
The Contribution of Muscle Innate Immunity to Uremic Cachexia
 , and
, and 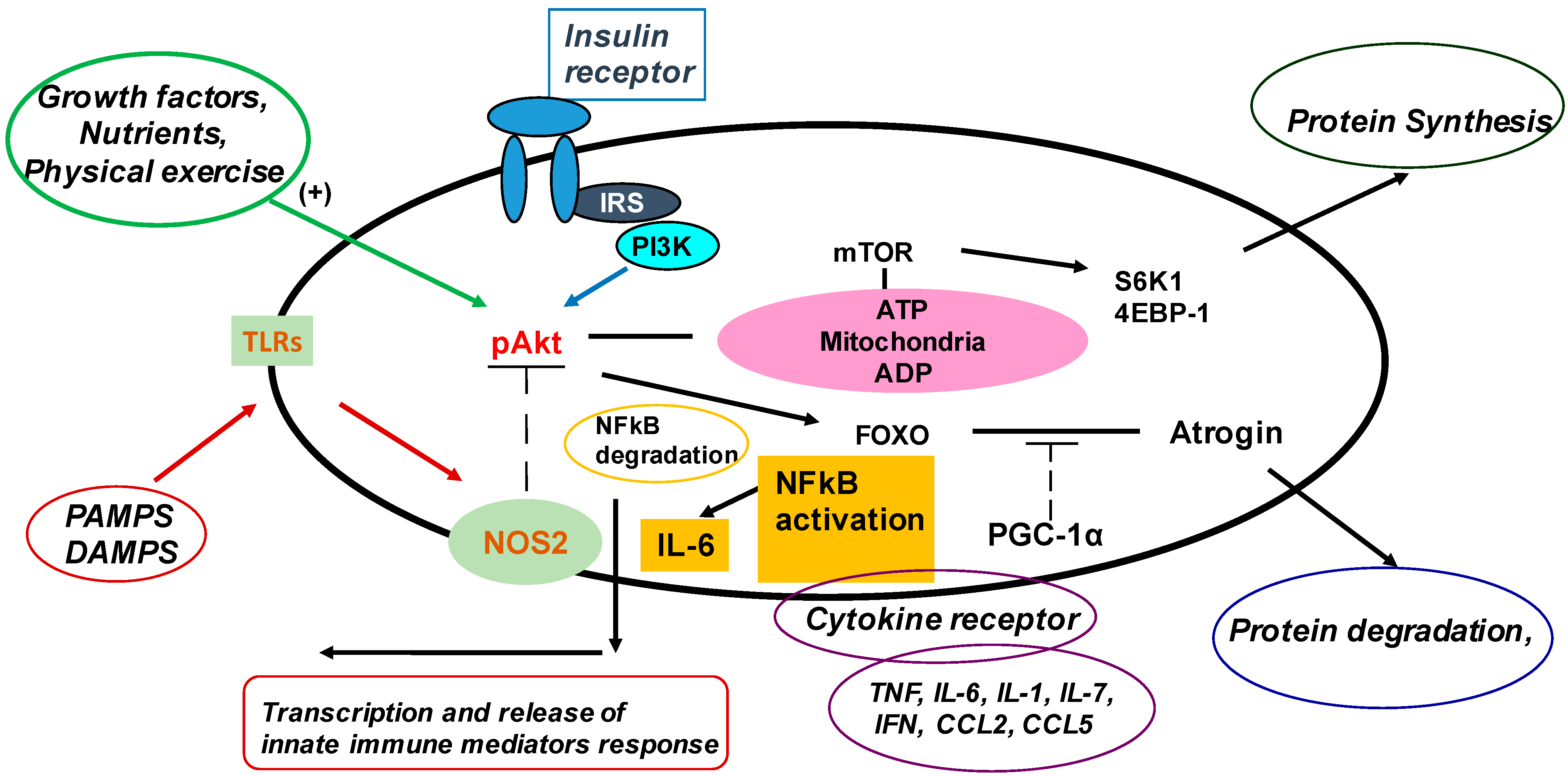{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Inflammation in CKD
3. Mechanisms and Mediators Contributing to Inflammation in Patients with CKD
4. The Innate Immune System in Muscle

5. Effects of Innate Immunity on Muscle Protein Metabolism
6. Patients with CKD5 Have an Activated Canonical TLR4–NF-κB–IL-6 Pathway in Muscle
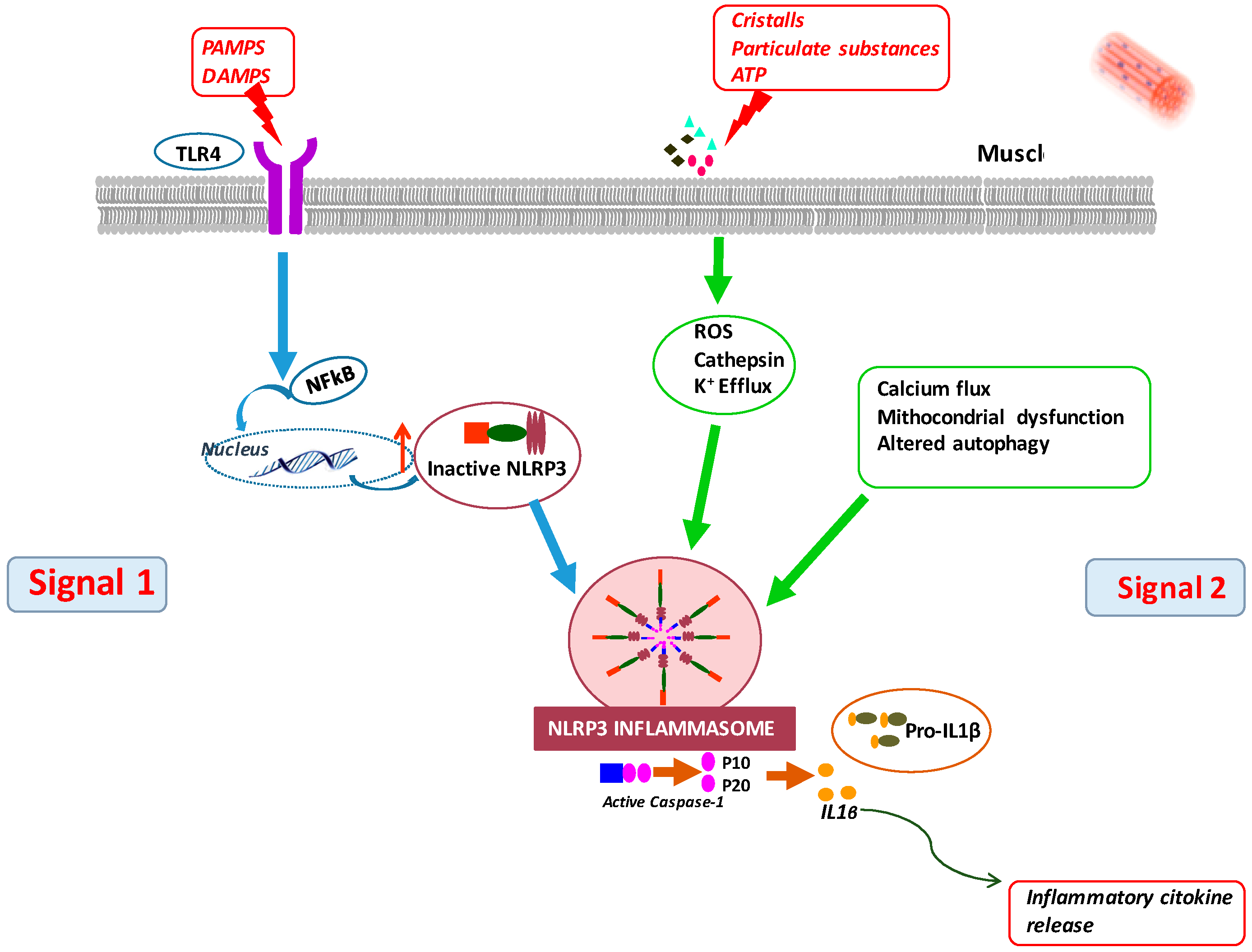
7. A TLR4–NLRP3 Inflammasome Pathway Is Also Activated in Muscle of CKD5 Patients
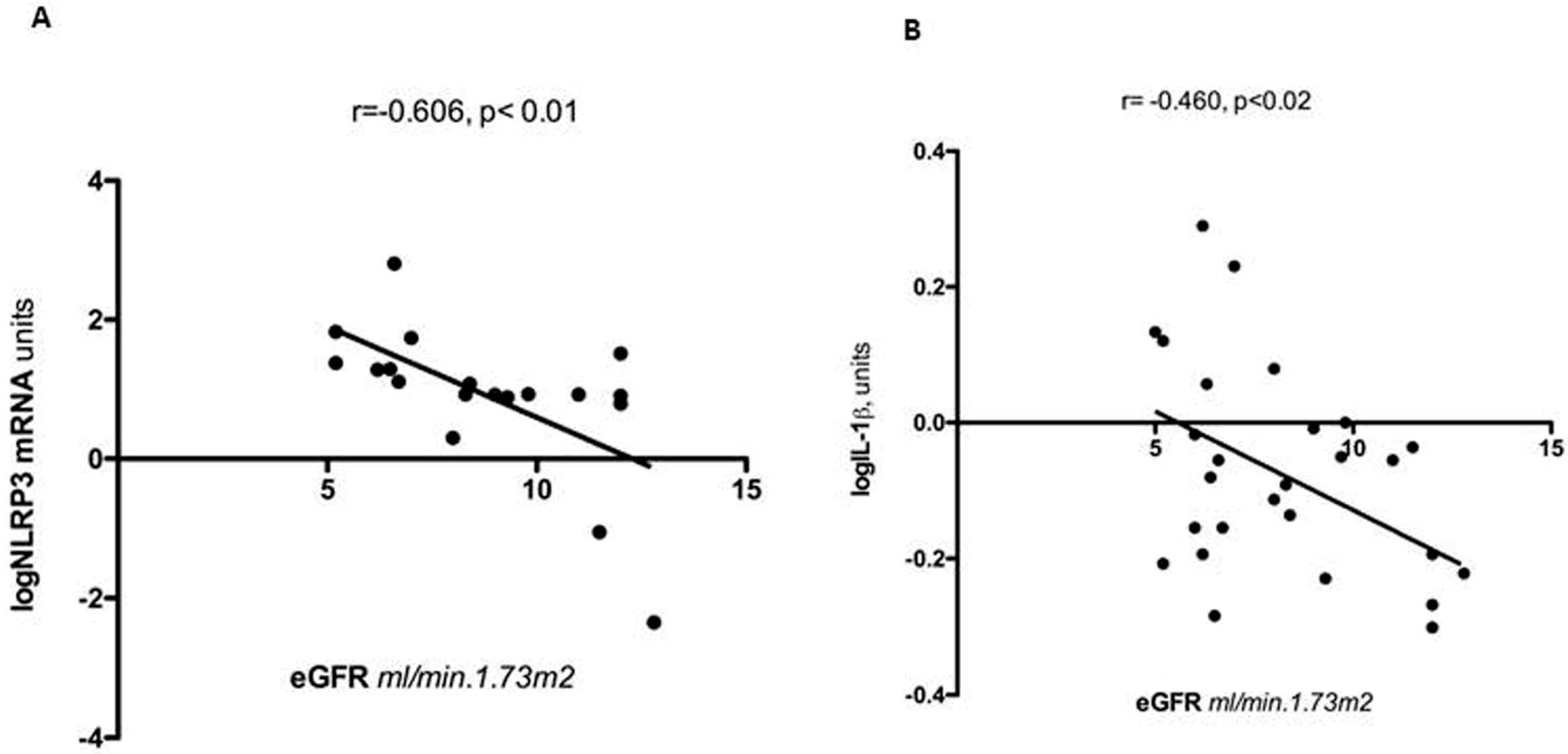
8. Clinical Associations of Muscle Inflammation in Patients with CKD5
9. Inflammation Up-Regulates Myostatin in Muscle of CKD5 Patients
10. Accelerated Cell Senescence in Uremic Muscle: A Novel Mechanism of Sterile Muscle Inflammation
11. Targeting Muscle Inflammation
11.1. Healthy Diets and Modulation of the Microbiome
11.2. Physical Exercise
11.3. Targeting Cell Senescence
11.4. Nuclear-Factor-Erythroid-2-Related Factor 2 Agonists
11.5. Targeting Inflammation to Treat Cardiovascular Disease in Patients with CKD
11.6. Targeting Inflammation to Treat PEW
12. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Carrero, J.J.; Thomas, F.; Nagy, K.; Arogundade, F.; Avesani, C.M.; Chan, M.; Chmielewski, M.; Cordeiro, A.C.; Espinosa-Cuevas, A.; Fiaccadori, E.; et al. Global Prevalence of Protein-Energy Wasting in Kidney Disease: A Meta-analysis of Contemporary Observational Studies from the International Society of Renal Nutrition and Metabolism. J. Ren. Nutr. 2018, 28, 380–392. [Google Scholar] [CrossRef] [PubMed]
- Tripepi, G.; Mallamaci, F.; Zoccali, C. Inflammation markers, adhesion molecules, and all-cause and cardiovascular mortality in patients with ESRD: Searching for the best risk marker by multivariate modeling. J. Am. Soc. Nephrol. 2005, 16 (Suppl. S1), S83–S88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herselman, M.; Esau, N.; Kruger, J.M.; Labadarios, D.; Moosa, R. Serum Protein and Mortality in Adults on Long-Term Hemodialysis: Exhaustive Review and Meta-Analysis. Nutrition 2010, 26, 10–32. [Google Scholar] [CrossRef]
- Hanna, R.M.; Ghobry, L.; Wassef, O.; Rhee, C.M.; Kalantar-Zadeh, K.A. Practical Approach to Nutrition, Protein-Energy Wasting, Sarcopenia, and Cachexia in Patients with Chronic Kidney Disease. Blood Purif. 2020, 49, 202–211. [Google Scholar] [CrossRef] [PubMed]
- Price, S.R.; Mitch, W.E.; Garibotto, G. Muscle Atrophy in CKD: A Historical Perspective of Advancements in Its Understanding. J. Ren. Nutr. 2022, S1051–S2276. [Google Scholar] [CrossRef] [PubMed]
- den Hoedt, C.H.; Bots, M.L.; Grooteman, M.P.C.; van der Weerd, N.C.; Penne, E.L.; Mazairac, A.H.A.; Levesque, R.; Blankestijn, P.J.; Nubeé, M.J.; ter Wee, P.M.; et al. Clinical Predictors of Decline in Nutritional Parameters over Time in ESRD. Clin. J. Am. Soc. Nephrol. 2014, 9, 318–329. [Google Scholar] [CrossRef] [Green Version]
- Amdur, R.L.; Feldman, H.I.; Gupta, J.; Yang, W.; Kanetsky, P.; Shlipak, M.; Rahman, M.; Lash, J.P.; Townsend, R.R.; Ojo, A.; et al. Inflammation and progression of CKD: The CRIC Study. Clin. J. Am. Soc. Nephrol. 2016, 11, 1546–1556. [Google Scholar] [CrossRef] [Green Version]
- Go, A.S.; Chertow, G.M.; Fan, D.; McCulloch, C.E.; Hsu, C.Y. Chronic kidney disease and the risks of death, cardiovascular events, and hospitalization. N. Engl. J. Med. 2004, 351, 1296–1305. [Google Scholar] [CrossRef]
- Ridker, P.M.; Cushman, M.; Stampfer, M.J.; Tracy, R.P.; Hennekens, C.H. Inflammation, aspirin, and the risk of cardiovascular disease in apparently healthy men. N. Engl. J. Med. 1997, 336, 973–979. [Google Scholar] [CrossRef]
