. 2020, 99, 6410–6421. [Google Scholar] [CrossRef] [PubMed]
- Wan, R.; Ding, Z.; Xia, S.; Zheng, L.; Lu, J. Effects Of PPARgamma2 Pro12Ala Variant On Adipocyte Phenotype Dependent Of DHA. Diabetes Metab. Syndr. Obes. 2019, 12, 2273–2279. [Google Scholar] [CrossRef]
- Wu, Z.; Rosen, E.D.; Brun, R.; Hauser, S.; Adelmant, G.; Troy, A.E.; McKeon, C.; Darlington, G.J.; Spiegelman, B.M. Cross-regulation of C/EBP alpha and PPAR gamma controls the transcriptional pathway of adipogenesis and insulin sensitivity. Mol. Cell 1999, 3, 151–158. [Google Scholar] [CrossRef]
- Jakab, J.; Miskic, B.; Miksic, S.; Juranic, B.; Cosic, V.; Schwarz, D.; Vcev, A. Adipogenesis as a Potential Anti-Obesity Target: A Review of Pharmacological Treatment and Natural Products. Diabetes Metab. Syndr. Obes. 2021, 14, 67–83. [Google Scholar] [CrossRef]
- Hallenborg, P.; Petersen, R.K.; Feddersen, S.; Sundekilde, U.; Hansen, J.B.; Blagoev, B.; Madsen, L.; Kristiansen, K. PPARgamma ligand production is tightly linked to clonal expansion during initiation of adipocyte differentiation. J. Lipid Res. 2014, 55, 2491–2500. [Google Scholar] [CrossRef]
- Park, S.H.; Choi, H.J.; Yang, H.; Do, K.H.; Kim, J.; Lee, D.W.; Moon, Y. Endoplasmic reticulum stress-activated C/EBP homologous protein enhances nuclear factor-kappaB signals via repression of peroxisome proliferator-activated receptor gamma. J. Biol. Chem. 2010, 285, 35330–35339. [Google Scholar] [CrossRef]
- Cucinotta, M.; Visalli, M.; Aguennouz, M.; Valenti, A.; Loddo, S.; Altucci, L.; Teti, D. Regulation of interleukin-8 gene at a distinct site of its promoter by CCAAT enhancer-binding protein homologous protein in prostaglandin E2-treated human T cells. J. Biol. Chem. 2008, 283, 29760–29769. [Google Scholar] [CrossRef] [PubMed]
- Hayakawa, K.; Nakajima, S.; Hiramatsu, N.; Okamura, M.; Huang, T.; Saito, Y.; Tagawa, Y.; Tamai, M.; Takahashi, S.; Yao, J.; et al. ER stress depresses NF-kappaB activation in mesangial cells through preferential induction of C/EBP beta. J. Am. Soc. Nephrol. 2010, 21, 73–81. [Google Scholar] [CrossRef] [PubMed]
- Weidemann, A.; Lovas, A.; Rauch, A.; Andreas, N.; von Maltzahn, J.; Riemann, M.; Weih, F. Classical and alternative NF-kappaB signaling cooperate in regulating adipocyte differentiation and function. Int. J. Obes. 2016, 40, 452–459. [Google Scholar] [CrossRef] [PubMed]
- Griffin, M.J. On the Immunometabolic Role of NF-kappaB in Adipocytes. Immunometabolism 2022, 4, e220003. [Google Scholar] [CrossRef] [PubMed]
- Siersbaek, R.; Nielsen, R.; Mandrup, S. PPARgamma in adipocyte differentiation and metabolism—novel insights from genome-wide studies. FEBS Lett. 2010, 584, 3242–3249. [Google Scholar] [CrossRef] [PubMed]
- Accattatis, F.M.; Caruso, A.; Carleo, A.; Del Console, P.; Gelsomino, L.; Bonofiglio, D.; Giordano, C.; Barone, I.; Ando, S.; Bianchi, L.; et al. CEBP-beta and PLK1 as Potential Mediators of the Breast Cancer/Obesity Crosstalk: In Vitro and In Silico Analyses. Nutrients 2023, 15, 2839. [Google Scholar] [CrossRef] [PubMed]
- DeZwaan-McCabe, D.; Riordan, J.D.; Arensdorf, A.M.; Icardi, M.S.; Dupuy, A.J.; Rutkowski, D.T. The stress-regulated transcription factor CHOP promotes hepatic inflammatory gene expression, fibrosis, and oncogenesis. PLoS Genet. 2013, 9, e1003937. [Google Scholar] [CrossRef] [PubMed]
- Fujita, E.; Dai, H.; Tanabe, Y.; Zhiling, Y.; Yamagata, T.; Miyakawa, T.; Tanokura, M.; Momoi, M.Y.; Momoi, T. Autism spectrum disorder is related to endoplasmic reticulum stress induced by mutations in the synaptic cell adhesion molecule, CADM1. Cell Death Dis. 2010, 1, e47. [Google Scholar] [CrossRef]
- Li, Y.; Jiang, W.; Niu, Q.; Sun, Y.; Meng, C.; Tan, L.; Song, C.; Qiu, X.; Liao, Y.; Ding, C. eIF2alpha-CHOP-BCl-2/JNK and IRE1alpha-XBP1/JNK signaling promote apoptosis and inflammation and support the proliferation of Newcastle disease virus. Cell Death Dis. 2019, 10, 891. [Google Scholar] [CrossRef]
- Yang, Y.; Liu, L.; Naik, I.; Braunstein, Z.; Zhong, J.; Ren, B. Transcription Factor C/EBP Homologous Protein in Health and Diseases. Front. Immunol. 2017, 8, 1612. [Google Scholar] [CrossRef]
- Allagnat, F.; Fukaya, M.; Nogueira, T.C.; Delaroche, D.; Welsh, N.; Marselli, L.; Marchetti, P.; Haefliger, J.A.; Eizirik, D.L.; Cardozo, A.K. C/EBP homologous protein contributes to cytokine-induced pro-inflammatory responses and apoptosis in beta-cells. Cell Death Differ. 2012, 19, 1836–1846. [Google Scholar] [CrossRef]
- Wang, P.; Qian, H.; Xiao, M.; Lv, J. Role of signal transduction pathways in IL-1beta-induced apoptosis: Pathological and therapeutic aspects. Immun. Inflamm. Dis. 2023, 11, e762. [Google Scholar] [CrossRef] [PubMed]
- Frith, J.; Genever, P. Transcriptional control of mesenchymal stem cell differentiation. Transfus. Med. Hemotherapy 2008, 35, 216–227. [Google Scholar] [CrossRef]
- Brenner, S.; Bercovich, Z.; Feiler, Y.; Keshet, R.; Kahana, C. Dual Regulatory Role of Polyamines in Adipogenesis. J. Biol. Chem. 2015, 290, 27384–27392. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Lee, P.; Chisholm, D.J.; James, D.E. Control of adipocyte differentiation in different fat depots; implications for pathophysiology or therapy. Front. Endocrinol. 2015, 6, 1. [Google Scholar] [CrossRef] [PubMed]