- Stenvinkel, P.; Heimbürger, O.; Paultre, F.; Diczfalusy, U.; Wang, T.; Berglund, L.; Jogestrand, T. Strong association between malnutrition, inflammation, and atherosclerosis in chronic renal failure. Kidney Int. 1999, 55, 1899–1911. [Google Scholar] [CrossRef] [Green Version]
- Kalantar-Zadeh, K. Recent advances in understanding the malnutrition-inflammation-cachexia syndrome in chronic kidney disease patients: What is next? Semin. Dial. 2005, 18, 365–369. [Google Scholar] [CrossRef] [Green Version]
- Batra, G.; Lakic, T.G.; Lindback, J.; Held, C.; White, H.D.; Stewart, R.A.H.; Koenig, W.; Cannon, C.P.; Budaj, A.; Hagstrom, E.; et al. Interleukin 6 and cardiovascular outcomes in patients with chronic kidney disease and chronic coronary syndrome. JAMA Cardiol. 2021, 6, 1440–1445. [Google Scholar] [CrossRef]
- Sun, J.; Axelsson, J.; Machowska, A.; Heimburger, O.; Barany, P.; Lindholm, B.; Lindstrom, K.; Stenvinkel, P.; Qureshi, A.R. Biomarkers of cardiovascular disease and mortality risk in patients with advanced CKD. Clin. J. Am. Soc. Nephrol. 2016, 11, 1163–1172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schunk, S.J.; Triem, S.; Schmit, D.; Zewinger, S.; Sarapki, T.; Becker, E.; Hutter, G.; Wrublewsky, S.; Kuting, F.; Hohl, M.; et al. Interleukin-1α is a central regulator of leukocyte-endothelial adhesion in myocardial infarction and in chronic kidney disease. Circulation 2021, 144, 893–908. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M.; Tuttle, K.R.; Perkovic, V.; Libby, P.; MacFadyen, J.G. Inflammation drives residual risk in chronic kidney disease: A CANTOS substudy. Eur. Heart J. 2022, 43, 4832–4844. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Dong, J.; Verzola, D.; Hruska, K.; Garibotto, G.; Hu, Z.; Mitch, W.E.; Thomas, S.S. Signal regulatory protein alpha initiates cachexia through muscle to adipose tissue crosstalk. J. Cachex.-Sarcopenia Muscle 2019, 10, 1210–1227. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Pan, J.; Dong, Y.; Tweardy, D.J.; Dong, Y.; Garibotto, G.; Mitch, W.E. Stat3 activation links a C/EBPδ to myostatin pathway to stimulate loss of muscle mass. Cell Metab. 2013, 18, 368–379. [Google Scholar] [CrossRef] [Green Version]
- Garibotto, G.; Sofia, A.; Procopio, V.; Villaggio, B.; Tarroni, A.; Di Martino, M.; Cappelli, V.; Gandolfo, M.T.; Aloisi, F.; De Cian, F.; et al. Peripheral tissue release of interleukin-6 in patients with chronic kidney diseases: Effects of end-stage renal disease and microinflammatory state. Kidney Int. 2006, 70, 384–390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deger, S.M.; Hung, A.M.; Gamboa, J.L.; Siew, E.D.; Ellis, C.D.; Booker, C.; Sha, F.; Li, H.; Bian, A.; Stewart, T.G.; et al. Systemic inflammation is associated with exaggerated skeletal muscle protein catabolism in maintenance hemodialysis patients. J. Clin. Investig. 2017, 2, e95185. [Google Scholar] [CrossRef] [Green Version]
- Torres, F.G.; Molina, M.; Soler-Majoral, J.; Romero-González, G.; Chitiva, N.R.; Troya-Saborido, M.; Rullan, G.S.; Burgos, E.; Martínez, J.P.; Jou, M.U.; et al. Evolving Concepts on Inflammatory Biomarkers and Malnutrition in Chronic Kidney Disease. Nutrients 2022, 14, 4297. [Google Scholar] [CrossRef]
- Stenvinkel, P.; Ketteler, M.; Johnson, R.J.; Lindholm, B.; Pecoits-Filho, R.; Riella, M.; Heimburger, O.; Cederholm, T.; Girndt, M. IL-10, IL-6, and TNF-alpha: Central factors in the altered cytokine network of uremia--the good, the bad, and the ugly. Kidney Int. 2005, 67, 1216–1233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gbd 2015 Mortality and Causes of Death Collaborators. Global, regional, and national life expectancy, all-cause mortality, and cause-specific mortality for 249 causes of death, 1980-2015: A systematic analysis for the global burden of disease study 2015. Lancet 2016, 388, 1459–1544. [Google Scholar] [CrossRef] [Green Version]