- Hosogai, N.; Fukuhara, A.; Oshima, K.; Miyata, Y.; Tanaka, S.; Segawa, K.; Furukawa, S.; Tochino, Y.; Komuro, R.; Matsuda, M.; et al. Adipose tissue hypoxia in obesity and its impact on adipocytokine dysregulation. Diabetes 2007, 56, 901–911. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Murthy, R.; Wood, B.; Song, B.; Wang, S.; Sun, B.; Malhi, H.; Kaufman, R.J. ER stress signalling through eIF2alpha and CHOP, but not IRE1alpha, attenuates adipogenesis in mice. Diabetologia 2013, 56, 911–924. [Google Scholar] [CrossRef] [PubMed]
- Cawthorn, W.P.; Scheller, E.L.; MacDougald, O.A. Adipose tissue stem cells meet preadipocyte commitment: Going back to the future. J. Lipid Res. 2012, 53, 227–246. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Niu, X.; Wang, M.; Li, Z.; Jiang, H.K.; Li, C.; Caton, S.J.; Bai, Y. Endoplasmic reticulum stress may be involved in insulin resistance and lipid metabolism disorders of the white adipose tissues induced by high-fat diet containing industrial trans-fatty acids. Diabetes Metab. Syndr. Obes. 2019, 12, 1625–1638. [Google Scholar] [CrossRef]
- Lemmer, I.L.; Willemsen, N.; Hilal, N.; Bartelt, A. A guide to understanding endoplasmic reticulum stress in metabolic disorders. Mol. Metab. 2021, 47, 101169. [Google Scholar] [CrossRef] [PubMed]
- Almanza, A.; Carlesso, A.; Chintha, C.; Creedican, S.; Doultsinos, D.; Leuzzi, B.; Luis, A.; McCarthy, N.; Montibeller, L.; More, S.; et al. Endoplasmic reticulum stress signalling—From basic mechanisms to clinical applications. FEBS J. 2019, 286, 241–278. [Google Scholar] [CrossRef]
- Mansour, S.Z.; Moustafa, E.M.; Moawed, F.S.M. Modulation of endoplasmic reticulum stress via sulforaphane-mediated AMPK upregulation against nonalcoholic fatty liver disease in rats. Cell Stress Chaperones 2022, 27, 499–511. [Google Scholar] [CrossRef] [PubMed]
- Guan, B.J.; Krokowski, D.; Majumder, M.; Schmotzer, C.L.; Kimball, S.R.; Merrick, W.C.; Koromilas, A.E.; Hatzoglou, M. Translational control during endoplasmic reticulum stress beyond phosphorylation of the translation initiation factor eIF2alpha. J. Biol. Chem. 2014, 289, 12593–12611. [Google Scholar] [CrossRef]
- Cnop, M.; Toivonen, S.; Igoillo-Esteve, M.; Salpea, P. Endoplasmic reticulum stress and eIF2alpha phosphorylation: The Achilles heel of pancreatic beta cells. Mol. Metab. 2017, 6, 1024–1039. [Google Scholar] [CrossRef]
- Leitman, J.; Barak, B.; Benyair, R.; Shenkman, M.; Ashery, U.; Hartl, F.U.; Lederkremer, G.Z. ER stress-induced eIF2-alpha phosphorylation underlies sensitivity of striatal neurons to pathogenic huntingtin. PLoS ONE 2014, 9, e90803. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Lim, Y.; Lee, D.; Elvira, R.; Lee, J.M.; Lee, M.R.; Han, J. Modulation of Protein Synthesis by eIF2alpha Phosphorylation Protects Cell from Heat Stress-Mediated Apoptosis. Cells 2018, 7, 254. [Google Scholar] [CrossRef] [PubMed]
- Kasinath, B.S.; Mariappan, M.M.; Sataranatarajan, K.; Lee, M.J.; Ghosh Choudhury, G.; Feliers, D. Novel mechanisms of protein synthesis in diabetic nephropathy—role of mRNA translation. Rev. Endocr. Metab. Disord. 2008, 9, 255–266. [Google Scholar] [CrossRef] [PubMed]
- Lang, C.H.; Frost, R.A.; Nairn, A.C.; MacLean, D.A.; Vary, T.C. TNF-alpha impairs heart and skeletal muscle protein synthesis by altering translation initiation. Am. J. Physiol. Endocrinol. Metab. 2002, 282, E336–E347. [Google Scholar] [CrossRef]
- Bagchi, D.P.; Nishii, A.; Li, Z.; DelProposto, J.B.; Corsa, C.A.; Mori, H.; Hardij, J.; Learman, B.S.; Lumeng, C.N.; MacDougald, O.A. Wnt/beta-catenin signaling regulates adipose tissue lipogenesis and adipocyte-specific loss is rigorously defended by neighboring stromal-vascular cells. Mol. Metab. 2020, 42, 101078. [Google Scholar] [CrossRef]
- Prestwich, T.C.; Macdougald, O.A. Wnt/beta-catenin signaling in adipogenesis and metabolism. Curr. Opin. Cell Biol. 2007, 19, 612–617. [Google Scholar] [CrossRef] [PubMed]
- Palsgaard, J.; Emanuelli, B.; Winnay, J.N.; Sumara, G.; Karsenty, G.; Kahn, C.R. Cross-talk between insulin and Wnt signaling in preadipocytes: Role of Wnt co-receptor low density lipoprotein receptor-related protein-5 (LRP5). J. Biol. Chem. 2012, 287, 12016–12026. [Google Scholar] [CrossRef]
- Bennett, C.N.; Ross, S.E.; Longo, K.A.; Bajnok, L.; Hemati, N.; Johnson, K.W.; Harrison, S.D.; MacDougald, O.A. Regulation of Wnt signaling during adipogenesis. J. Biol. Chem. 2002, 277, 30998–31004. [Google Scholar] [CrossRef] [PubMed]
- Corda, G.; Sala, A. Non-canonical WNT/PCP signalling in cancer: Fzd6 takes centre stage. Oncogenesis 2017, 6, e364. [Google Scholar] [CrossRef]
- Li, F.; Chong, Z.Z.; Maiese, K. Vital elements of the Wnt-Frizzled signaling pathway in the nervous system. Curr. Neurovasc. Res. 2005, 2, 331–340. [Google Scholar] [CrossRef]
- MacDonald, B.T.; Tamai, K.; He, X. Wnt/beta-catenin signaling: Components, mechanisms, and diseases. Dev. Cell 2009, 17, 9–26. [Google Scholar] [CrossRef] [PubMed]