- Garibotto, G.; Sofia, A.; Balbi, M.; Procopio, V.; Villaggio, B.; Tarroni, A.; Di Martino, M.; Cappelli, V.; Gandolfo, M.T.; Valli, A.; et al. Kidney and splanchnic handling of interleukin-6 in humans. Cytokine 2007, 37, 51–54. [Google Scholar] [CrossRef] [PubMed]
- Dubuisson, N.; Versele, R.; Davis-López de Carrizosa, M.A.; Selvais, C.M.; Brichard, S.M.; Abou-Samra, M. Walking down Skeletal Muscle Lane: From Inflammasome to Disease. Cells 2021, 10, 3023. [Google Scholar] [CrossRef] [PubMed]
- Lang, C.H. Importance of the innate immune response in skeletal muscle to sepsis induced alterations in protein balance. Shock 2023, 59, 214–223. [Google Scholar] [CrossRef]
- Wada, J.; Makino, H. Innate immunity in diabetes and diabetic nephropathy. Nat. Rev. Nephrol. 2016, 12, 13–26. [Google Scholar] [CrossRef]
- Godkowicz, M.; Druszczyńska, M. NOD1,NOD2, and NLRC5 Receptors in Antiviral and Antimycobacterial Immunity. Vaccines 2022, 10, 1487. [Google Scholar] [CrossRef]
- Frost, R.A.; Lang, C.H. Regulation of muscle growth by pathogen-associated molecules. J. Anim. Sci. 2008, 86 (Suppl. S14), E84–E93. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Moylan, J.S.; Chambers, M.A.; Smith, J.; Reid, M.B. Interleukin-1 stimulates catabolism in C2C12 myotubes. Am. J. Physiol. Cell Physiol. 2009, 297, C706–C714. [Google Scholar] [CrossRef] [Green Version]
- Braun, T.P.; Zhu, X.; Szumowski, M.; Scott, G.D.; Grossberg, A.J.; Levasseur, P.R.; Graham, K.; Khan, S.; Damaraju, S.; Colmers, W.F.; et al. Central nervous system inflammation induces muscle atrophy via activation of the hypothalamic-pituitary-adrenal axis. J. Exp. Med. 2011, 208, 2449–2463. [Google Scholar] [CrossRef]
- Garibotto, G.; Saio, M.; Aimasso, F.; Russo, E.; Picciotto, D.; Viazzi, F.; Verzola, D.; Laudon, A.; Esposito, P.; Brunori, G. How to overcome anabolic resistance in Dialysis-treated patients? Front. Nutr. 2021, 8, 701386. [Google Scholar] [CrossRef] [PubMed]
- Sartori, R.; Romanello, V.; Sandri, M. Mechanisms of muscle atrophy and hypertrophy: Implications in health and disease. Nat. Commun. 2021, 12, 330–350. [Google Scholar] [CrossRef] [PubMed]
- Verzola, D.; Bonanni, A.; Sofia, A.; Montecucco, F.; D’Amato, E.; Cademartori, V.; Parodi, E.L.; Viazzi, F.; Venturelli, C.; Brunori, G.; et al. Toll-like receptor 4 signalling mediates inflammation in skeletal muscle of patients with chronic kidney disease. J. Cachexia Sarcopenia Muscle 2017, 8, 131–144. [Google Scholar] [CrossRef] [PubMed]
- Thomas, S.S.; Zhang, L.; Mitch, W.E. Molecular Mechanisms of insulin resistance in chronic kidney disease. Kidney Int. 2015, 88, 1233–1239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.H.; Mitch, W.E. Muscle wasting from kidney failure-a model for catabolic conditions. Int. J. Biochem. Cell Biol. 2013, 45, 2230–2238. [Google Scholar] [CrossRef] [Green Version]
- Lysakova-Devine, T.; Keogh, B.; Harrington, B.; Nagpal, K.; Halle, A.; Golenbock, D.T.; Monie, T.; Bowie, A.G. Viral inhibitory peptide of TLR4, a peptide derived from vaccinia protein A46 specifically inhibits TLR4 by directly targeting MyD88 adaptor-like and TRIF-related adaptor molecule. J. Immunol. 2010, 185, 4261–4271. [Google Scholar] [CrossRef] [Green Version]
- Kumar, A.; Takada, Y.; Boriek, A.M.; Aggarwal, B.B. Nuclear factor-kB: Its role in health and disease. J. Mol. Med. 2004, 82, 434–448. [Google Scholar] [CrossRef]