- Tran, B.M.; Flanagan, D.J.; Phesse, T.J.; Vincan, E. Frizzled(7) Activates beta-Catenin-Dependent and beta-Catenin-Independent Wnt Signalling Pathways During Developmental Morphogenesis: Implications for Therapeutic Targeting in Colorectal Cancer. Handb. Exp. Pharmacol. 2021, 269, 251–277. [Google Scholar] [PubMed]
- Longo, K.A.; Wright, W.S.; Kang, S.; Gerin, I.; Chiang, S.H.; Lucas, P.C.; Opp, M.R.; MacDougald, O.A. Wnt10b inhibits development of white and brown adipose tissues. J. Biol. Chem. 2004, 279, 35503–35509. [Google Scholar] [CrossRef] [PubMed]
- Calejman, C.M.; Doxsey, W.G.; Fazakerley, D.J.; Guertin, D.A. Integrating adipocyte insulin signaling and metabolism in the multi-omics era. Trends Biochem. Sci. 2022, 47, 531–546. [Google Scholar] [CrossRef] [PubMed]
- Caron, A.; Richard, D.; Laplante, M. The Roles of mTOR Complexes in Lipid Metabolism. Annu. Rev. Nutr. 2015, 35, 321–348. [Google Scholar] [CrossRef]
- Kato, H.; Nakajima, S.; Saito, Y.; Takahashi, S.; Katoh, R.; Kitamura, M. mTORC1 serves ER stress-triggered apoptosis via selective activation of the IRE1-JNK pathway. Cell Death Differ. 2012, 19, 310–320. [Google Scholar] [CrossRef]
- Heberle, A.M.; Prentzell, M.T.; van Eunen, K.; Bakker, B.M.; Grellscheid, S.N.; Thedieck, K. Molecular mechanisms of mTOR regulation by stress. Mol. Cell Oncol. 2015, 2, e970489. [Google Scholar] [CrossRef]
- Boss, M.; Newbatt, Y.; Gupta, S.; Collins, I.; Brune, B.; Namgaladze, D. AMPK-independent inhibition of human macrophage ER stress response by AICAR. Sci. Rep. 2016, 6, 32111. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Moon, S.Y.; Kim, J.S.; Baek, C.H.; Kim, M.; Min, J.Y.; Lee, S.K. Activation of AMP-activated protein kinase inhibits ER stress and renal fibrosis. Am. J. Physiol. Renal. Physiol. 2015, 308, F226–F236. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Min, Q.; Ouyang, C.; Lee, J.; He, C.; Zou, M.H.; Xie, Z. AMPK activation prevents excess nutrient-induced hepatic lipid accumulation by inhibiting mTORC1 signaling and endoplasmic reticulum stress response. Biochim. Biophys. Acta 2014, 1842, 1844–1854. [Google Scholar] [CrossRef] [PubMed]
- Steinberg, G.R.; Hardie, D.G. New insights into activation and function of the AMPK. Nat. Rev. Mol. Cell Biol. 2023, 24, 255–272. [Google Scholar] [CrossRef]
- Gwinn, D.M.; Shackelford, D.B.; Egan, D.F.; Mihaylova, M.M.; Mery, A.; Vasquez, D.S.; Turk, B.E.; Shaw, R.J. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol. Cell 2008, 30, 214–226. [Google Scholar] [CrossRef]
- Hindupur, S.K.; Gonzalez, A.; Hall, M.N. The opposing actions of target of rapamycin and AMP-activated protein kinase in cell growth control. Cold Spring Harb. Perspect. Biol. 2015, 7, a019141. [Google Scholar] [CrossRef]
- Peterson, T.R.; Sengupta, S.S.; Harris, T.E.; Carmack, A.E.; Kang, S.A.; Balderas, E.; Guertin, D.A.; Madden, K.L.; Carpenter, A.E.; Finck, B.N.; et al. mTOR complex 1 regulates lipin 1 localization to control the SREBP pathway. Cell 2011, 146, 408–420. [Google Scholar] [CrossRef]
- Qin, L.; Wang, Z.; Tao, L.; Wang, Y. ER stress negatively regulates AKT/TSC/mTOR pathway to enhance autophagy. Autophagy 2010, 6, 239–247. [Google Scholar] [CrossRef]
- Ohoka, N.; Yoshii, S.; Hattori, T.; Onozaki, K.; Hayashi, H. TRB3, a novel ER stress-inducible gene, is induced via ATF4-CHOP pathway and is involved in cell death. EMBO J. 2005, 24, 1243–1255. [Google Scholar] [CrossRef]
- Lee, P.L.; Jung, S.M.; Guertin, D.A. The Complex Roles of Mechanistic Target of Rapamycin in Adipocytes and Beyond. Trends Endocrinol. Metab. 2017, 28, 319–339. [Google Scholar] [CrossRef]
- Chen, C.H.; Shaikenov, T.; Peterson, T.R.; Aimbetov, R.; Bissenbaev, A.K.; Lee, S.W.; Wu, J.; Lin, H.K.; Sarbassov, D.D. ER stress inhibits mTORC2 and Akt signaling through GSK-3beta-mediated phosphorylation of rictor. Sci. Signal. 2011, 4, ra10. [Google Scholar] [CrossRef]
- Ozcan, U.; Yilmaz, E.; Ozcan, L.; Furuhashi, M.; Vaillancourt, E.; Smith, R.O.; Gorgun, C.Z.; Hotamisligil, G.S. Chemical chaperones reduce ER stress and restore glucose homeostasis in a mouse model of type 2 diabetes. Science 2006, 313, 1137–1140. [Google Scholar] [CrossRef]
- Ozcan, U.; Cao, Q.; Yilmaz, E.; Lee, A.H.; Iwakoshi, N.N.; Ozdelen, E.; Tuncman, G.; Gorgun, C.; Glimcher, L.H.; Hotamisligil, G.S. Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science 2004, 306, 457–461. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, B.J. Insulin resistance as the core defect in type 2 diabetes mellitus. Am. J. Cardiol. 2002, 90, 3G–10G. [Google Scholar] [CrossRef] [PubMed]
- Korbecki, J.; Bajdak-Rusinek, K. The effect of palmitic acid on inflammatory response in macrophages: An overview of molecular mechanisms. Inflamm. Res. 2019, 68, 915–932. [Google Scholar] [CrossRef]
- Shi, F.; Collins, S. Regulation of mTOR Signaling: Emerging Role of Cyclic Nucleotide-Dependent Protein Kinases and Implications for Cardiometabolic Disease. Int. J. Mol. Sci. 2023, 24, 11497. [Google Scholar] [CrossRef] [PubMed]
- Jayaraman, S.; Devarajan, N.; Rajagopal, P.; Babu, S.; Ganesan, S.K.; Veeraraghavan, V.P.; Palanisamy, C.P.; Cui, B.; Periyasamy, V.; Chandrasekar, K. β-Sitosterol Circumvents Obesity Induced Inflammation and Insulin Resistance by down-Regulating IKKbeta/NF-kappaB and JNK Signaling Pathway in Adipocytes of Type 2 Diabetic Rats. Molecules 2021, 26, 2101. [Google Scholar] [CrossRef] [PubMed]
- Shi, C.; Zhu, L.; Chen, X.; Gu, N.; Chen, L.; Zhu, L.; Yang, L.; Pang, L.; Guo, X.; Ji, C.; et al. IL-6 and TNF-alpha induced obesity-related inflammatory response through transcriptional regulation of miR-146b. J. Interferon Cytokine Res. 2014, 34, 342–348. [Google Scholar] [CrossRef]
- Roy, P.K.; Islam, J.; Lalhlenmawia, H. Prospects of potential adipokines as therapeutic agents in obesity-linked atherogenic dyslipidemia and insulin resistance. Egypt Heart J. 2023, 75, 24. [Google Scholar] [CrossRef]
- Jayawardena, T.U.; Kim, S.Y.; Jeon, Y.J. Sarcopenia; functional concerns, molecular mechanisms involved, and seafood as a nutritional intervention—Review article. Crit. Rev. Food Sci. Nutr. 2023, 63, 1983–2003. [Google Scholar] [CrossRef]
- Yung, J.H.M.; Giacca, A. Role of c-Jun N-terminal Kinase (JNK) in Obesity and Type 2 Diabetes. Cells 2020, 9, 706. [Google Scholar] [CrossRef] [PubMed]
- Handschumacher, R.E.; Harding, M.W.; Rice, J.; Drugge, R.J.; Speicher, D.W. Cyclophilin: A specific cytosolic binding protein for cyclosporin A. Science 1984, 226, 544–547. [Google Scholar] [CrossRef]
- Lee, J.M. Nuclear Receptors Resolve Endoplasmic Reticulum Stress to Improve Hepatic Insulin Resistance. Diabetes Metab. J. 2017, 41, 10–19. [Google Scholar] [CrossRef] [PubMed]
- Djedaini, M.; Peraldi, P.; Drici, M.D.; Darini, C.; Saint-Marc, P.; Dani, C.; Ladoux, A. Lopinavir co-induces insulin resistance and ER stress in human adipocytes. Biochem. Biophys. Res. Commun. 2009, 386, 96–100. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.; Dainty, S.; Strudwick, N.; Mihai, A.D.; Watson, J.N.; Dendooven, R.; Paton, A.W.; Paton, J.C.; Schroder, M. Endoplasmic reticulum stress causes insulin resistance by inhibiting delivery of newly synthesized insulin receptors to the cell surface. Mol. Biol. Cell 2020, 31, 2597–2629. [Google Scholar] [CrossRef] [PubMed]
- Eizirik, D.L.; Cardozo, A.K.; Cnop, M. The role for endoplasmic reticulum stress in diabetes mellitus. Endocr. Rev. 2008, 29, 42–61. [Google Scholar] [CrossRef]
- Tong, Y.; Xu, S.; Huang, L.; Chen, C. Obesity and insulin resistance: Pathophysiology and treatment. Drug Discov. Today 2022, 27, 822–830. [Google Scholar] [CrossRef]
- Liang, L.; Chen, J.; Zhan, L.; Lu, X.; Sun, X.; Sui, H.; Zheng, L.; Xiang, H.; Zhang, F. Endoplasmic reticulum stress impairs insulin receptor signaling in the brains of obese rats. PLoS ONE 2015, 10, e0126384. [Google Scholar] [CrossRef]
- Solinas, G.; Becattini, B. JNK at the crossroad of obesity, insulin resistance, and cell stress response. Mol. Metab. 2017, 6, 174–184. [Google Scholar] [CrossRef]
- Chen, Y.; Wu, Z.; Zhao, S.; Xiang, R. Chemical chaperones reduce ER stress and adipose tissue inflammation in high fat diet-induced mouse model of obesity. Sci. Rep. 2016, 6, 27486. [Google Scholar] [CrossRef]
- Wu, J.; Wu, D.; Zhang, L.; Lin, C.; Liao, J.; Xie, R.; Li, Z.; Wu, S.; Liu, A.; Hu, W.; et al. NK cells induce hepatic ER stress to promote insulin resistance in obesity through osteopontin production. J. Leukoc. Biol. 2020, 107, 589–596. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.; Wang, C.H. ER stress in adipocytes and insulin resistance: Mechanisms and significance (Review). Mol. Med. Rep. 2014, 10, 2234–2240. [Google Scholar] [CrossRef]
- Copps, K.D.; White, M.F. Regulation of insulin sensitivity by serine/threonine phosphorylation of insulin receptor substrate proteins IRS1 and IRS2. Diabetologia 2012, 55, 2565–2582. [Google Scholar] [CrossRef]
- Mathew, D.; Barillas-Cerritos, J.; Nedeljkovic-Kurepa, A.; Abraham, M.; Taylor, M.D.; Deutschman, C.S. Phosphorylation of insulin receptor substrates (IRS-1 and IRS-2) is attenuated following cecal ligation and puncture in mice. Mol. Med. 2023, 29, 106. [Google Scholar] [CrossRef]
- Sadeghi, A.; Niknam, M.; Momeni-Moghaddam, M.A.; Shabani, M.; Aria, H.; Bastin, A.; Teimouri, M.; Meshkani, R.; Akbari, H. Crosstalk between autophagy and insulin resistance: Evidence from different tissues. Eur. J. Med. Res. 2023, 28, 456. [Google Scholar] [CrossRef]
- Dixon, E.D.; Nardo, A.D.; Claudel, T.; Trauner, M. The Role of Lipid Sensing Nuclear Receptors (PPARs and LXR) and Metabolic Lipases in Obesity, Diabetes and NAFLD. Genes 2021, 12, 645. [Google Scholar] [CrossRef]
- Zhao, Y.; Ma, D.X.; Wang, H.G.; Li, M.Z.; Talukder, M.; Wang, H.R.; Li, J.L. Lycopene Prevents DEHP-Induced Liver Lipid Metabolism Disorder by Inhibiting the HIF-1alpha-Induced PPARalpha/PPARgamma/FXR/LXR System. J. Agric. Food Chem. 2020, 68, 11468–11479. [Google Scholar] [CrossRef]
- Lee, S.D.; Tontonoz, P. Liver X receptors at the intersection of lipid metabolism and atherogenesis. Atherosclerosis 2015, 242, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Zhao, Y.; You, Z.; Li, X.; Xiong, M.; Li, H.; Yan, N. Endoplasmic Reticulum Stress Affects Cholesterol Homeostasis by Inhibiting LXRalpha Expression in Hepatocytes and Macrophages. Nutrients 2020, 12, 3088. [Google Scholar] [CrossRef] [PubMed]
- Bilotta, M.T.; Petillo, S.; Santoni, A.; Cippitelli, M. Liver X Receptors: Regulators of Cholesterol Metabolism, Inflammation, Autoimmunity, and Cancer. Front. Immunol. 2020, 11, 584303. [Google Scholar] [CrossRef]