- Ping Li, Y.; Chen, Y.; John, J.; Moylan, J.; Jin, B.; Mann, D.L.; Reid, M. TNF-α acts via p38 MAPK to stimulate expression of the ubiquitin ligase atrogin1/MAFbx in skeletal muscle. FASEB J. 2005, 19, 362–370. [Google Scholar]
- Williamson, D.; Gallagher, P.; Harber, M. Mitogen-activated protein kinase [MAPK] pathway activation: Effects of age and acute exercise on human skeletal muscle. J. Physiol. 2003, 547, 977–987. [Google Scholar] [CrossRef]
- Koistinen, H.A.; Chibalin, A.V.; Zierath, J.R. Aberrant p38 mitogen-activated protein kinase signaling in skeletal muscle from Type 2 diabetic patients. Diabetologia 2003, 46, 1324–1328. [Google Scholar] [CrossRef] [Green Version]
- Childs, T.E.; Spangenburg, E.E.; Vyas, D.R.; Botth, F.W. Temporal alterations in protein signaling cascades during recovery from muscle atrophy. Am. J. Physiol.-Cell Physiol. 2003, 285, C391–C398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Giovanni, S.; Molon, A.; Broccolini, A.; Melco, G.; Mirabella, M.; Hoffman, E.; Servidei, S. Constitutive activation of MAPK cascade in acute quadriplegic myopathy. Ann. Neurol. 2004, 55, 195–206. [Google Scholar] [CrossRef] [PubMed]
- Eggelbusch, M.; Shi, A.; Broeksma, B.C.; Vázquez-Cruz, M.; Soares, M.; de Wit, G.M.J.; Everts, B.; Jaspers, R.T.; Wust, R.C. The NLRP3 inflammasome contributes to inflammation-induced morphological and metabolic alterations in skeletal muscle. J. Cachexia Sarcopenia Muscle 2022, 13, 3048–3061. [Google Scholar] [CrossRef] [PubMed]
- Youm, Y.H.; Grant, R.W.; McCabe, L.R.; Albarado, D.C.; Nguyen, K.Y.; Ravussin, A.; Pistell, P.; Newman, S.; Carter, R.; Laque, A.; et al. Canonical Nlrp3 inflammasome links systemic low-grade inflammation to functional decline in aging. Cell Metab. 2013, 18, 519–532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, N.; Kny, M.; Riediger, F.; Busch, K.; Schmidt, S.; Luft, F.C.; Slevogt, H.; Fielitz, J. Deletion of Nlrp3 protects from inflammation-induced skeletal muscle atrophy. Intensive Care Med. Exp. 2017, 5, 3–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boursereau, R.; Abou-Samra, M.; Lecompte, S.; Noel, L.; Brichard, S. Downregulation of the NLRP3 inflammasome by adiponectin rescues Duchenne muscular dystrophy. BMC Biol. 2018, 16, 33. [Google Scholar] [CrossRef] [Green Version]
- Hi, X.; Tan, S.; Tan, S. NLRP3 inflammasome in sepsis. Mol. Med. Rep. 2021, 24, 514–524. [Google Scholar]
- Quiao, L.; Ma, J.; Zhang, Z.; Sui, W.; Zhai, C.; Xu, D.; Wang, Z.; Liu, H.; Zhang, M.; Zhang, C.; et al. Deficient chaperone-mediated autophagy promotes inflammation and atherosclerosis. Circ. Res. 2021, 129, 1141–1157. [Google Scholar] [CrossRef]
- Kooman, J.P.; Dekker, M.J.; Usvyat, L.A.; Kotanko, P.; van der Sande, F.; Schalkwijk, C.G.; Shiels, P.G.; Stenvinkel, P. Inflammation and premature aging in advanced chronic kidney disease. Am. J. Physiol. Renal. Physiol. 2017, 313, F938–F950. [Google Scholar] [CrossRef] [Green Version]
- Verzola, D.; Esposito, P.; Milanesi, S.; Saio, M.; Picciotto, D.; Frascio, M.; Laudon, A.; Garibotto, G.; Brunori, G.; Viazzi, F. A Toll-like receptor-4/NLRP3 inflammasome pathway promotes inflammation in skeletal muscle of chronic kidney disease patients. JCSM Rapid Commun. 2023, 6, 50–61. [Google Scholar] [CrossRef]
- Banerjee, T.; Sebastian, A.; Frassetto, L. Association of Diet-dependent Systemic Acid Load, Renal Function, and Serum Albumin Concentration. J. Ren. Nutr. 2023, 3, 428–434. [Google Scholar] [CrossRef] [PubMed]