- Zhu, R.; Ou, Z.; Ruan, X.; Gong, J. Role of liver X receptors in cholesterol efflux and inflammatory signaling (review). Mol. Med. Rep. 2012, 5, 895–900. [Google Scholar] [CrossRef]
- Kalaany, N.Y.; Gauthier, K.C.; Zavacki, A.M.; Mammen, P.P.; Kitazume, T.; Peterson, J.A.; Horton, J.D.; Garry, D.J.; Bianco, A.C.; Mangelsdorf, D.J. LXRs regulate the balance between fat storage and oxidation. Cell Metab. 2005, 1, 231–244. [Google Scholar] [CrossRef]
- Alnaaim, S.A.; Al-Kuraishy, H.M.; Alexiou, A.; Papadakis, M.; Saad, H.M.; Batiha, G.E. Role of Brain Liver X Receptor in Parkinson’s Disease: Hidden Treasure and Emerging Opportunities. Mol. Neurobiol. 2023, 60, 1–17. [Google Scholar] [CrossRef]
- Tavoosi, Z.; Moradi-Sardareh, H.; Saidijam, M.; Yadegarazari, R.; Borzuei, S.; Soltanian, A.; Goodarzi, M.T. Cholesterol Transporters ABCA1 and ABCG1 Gene Expression in Peripheral Blood Mononuclear Cells in Patients with Metabolic Syndrome. Cholesterol 2015, 2015, 682904. [Google Scholar] [CrossRef] [PubMed]
- He, P.; Gelissen, I.C.; Ammit, A.J. Regulation of ATP binding cassette transporter A1 (ABCA1) expression: Cholesterol-dependent and—Independent signaling pathways with relevance to inflammatory lung disease. Respir. Res. 2020, 21, 250. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Tang, C. Regulation of ABCA1 functions by signaling pathways. Biochim. Biophys. Acta 2012, 1821, 522–529. [Google Scholar] [CrossRef] [PubMed]
- Rezaei, F.; Farhat, D.; Gursu, G.; Samnani, S.; Lee, J.Y. Snapshots of ABCG1 and ABCG5/G8: A Sterol’s Journey to Cross the Cellular Membranes. Int. J. Mol. Sci. 2022, 24, 484. [Google Scholar] [CrossRef]
- Liu, M.; Chen, Y.; Zhang, L.; Wang, Q.; Ma, X.; Li, X.; Xiang, R.; Zhu, Y.; Qin, S.; Yu, Y.; et al. Regulation of Hepatic Cholesteryl Ester Transfer Protein Expression and Reverse Cholesterol Transport by Inhibition of DNA Topoisomerase II. J. Biol. Chem. 2015, 290, 14418–14429. [Google Scholar] [CrossRef] [PubMed]
- Jalil, A.; Bourgeois, T.; Menegaut, L.; Lagrost, L.; Thomas, C.; Masson, D. Revisiting the Role of LXRs in PUFA Metabolism and Phospholipid Homeostasis. Int. J. Mol. Sci. 2019, 20, 3787. [Google Scholar] [CrossRef] [PubMed]
- Rong, X.; Albert, C.J.; Hong, C.; Duerr, M.A.; Chamberlain, B.T.; Tarling, E.J.; Ito, A.; Gao, J.; Wang, B.; Edwards, P.A.; et al. LXRs regulate ER stress and inflammation through dynamic modulation of membrane phospholipid composition. Cell Metab. 2013, 18, 685–697. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Tontonoz, P. Phospholipid Remodeling in Physiology and Disease. Annu. Rev. Physiol. 2019, 81, 165–188. [Google Scholar] [CrossRef] [PubMed]
- Shao, G.; Qian, Y.; Lu, L.; Liu, Y.; Wu, T.; Ji, G.; Xu, H. Research progress in the role and mechanism of LPCAT3 in metabolic related diseases and cancer. J. Cancer 2022, 13, 2430–2439. [Google Scholar] [CrossRef]
- Sun, M.M.; Beier, F. Liver X Receptor activation regulates genes involved in lipid homeostasis in developing chondrocytes. Osteoarthr. Cartil. Open 2020, 2, 100030. [Google Scholar] [CrossRef]
- Feng, C.; Lou, B.; Dong, J.; Li, Z.; Chen, Y.; Li, Y.; Zhang, X.; Jiang, X.C.; Ding, T. Lysophosphatidylcholine acyltransferase 3 deficiency impairs 3T3L1 cell adipogenesis through activating Wnt/beta-catenin pathway. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2018, 1863, 834–843. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.Q.; Xu, C.F.; Yu, C.H.; Chen, W.X.; Li, Y.M. Role of endoplasmic reticulum stress in the pathogenesis of nonalcoholic fatty liver disease. World J. Gastroenterol. 2014, 20, 1768–1776. [Google Scholar] [CrossRef] [PubMed]
- De Luca, M.; Mandala, M.; Rose, G. Towards an understanding of the mechanoreciprocity process in adipocytes and its perturbation with aging. Mech. Ageing Dev. 2021, 197, 111522. [Google Scholar] [CrossRef] [PubMed]
- Zha, B.S.; Zhou, H. ER Stress and Lipid Metabolism in Adipocytes. Biochem. Res. Int. 2012, 2012, 312943. [Google Scholar] [CrossRef]
- Amen, O.M.; Sarker, S.D.; Ghildyal, R.; Arya, A. Endoplasmic Reticulum Stress Activates Unfolded Protein Response Signaling and Mediates Inflammation, Obesity, and Cardiac Dysfunction: Therapeutic and Molecular Approach. Front. Pharmacol. 2019, 10, 977. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Heitman, J. The cyclophilins. Genome Biol. 2005, 6, 226. [Google Scholar]
- Takahashi, N.; Hayano, T.; Suzuki, M. Peptidyl-prolyl cis-trans isomerase is the cyclosporin A-binding protein cyclophilin. Nature 1989, 337, 473–475. [Google Scholar] [CrossRef] [PubMed]
- Gegunde, S.; Alfonso, A.; Alvarino, R.; Alonso, E.; Botana, L.M. Cyclophilins A, B, and C Role in Human T Lymphocytes Upon Inflammatory Conditions. Front. Immunol. 2021, 12, 609196. [Google Scholar] [CrossRef]
- Jin, Z.G.; Lungu, A.O.; Xie, L.; Wang, M.; Wong, C.; Berk, B.C. Cyclophilin A is a proinflammatory cytokine that activates endothelial cells. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 1186–1191. [Google Scholar] [CrossRef]
- Dawar, F.U.; Xiong, Y.; Khattak, M.N.K.; Li, J.; Lin, L.; Mei, J. Potential role of cyclophilin A in regulating cytokine secretion. J. Leukoc. Biol. 2017, 102, 989–992. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Choi, T.G.; Ding, Y.; Kim, Y.; Ha, K.S.; Lee, K.H.; Kang, I.; Ha, J.; Kaufman, R.J.; Lee, J.; et al. Overexpressed cyclophilin B suppresses apoptosis associated with ROS and Ca2+ homeostasis after ER stress. J Cell Sci 2008, 121 Pt 21, 3636–3648. [Google Scholar] [CrossRef]
- Bernasconi, R.; Solda, T.; Galli, C.; Pertel, T.; Luban, J.; Molinari, M. Cyclosporine A-sensitive, cyclophilin B-dependent endoplasmic reticulum-associated degradation. PLoS ONE 2010, 5, e13008. [Google Scholar] [CrossRef] [PubMed]
- Stocki, P.; Chapman, D.C.; Beach, L.A.; Williams, D.B. Depletion of cyclophilins B and C leads to dysregulation of endoplasmic reticulum redox homeostasis. J. Biol. Chem. 2014, 289, 23086–23096. [Google Scholar] [CrossRef]
- Bagola, K.; Mehnert, M.; Jarosch, E.; Sommer, T. Protein dislocation from the ER. Biochim. Biophys. Acta 2011, 1808, 925–936. [Google Scholar] [CrossRef] [PubMed]
- DeBoer, J.; Madson, C.J.; Belshan, M. Cyclophilin B enhances HIV-1 infection. Virology 2016, 489, 282–291. [Google Scholar] [CrossRef]
- Khong, M.L.; Li, L.; Solesio, M.E.; Pavlov, E.V.; Tanner, J.A. Inorganic polyphosphate controls cyclophilin B-mediated collagen folding in osteoblast-like cells. FEBS J. 2020, 287, 4500–4524. [Google Scholar] [CrossRef]
- Zhang, L.; Li, Z.; Zhang, B.; He, H.; Bai, Y. PPIA is a novel adipogenic factor implicated in obesity. Obesity 2015, 23, 2093–2100. [Google Scholar] [CrossRef]
- Qu, Y.; Lin, Q.; Yuan, Y.; Sun, Z.; Li, P.; Wang, F.; Jiang, H.; Chen, T. Cyclosporin A inhibits adipogenic differentiation and regulates immunomodulatory functions of murine mesenchymal stem cells. Biochem. Biophys. Res. Commun. 2018, 498, 516–522. [Google Scholar] [CrossRef] [PubMed]
- Rosen, E.D. The transcriptional basis of adipocyte development. Prostaglandins Leukot. Essent. Fat. Acids 2005, 73, 31–34. [Google Scholar] [CrossRef]
- Banerjee, D.; Patra, D.; Sinha, A.; Roy, S.; Pant, R.; Sarmah, R.; Dutta, R.; Kanta Bhagabati, S.; Tikoo, K.; Pal, D.; et al. Lipid-induced monokine cyclophilin-A promotes adipose tissue dysfunction implementing insulin resistance and type 2 diabetes in zebrafish and mice models of obesity. Cell Mol. Life Sci. 2022, 79, 282. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Li, H.; Wang, K.; Xu, B.; Chen, Z.N.; Bian, H. Deficiency of CD147 Attenuated Non-alcoholic Steatohepatitis Progression in an NLRP3-Dependent Manner. Front. Cell Dev. Biol. 2020, 8, 784. [Google Scholar] [CrossRef] [PubMed]
- Price, E.R.; Zydowsky, L.D.; Jin, M.J.; Baker, C.H.; McKeon, F.D.; Walsh, C.T. Human cyclophilin B: A second cyclophilin gene encodes a peptidyl-prolyl isomerase with a signal sequence. Proc. Natl. Acad. Sci. USA 1991, 88, 1903–1907. [Google Scholar] [CrossRef]
- Fang, F.; Zheng, J.; Galbaugh, T.L.; Fiorillo, A.A.; Hjort, E.E.; Zeng, X.; Clevenger, C.V. Cyclophilin B as a co-regulator of prolactin-induced gene expression and function in breast cancer cells. J. Mol. Endocrinol. 2010, 44, 319–329. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.W.; Sutor, S.L.; Lindquist, L.; Evans, G.L.; Madden, B.J.; Bergen, H.R., 3rd; Hefferan, T.E.; Yaszemski, M.J.; Bram, R.J. Severe osteogenesis imperfecta in cyclophilin B-deficient mice. PLoS Genet. 2009, 5, e1000750. [Google Scholar] [CrossRef]
- Yoon, J.S.; Kim, S.S.; Ha, J.; Kang, I.; Choe, W. Cyclophilin B, a molecule chaperone, promotes adipogenesis in 3T3-L1 preadipocytes via AKT/mTOR pathway. Int. J. Mol. Med. 2023, 51, 6. [Google Scholar] [CrossRef]
- Jeong, K.; Kim, H.; Kim, K.; Kim, S.J.; Hahn, B.S.; Jahng, G.H.; Yoon, K.S.; Kim, S.S.; Ha, J.; Kang, I.; et al. Cyclophilin B is involved in p300-mediated degradation of CHOP in tumor cell adaptation to hypoxia. Cell Death Differ. 2014, 21, 438–450. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Fan, Q.; Xie, H.; Lu, L.; Tao, R.; Wang, F.; Xi, R.; Hu, J.; Chen, Q.; Shen, W.; et al. Elevated Serum Cyclophilin B Levels Are Associated with the Prevalence and Severity of Metabolic Syndrome. Front. Endocrinol. 2017, 8, 360. [Google Scholar] [CrossRef]
- Tang, Q.Q.; Lane, M.D. Role of C/EBP homologous protein (CHOP-10) in the programmed activation of CCAAT/enhancer-binding protein-beta during adipogenesis. Proc. Natl. Acad. Sci. USA 2000, 97, 12446–12450. [Google Scholar] [CrossRef]
- Fu, S.; Watkins, S.M.; Hotamisligil, G.S. The role of endoplasmic reticulum in hepatic lipid homeostasis and stress signaling. Cell Metab. 2012, 15, 623–634. [Google Scholar] [CrossRef]
- Han, J.; Kaufman, R.J. The role of ER stress in lipid metabolism and lipotoxicity. J. Lipid Res. 2016, 57, 1329–1338. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Liu, R. ER stress and hepatic lipid metabolism. Front Genet. 2014, 5, 112. [Google Scholar] [CrossRef] [PubMed]
- Basseri, S.; Austin, R.C. Endoplasmic reticulum stress and lipid metabolism: Mechanisms and therapeutic potential. Biochem. Res. Int. 2012, 2012, 841362. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Goodman, J.M. The collaborative work of droplet assembly. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2017, 1862 Pt B, 1205–1211. [Google Scholar] [CrossRef]