- Takkavatakarn, K.; Wuttiputinun, T.; Phannajit, J.; Praditpornsilpa, K.; Eiam-Ong, S.; Susantitaphong, P. Protein-bound uremic toxin lowering strategies in chronic kidney disease: A systematic review and metaanalysis. J. Nephrol. 2021, 34, 1805–1817. [Google Scholar] [CrossRef] [PubMed]
- Avin, K.G.; Chen, N.X.; Organ, J.M.; Zarse, C.; O’Neill, K.; Conway, R.G.; Konrad, R.J.; Bacallao, R.L.; Allen, M.R.; Moe, S.M. Skeletal muscle regeneration and oxidative stress are altered in chronic kidney disease. PLoS ONE 2016, 11, e0159411. [Google Scholar] [CrossRef] [Green Version]
- Thome, T.; Kumar, R.A.; Burke, S.K.; Khattri, R.B.; Salyers, Z.R.; Kelley, R.C.; Coleman, M.D.; Christou, D.D.; Hepple, R.T.; Scali, S.T.; et al. Impaired muscle mitochondrial energetics is associated with uremic metabolite accumulation in chronic kidney disease. JCI Insight 2021, 6, e139826. [Google Scholar] [CrossRef]
- Adesso, S.; Popolo, A.; Bianco, G.; Sorrentino, R.; Pinto, A.; Autore, G.; Marzocco, S. The Uremic Toxin Indoxyl Sulphate Enhances Macrophage Response to LPS. PLoS ONE 2013, 8, e76778. [Google Scholar] [CrossRef] [Green Version]
- Matsuo, K.; Yamamoto, S.; Wakamatsu, T.; Takahashi, Y.; Kawamura, K.; Kaneko, Y.; Goto, S.; Kazama, J.J.; Narita, I. Increased Proinflammatory Cytokine Production and Decreased Cholesterol Efflux Due to Downregulation of ABCG1 in Macrophages Exposed to Indoxyl Sulfate. Toxins 2015, 7, 3155–3166. [Google Scholar] [CrossRef] [Green Version]
- Yamaguchi, K.; Yisireyili, M.; Goto, S.; Cheng, X.W.; Nakayama, T.; Matsushita, T.; Niwa, T.; Murohara, T.; Takeshita, K. Indoxyl Sulfate Activates NLRP3 Inflammasome to Induce Cardiac Contractile Dysfunction Accompanied by Myocardial Fibrosis and Hypertrophy. Cardiovasc. Toxicol. 2022, 22, 365–377. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, M.; Kasamatsu, S.; Shinozaki, S.; Yasuhara, S.; Masao Kaneki, M. Myostatin deficiency not only prevents muscle wasting but also improves survival in septic mice. Am. J. Physiol. Endocrinol. Metab. 2021, 320, E150–E159. [Google Scholar] [CrossRef]
- Verzola, D.; Barisone, C.; Picciotto, D.; Garibotto, G.; Koppe, L. Emerging role of myostatin and its inhibition in the setting of chronic kidney disease. Kidney Int. 2019, 95, 506–517. [Google Scholar] [CrossRef]
- Wang, X.H.; Mitch, W.E.; Price, S.R. Pathophysiological mechanisms leading to muscle loss in chronic kidney disease. Nat. Rev. Nephrol. 2022, 18, 138–152. [Google Scholar] [CrossRef]
- Huang, Y.; Wang, B.; Hassounah, F.; Price, S.R.; Klein, J.; Mohamed, T.M.A.; Wang, Y.; Park, J.; Cai, H.; Zhang, X.; et al. The impact of senescence on muscle wasting in chronic kidney disease. J. Cachexia Sarcopenia Muscle 2023, 14, 126–141. [Google Scholar] [CrossRef] [PubMed]
- Docherty, M.H.; O’ Sullivan, E.D.; Bonventre, J.V.; Ferenbach, D.A. Cellular Senescence in the Kidney. J. Am. Soc. Nephrol. 2019, 30, 726–736. [Google Scholar] [CrossRef] [PubMed]
- Tournadre, A.; Vial, G.; Capel, F.; Soubrier, M.; Boirie, Y. Sarcopenia. Jt. Bone Spine 2019, 86, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Larsson, L.; Degens, H.; Meishan, L.; Salviati, L.; Lee, Y.I.; Thompson, W.; Kirkland, J.L.; Sandri, M. Sarcopenia: Aging-Related Loss of Muscle Mass and Function. Physiol. Rev. 2019, 99, 427–511. [Google Scholar] [CrossRef] [PubMed]
- Shannon, O.M.; Mendes, I.; Köchl, C.; Mazidi, M.; Ashor, A.W.; Rubele, S.; Minihane, A.M.; Mathers, J.C.; Siervo, M. Mediterranean Diet Increases Endothelial Function in Adults: A Systematic Review and Meta-Analysis of Randomized Controlled Trials. J. Nutr. 2020, 150, 1151–1159. [Google Scholar] [CrossRef]
- Micheli, L.; Bertini, L.; Bonato, A.; Villanova, N.; Caruso, C.; Caruso, M.; Bernini, R.; Tirone, F. Role of Hydroxytyrosol and Oleuropein in the Prevention of Aging and Related Disorders: Focus on Neurodegeneration, Skeletal Muscle Dysfunction and Gut Microbiota. Nutrients 2023, 15, 1767. [Google Scholar] [PubMed]
- Jovanovich, A.; Isakova, T.; Stubbs, J. Microbiome and cardiovascular disease in CKD. Clin. J. Am. Soc. Nephrol. 2018, 13, 1598–1604. [Google Scholar] [CrossRef] [Green Version]
- Ramezani, A.; Raj, D.S. The gut microbiome, kidney disease, and targeted interventions. J. Am. Soc. Nephrol. 2014, 25, 657–670. [Google Scholar] [CrossRef] [Green Version]
- Schulman, G.; Berl, T.; Beck, G.J.; Remuzzi, G.; Ritz, E.; Arita, K.; Kato, A.; Shimizu, M. Randomized placebo-controlled EPPIC trials of AST-120 in CKD. J. Am. Soc. Nephrol. 2015, 26, 1732–1746. [Google Scholar] [CrossRef] [Green Version]
- Nicklas, J.M.; Sacks, F.M.; Smith, S.R.; Leboff, M.S.; Rood, J.C.; Bray, G.A.; Ridker, P.M. Effect of dietary composition of weight loss diets on high-sensitivity C-reactive protein: The randomized POUNDS LOST trial. Obesity 2013, 21, 681–689. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Lee, D.H.; Hu, J.; Tabung, F.K.; Li, Y.; Bhupathiraju, S.N.; Rimm, E.B.; Rexrode, K.M.; Manson, J.E.; Willett, W.C.; et al. Dietary inflammatory potential and risk of cardiovascular disease among men and women in the US. J. Am. Coll. Cardiol. 2020, 76, 2181–2193. [Google Scholar] [CrossRef] [PubMed]
- Estruch, R.; Ros, E.; Salas-Salvadó, J.; Covas, M.I.; Corella, D.; Arós, F.; Gómez-Gracia, E.; Ruiz-Gutiérrez, V.; Fiol, M.; Lapetra, J.; et al. Primary prevention of cardiovascular disease with a Mediterranean diet supplemented with extra-virgin olive oil or nuts. N. Engl. J. Med. 2018, 378, e34. [Google Scholar] [CrossRef] [PubMed]
- Koh, K.P.; Fassett, R.G.; Sharman, J.E.; Coombes, J.S.; Williams, A.D. Effect of intradialytic versus home-based aerobic exercise training on physical function and vascular parameters in hemodialysis patients: A randomized pilot study. Am. J. Kidney Dis. 2010, 55, 88–99. [Google Scholar]
- Kouidi, E.; Grekas, D.; Deligiannis, A. Effects of exercise training on noninvasive cardiac measures in patients undergoing long-term hemodialysis: A randomized controlled trial. Am. J. Kidney Dis. 2009, 54, 511–552. [Google Scholar] [PubMed]
- Bishop, N.C.; Burton, J.O.; Graham-Brown, M.P.M.; Stensel, D.J.; Viana, J.L.; Watson, E.L. Exercise and chronic kidney disease: Potential mechanisms underlying the physiological benefits. Nat. Rev. Nephrol. 2023, 19, 244–256. [Google Scholar] [PubMed]
- Koufaki, P.; Greenwood, S.; Painter, P.; Mercer, T. The BASES expert statement on exercise therapy for people with chronic kidney disease. J. Sport. Sci. 2015, 33, 1902–1907. [Google Scholar] [CrossRef]
- Smart, N.A.; Williams, A.D.; Levinger, I.; Seling, S.; Howden, E.; Coombes, J.S.; Fassettt, R.G. Exercise & Sports Science Australia [ESSA] position statement on exercise and chronic kidney disease. J. Sci. Med. Sport 2013, 16, 406–411. [Google Scholar] [PubMed]
- K/DOQI Workgroup. K/DOQI clinical practice guidelines for cardiovascular disease in dialysis patients. Am. J. Kidney Dis. 2012, 45 (Suppl. S3), S1–S153. [Google Scholar]
- Matsuzawa, R.; Hoshi, K.; Yoneki, K.; Harada, M.; Watanabe, T.; Shimoda, T.; Yamamoto, S.; Matsunaga, A. Exercise training in elderly people undergoing hemodialysis: A systematic review and meta-analysis. Kidney Int. Rep. 2017, 2, 1096–1110. [Google Scholar] [CrossRef] [Green Version]
- Labib, M.; Bohm, C.; MacRae, J.M.; Paul, N.; Bennett, P.N.; Wilund, K.; McAdams-DeMarco, M.; Jhamb, M.; Mustata, S.; Thompson, S. An international delphi survey on exercise priorities in CKD. Kidney Int. Rep. 2020, 6, 657–668. [Google Scholar] [CrossRef]