- Olzmann, J.A.; Carvalho, P. Dynamics and functions of lipid droplets. Nat. Rev. Mol. Cell Biol. 2019, 20, 137–155. [Google Scholar] [CrossRef]
- Miura, S.; Gan, J.W.; Brzostowski, J.; Parisi, M.J.; Schultz, C.J.; Londos, C.; Oliver, B.; Kimmel, A.R. Functional conservation for lipid storage droplet association among Perilipin, ADRP, and TIP47 (PAT)-related proteins in mammals, Drosophila, and Dictyostelium. J. Biol. Chem. 2002, 277, 32253–32257. [Google Scholar] [CrossRef] [PubMed]
- Al-Mansoori, L.; Al-Jaber, H.; Prince, M.S.; Elrayess, M.A. Role of Inflammatory Cytokines, Growth Factors and Adipokines in Adipogenesis and Insulin Resistance. Inflammation 2022, 45, 31–44. [Google Scholar] [CrossRef]
- Makki, K.; Froguel, P.; Wolowczuk, I. Adipose tissue in obesity-related inflammation and insulin resistance: Cells, cytokines, and chemokines. ISRN Inflamm. 2013, 2013, 139239. [Google Scholar] [CrossRef]
- Roszer, T. Metabolic impact of adipose tissue macrophages in the early postnatal life. J. Leukoc. Biol. 2022, 112, 1515–1524. [Google Scholar] [CrossRef] [PubMed]
- Weisberg, S.P.; McCann, D.; Desai, M.; Rosenbaum, M.; Leibel, R.L.; Ferrante, A.W., Jr. Obesity is associated with macrophage accumulation in adipose tissue. J. Clin. Invest. 2003, 112, 1796–1808. [Google Scholar] [CrossRef] [PubMed]
- Cinti, S.; Mitchell, G.; Barbatelli, G.; Murano, I.; Ceresi, E.; Faloia, E.; Wang, S.; Fortier, M.; Greenberg, A.S.; Obin, M.S. Adipocyte death defines macrophage localization and function in adipose tissue of obese mice and humans. J. Lipid Res. 2005, 46, 2347–2355. [Google Scholar] [CrossRef]
- Jia, Q.; Morgan-Bathke, M.E.; Jensen, M.D. Adipose tissue macrophage burden, systemic inflammation, and insulin resistance. Am. J. Physiol. Endocrinol. Metab. 2020, 319, E254–E264. [Google Scholar] [CrossRef]
- Martos-Rus, C.; Katz-Greenberg, G.; Lin, Z.; Serrano, E.; Whitaker-Menezes, D.; Domingo-Vidal, M.; Roche, M.; Ramaswamy, K.; Hooper, D.C.; Falkner, B.; et al. Macrophage and adipocyte interaction as a source of inflammation in kidney disease. Sci. Rep. 2021, 11, 2974. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Ortega, O.; Moreno-Corona, N.C.; Cruz-Holguin, V.J.; Garcia-Gonzalez, L.D.; Helguera-Repetto, A.C.; Romero-Valdovinos, M.; Arevalo-Romero, H.; Cedillo-Barron, L.; Leon-Juarez, M. The Immune Response in Adipocytes and Their Susceptibility to Infection: A Possible Relationship with Infectobesity. Int. J. Mol. Sci. 2022, 23, 6154. [Google Scholar] [CrossRef] [PubMed]
- Nance, S.A.; Muir, L.; Lumeng, C. Adipose tissue macrophages: Regulators of adipose tissue immunometabolism during obesity. Mol. Metab. 2022, 66, 101642. [Google Scholar] [CrossRef] [PubMed]
- Fischer-Posovszky, P.; Wang, Q.A.; Asterholm, I.W.; Rutkowski, J.M.; Scherer, P.E. Targeted deletion of adipocytes by apoptosis leads to adipose tissue recruitment of alternatively activated M2 macrophages. Endocrinology 2011, 152, 3074–3081. [Google Scholar] [CrossRef]
- Faria, S.S.; Correa, L.H.; Heyn, G.S.; de Sant’Ana, L.P.; Almeida, R.D.N.; Magalhaes, K.G. Obesity and Breast Cancer: The Role of Crown-Like Structures in Breast Adipose Tissue in Tumor Progression, Prognosis, and Therapy. J. Breast Cancer 2020, 23, 233–245. [Google Scholar] [CrossRef]
- Fuster, J.J.; Ouchi, N.; Gokce, N.; Walsh, K. Obesity-Induced Changes in Adipose Tissue Microenvironment and Their Impact on Cardiovascular Disease. Circ. Res. 2016, 118, 1786–1807. [Google Scholar] [CrossRef]
- Yao, J.; Wu, D.; Qiu, Y. Adipose tissue macrophage in obesity-associated metabolic diseases. Front. Immunol. 2022, 13, 977485. [Google Scholar] [CrossRef]
- Ou, M.Y.; Zhang, H.; Tan, P.C.; Zhou, S.B.; Li, Q.F. Adipose tissue aging: Mechanisms and therapeutic implications. Cell Death Dis. 2022, 13, 300. [Google Scholar] [CrossRef]
- Zuo, L.; Prather, E.R.; Stetskiv, M.; Garrison, D.E.; Meade, J.R.; Peace, T.I.; Zhou, T. Inflammaging and Oxidative Stress in Human Diseases: From Molecular Mechanisms to Novel Treatments. Int. J. Mol. Sci. 2019, 20, 4472. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Sharma, A.K.; Wolfrum, C. Novel insights into adipose tissue heterogeneity. Rev. Endocr. Metab. Disord. 2022, 23, 5–12. [Google Scholar] [CrossRef]
- Ruiz-Ojeda, F.J.; Mendez-Gutierrez, A.; Aguilera, C.M.; Plaza-Diaz, J. Extracellular Matrix Remodeling of Adipose Tissue in Obesity and Metabolic Diseases. Int. J. Mol. Sci. 2019, 20, 4888. [Google Scholar] [CrossRef]
- Ghosh, A.K.; Garg, S.K.; Mau, T.; O’Brien, M.; Liu, J.; Yung, R. Elevated Endoplasmic Reticulum Stress Response Contributes to Adipose Tissue Inflammation in Aging. J. Gerontol. A Biol. Sci. Med. Sci. 2015, 70, 1320–1329. [Google Scholar] [CrossRef]
- Weinstock, A.; Brown, E.J.; Garabedian, M.L.; Pena, S.; Sharma, M.; Lafaille, J.; Moore, K.J.; Fisher, E.A. Single-Cell RNA Sequencing of Visceral Adipose Tissue Leukocytes Reveals that Caloric Restriction Following Obesity Promotes the Accumulation of a Distinct Macrophage Population with Features of Phagocytic Cells. Immunometabolism 2019, 1, e190008. [Google Scholar] [CrossRef] [PubMed]