- Chiche, A.; Le Roux, I.; von Joest, M.; Sakai, H.; Aguin, S.B.; Cazin, C.; Salam, R.; Fiette, L.; Alegria, O.; Flamant, P.; et al. Injury-Induced Senescence Enables In Vivo Reprogramming in Skeletal Muscle. Cell Stem Cell 2017, 20, 407–414.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sugihara, H.; Teramoto, N.; Yamanouchi, K.; Matsuwaki, T.; Nishihara, M. Oxidative stress-mediated senescence in mesenchymal progenitor cells causes the loss of their fibro/adipogenic potential and abrogates myoblast fusion. Aging 2018, 10, 747–763. [Google Scholar] [PubMed] [Green Version]
- Chan, J.; Eide, I.A.; Tannæs, T.M.; Waldum-Grevbo, B.; Jenssen, T.; Svensson, M. Marine n-3 Polyunsaturated Fatty Acids and Cellular Senescence Markers in Incident Kidney Transplant Recipients: The Omega-3 Fatty Acids in Renal Transplantation [ORENTRA] Randomized Clinical Trial. Kidney Med. 2021, 3, 1041–1049. [Google Scholar] [CrossRef] [PubMed]
- Koppelstaetter, C.; Leierer, J.; Rudnicki, M.; Kerschbaum, J.; Kronbichler, A.; Melk, A.; Mayer, G.; Perco, P. Computational Drug Screening Identifies Compounds Targeting Renal Age-associated Molecular Profiles. Comput. Struct Biotechnol. J. 2019, 17, 843–853. [Google Scholar]
- Fang, Y.; Chen, B.; Liu, Z.; Gong, A.; Gunning, W.T.; Ge, Y.; Malhotra, D.; Gohara, A.; Dworkin, L.D.; Gong, R. Age-related GSK3β overexpression drives podocyte senescence and glomerular aging. J. Clin. Investig. 2022, 132, e141848. [Google Scholar] [CrossRef]
- Kim, M.N.; Moon, J.H.; Cho, Y.M. Sodium-glucose cotransporter-2 inhibition reduces cellular senescence in the diabetic kidney by promoting ketone body-induced NRF2 activation. Diabetes Obes. Metab. 2021, 23, 2561–2571. [Google Scholar] [CrossRef]
- Zhu, Y.; Doornebal, E.J.; Pirtskhalava, T.; Giorgadze, N.; Wentworth, M.; Furmann-Stroissnig, H.; Niedernhofer, L.J.; Robbins, P.D.; Tchkonia, T.; Kirkland, J. New agents that target senescent cells: The flavone, fisetin, and the BCL-XL inhibitors, A1331852 and A1155463. Aging 2017, 9, 955–963. [Google Scholar] [CrossRef] [Green Version]
- Sun, C.C.; Li, S.J.; Yang, C.L.; Xue, R.L.; Xi, Y.Y.; Wang, L.; Zhao, Q.L.; Li, D.J. Sulforaphane Attenuates Muscle Inflammation in Dystrophin-deficient mdx Mice via NF-E2-related Factor 2 [Nrf2]-mediated Inhibition of NF-κB Signaling Pathway. J. Biol. Chem. 2015, 290, 17784–17795. [Google Scholar]
- Zhang, M.; Zhang, M.; Wang, L.; Yu, T.; Jiang, S.; Jiang, P.; Sun, Y.; Pi, J.; Zhao, R.; Guan, D. Activation of cannabinoid type 2 receptor protects skeletal muscle from ischemia-reperfusion injury partly via Nrf2 signaling. Life Sci. 2019, 230, 55–67. [Google Scholar] [CrossRef]
- Liu, C.; Gidlund, E.K.; Witasp, A.; Qureshi, A.R.; Söderberg, M.; Thorell, A.; Nader, G.A.; Barany, P.; Stenvinkel, P.; von Walden, F. Reduced skeletal muscle expression of mitochondrial-derived peptides humanin and MOTS-C and Nrf2 in chronic kidney disease. Am. J. Physiol. Renal. Physiol. 2019, 317, F1122–F1131. [Google Scholar]
- Pergola, P.E.; Raskin, P.; Toto, R.D.; Meyer, C.J.; Huff, J.W.; Grossman, E.B.; Krauth, M.; Ruiz, S.; Audhya, P.; Christ-Schmidt, H.; et al. BEAM Study Investigators. Bardoxolone methyl and kidney function in CKD with type 2 diabetes. N. Engl. J. Med. 2011, 365, 327–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Zeeuw, D.; Akizawa, T.; Audhya, P.; Bakris, G.L.; Chin, M.; Christ-Schmidt, H.; Goldsberry, A.; Houser, M.; Krauth, M.; Lambers Heerspink, H.J.; et al. BEACON Trial Investigators. Bardoxolone methyl in type 2 diabetes and stage 4 chronic kidney disease. N. Engl. J. Med. 2013, 369, 2492–2503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Speer, T.; Dimmeler, S.; Schunk, S.J.; Fliser, D.; Ridkers, P.M. Targeting innate immunity-driven inflammation in CKD and cardiovascular disease. Nat. Rev. Nephrol. 2022, 18, 762–778. [Google Scholar] [PubMed]