- Auger, C.; Kajimura, S. Adipose Tissue Remodeling in Pathophysiology. Annu. Rev. Pathol. 2023, 18, 71–93. [Google Scholar] [CrossRef]
- Tchkonia, T.; Pirtskhalava, T.; Thomou, T.; Cartwright, M.J.; Wise, B.; Karagiannides, I.; Shpilman, A.; Lash, T.L.; Becherer, J.D.; Kirkland, J.L. Increased TNFalpha and CCAAT/enhancer-binding protein homologous protein with aging predispose preadipocytes to resist adipogenesis. Am. J. Physiol. Endocrinol. Metab. 2007, 293, E1810–E1819. [Google Scholar] [CrossRef]
- Chung, K.W.; Chung, H.Y. The Effects of Calorie Restriction on Autophagy: Role on Aging Intervention. Nutrients 2019, 11, 2923. [Google Scholar] [CrossRef]
- Cui, J.; Bai, X.Y.; Shi, S.; Cui, S.; Hong, Q.; Cai, G.; Chen, X. Age-related changes in the function of autophagy in rat kidneys. Age 2012, 34, 329–339. [Google Scholar] [CrossRef] [PubMed]
- Melendez, A.; Talloczy, Z.; Seaman, M.; Eskelinen, E.L.; Hall, D.H.; Levine, B. Autophagy genes are essential for dauer development and life-span extension in C. elegans. Science 2003, 301, 1387–1391. [Google Scholar] [CrossRef] [PubMed]
- Sorrenti, V.; Davinelli, S.; Scapagnini, G.; Willcox, B.J.; Allsopp, R.C.; Willcox, D.C. Astaxanthin as a Putative Geroprotector: Molecular Basis and Focus on Brain Aging. Mar. Drugs 2020, 18, 351. [Google Scholar] [CrossRef]
- Gao, L.; Liu, X.; Luo, X.; Lou, X.; Li, P.; Li, X.; Liu, X. Antiaging effects of dietary supplements and natural products. Front. Pharmacol. 2023, 14, 1192714. [Google Scholar] [CrossRef] [PubMed]
- Aman, Y.; Schmauck-Medina, T.; Hansen, M.; Morimoto, R.I.; Simon, A.K.; Bjedov, I.; Palikaras, K.; Simonsen, A.; Johansen, T.; Tavernarakis, N.; et al. Autophagy in healthy aging and disease. Nat. Aging 2021, 1, 634–650. [Google Scholar] [CrossRef]
- Wang, M.; Miller, R.A. Fibroblasts from long-lived mutant mice exhibit increased autophagy and lower TOR activity after nutrient deprivation or oxidative stress. Aging Cell 2012, 11, 668–674. [Google Scholar] [CrossRef]
- Kitada, M.; Koya, D. Autophagy in metabolic disease and ageing. Nat. Rev. Endocrinol. 2021, 17, 647–661. [Google Scholar] [CrossRef]
- Mau, T.; Yung, R. Adipose tissue inflammation in aging. Exp. Gerontol. 2018, 105, 27–31. [Google Scholar] [CrossRef]
- Ghosh, A.K.; Mau, T.; O’Brien, M.; Garg, S.; Yung, R. Impaired autophagy activity is linked to elevated ER-stress and inflammation in aging adipose tissue. Aging 2016, 8, 2525–2537. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Modulators | Full Name | Roles in Lipogenesis |
|---|---|---|
| FAS | Fatty acid synthase | XBP1s promotes the expression of the FAS gene, contributing to fatty acid synthesis. FAS is an enzyme responsible for generating fatty acids and plays a crucial role in lipid metabolism. |
| SREBP1c | Sterol regulatory element-binding protein 1c | XBP1s regulates the expression of the SREBP1c gene, facilitating lipid synthesis. SREBP1c also activates other important genes related to lipid metabolism. |
| ACC | Acetyl-CoA carboxylase | XBP1s enhances the expression of the ACC gene, increasing the conversion of acetyl-CoA into fatty acids. This process is essential in fatty acid synthesis and is one of the key steps. |
| DGAT | Diacylglycerol O-Acyltransferase | XBP1s regulates the expression of the DGAT gene, promoting processes related to lipid droplets. This is associated with lipid storage |
| ChREBP | Carbohydrate-responsive element-binding protein | XBP1s controls the expression of the ChREBP gene, regulating the interaction between carbohydrate metabolism and fatty acid synthesis. |
| PLIN | Perilipin | XBP1s controls the expression of the PLIN gene, facilitating the perilipin protein found on the surface of lipid droplets. Perilipin stabilizes lipid droplets and regulates lipid storage and movement processes. |
| CIDE | Cell death-inducing DFFA-like effector | XBP1s contributes to the dynamics of lipid droplets by regulating the expression of certain genes within the CIDE gene family. These genes play a role in modulating the structure and function of lipid droplets. |
| ATGL | Adipose triglyceride lipase | XBP1s regulates the expression of the ATGL gene, controlling the breakdown of triglycerides in neutral fat. This process is associated with the movement of lipids within lipid droplets. |
| HSL | Hormone-sensitive lipase | XBP1s further regulates the breakdown of triglycerides in neutral fat by controlling the expression of the HSL gene. This process is related to energy metabolism. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, G.; Lee, J.; Ha, J.; Kang, I.; Choe, W. Endoplasmic Reticulum Stress and Its Impact on Adipogenesis: Molecular Mechanisms Implicated. Nutrients 2023, 15, 5082. https://doi.org/10.3390/nu15245082
Kim G, Lee J, Ha J, Kang I, Choe W. Endoplasmic Reticulum Stress and Its Impact on Adipogenesis: Molecular Mechanisms Implicated. Nutrients. 2023; 15(24):5082. https://doi.org/10.3390/nu15245082
Chicago/Turabian StyleKim, Gyuhui, Jiyoon Lee, Joohun Ha, Insug Kang, and Wonchae Choe. 2023. "Endoplasmic Reticulum Stress and Its Impact on Adipogenesis: Molecular Mechanisms Implicated" Nutrients 15, no. 24: 5082. https://doi.org/10.3390/nu15245082