- Gimenez, F.; Bhela, S.; Dogra, P.; Harvey, L.; Varanasi, S.K.; Jaggi, U.; Rouse, B.T. The Inflammasome NLRP3 Plays a Protective Role against a Viral Immunopathological Lesion. J. Leukoc. Biol. 2016, 99, 647–657. [Google Scholar]
- Toldo, S.; Mezzaroma, E.; Buckley, L.F.; Potere, N.; Di Nisio, M.; Biondi-Zoccai, G.; Van Tassell, B.W.; Abbate, A. Targeting the NLRP3 inflammasome in cardiovascular diseases. Pharmacol. Ther. 2022, 236, 108053. [Google Scholar]
- Dinarello, C.A. Overview of the IL-1 family in innate inflammation and acquired immunity. Immunol. Rev. 2018, 281, 8–27. [Google Scholar]
- Benny Klimek, M.E.; Sali, A.; Rayavarapu, S.; Van der Meulen, J.H.; Nagaraju, K. Effect of the IL-1 receptor antagonist Kineret[R] on disease phenotype in mdx mice. PLoS ONE 2016, 11, e0155944. [Google Scholar] [CrossRef] [Green Version]
- Hung, A.M.; Ellis, C.D.; Shintani, A.; Booker, C.; Ikizler, T.A. IL-1beta receptor antagonist reduces inflammation in hemodialysis patients. J. Am. Soc. Nephrol. 2011, 22, 437–449. [Google Scholar] [CrossRef] [Green Version]
- Dember, L.M.; Hung, A.; Mehrotra, R.; Hsu, J.Y.; Raj, D.S.; Charytan, D.M.; Mc Causland, F.R.; Regunathan-Shenk, R.; Landis, J.R.; Kimmel, P.L.; et al. Hemodialysis Novel Therapies Consortium. A randomized controlled pilot trial of anakinra for hemodialysis inflammation. Kidney Int. 2022, 102, 1178–1187. [Google Scholar] [CrossRef]
- Cheung, W.W.; Zheng, R.; Hao, S.; Wang, Z.; Gonzalez, A.; Zhou, P.; Hoffman, H.M.; Mak, R.H. The role of IL-1 in adipose browning and muscle wasting in CKD-associated cachexia. Sci. Rep. 2021, 11, 15141. [Google Scholar]
- Matsunaga, N.; Tsuchimori, N.; Matsumoto, T. TAK-242 [resatorvid], a small-molecule inhibitor of Toll-like receptor [TLR] 4 signaling, binds selectively t TLR4 and interferes with interactions betwee TLR4 and its adaptor molecules. Mol. Pharmacol. 2011, 79, 34–41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ono, Y.; Maejima, Y.; Saito, M.; Sakamoto, K.; Horita, S.; Shimomura, K.; Inoue, S.; Kotani, J. TAK-242, aspecific inhibitor of Toll-like receptor 4 signalling, prevents endotoxemia-inducedskeletal muscle wasting in mice. Sci. Rep. 2020, 10, 694–706. [Google Scholar] [PubMed] [Green Version]
- Rice, T.W.; Wheeler, A.P.; Bernard, G.R.; Vincent, J.L.; Angus, D.C.; Aikawa, N.; Demeyer, I.; Sainati, S.; Amlot, N.; Charlie, C.; et al. A randomized, double-blind, placebo-controlled trial of TAK-242 for the treatment of severe sepsis. Crit. Care Med. 2010, 38, 1685–1694. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Esposito, P.; Verzola, D.; Saio, M.; Picciotto, D.; Frascio, M.; Laudon, A.; Zanetti, V.; Brunori, G.; Garibotto, G.; Viazzi, F. The Contribution of Muscle Innate Immunity to Uremic Cachexia. Nutrients 2023, 15, 2832. https://doi.org/10.3390/nu15132832
Esposito P, Verzola D, Saio M, Picciotto D, Frascio M, Laudon A, Zanetti V, Brunori G, Garibotto G, Viazzi F. The Contribution of Muscle Innate Immunity to Uremic Cachexia. Nutrients. 2023; 15(13):2832. https://doi.org/10.3390/nu15132832
Chicago/Turabian StyleEsposito, Pasquale, Daniela Verzola, Michela Saio, Daniela Picciotto, Marco Frascio, Alessandro Laudon, Valentina Zanetti, Giuliano Brunori, Giacomo Garibotto, and Francesca Viazzi. 2023. "The Contribution of Muscle Innate Immunity to Uremic Cachexia" Nutrients 15, no. 13: 2832. https://doi.org/10.3390/nu15132832