
Ascorbic Acid Reduces Neurotransmission, Synaptic Plasticity, and Spontaneous Hippocampal Rhythms in In Vitro Slices

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Chemicals
2.3. Multi-Electrode Array Recording
2.4. Statistical Analysis
3. Results
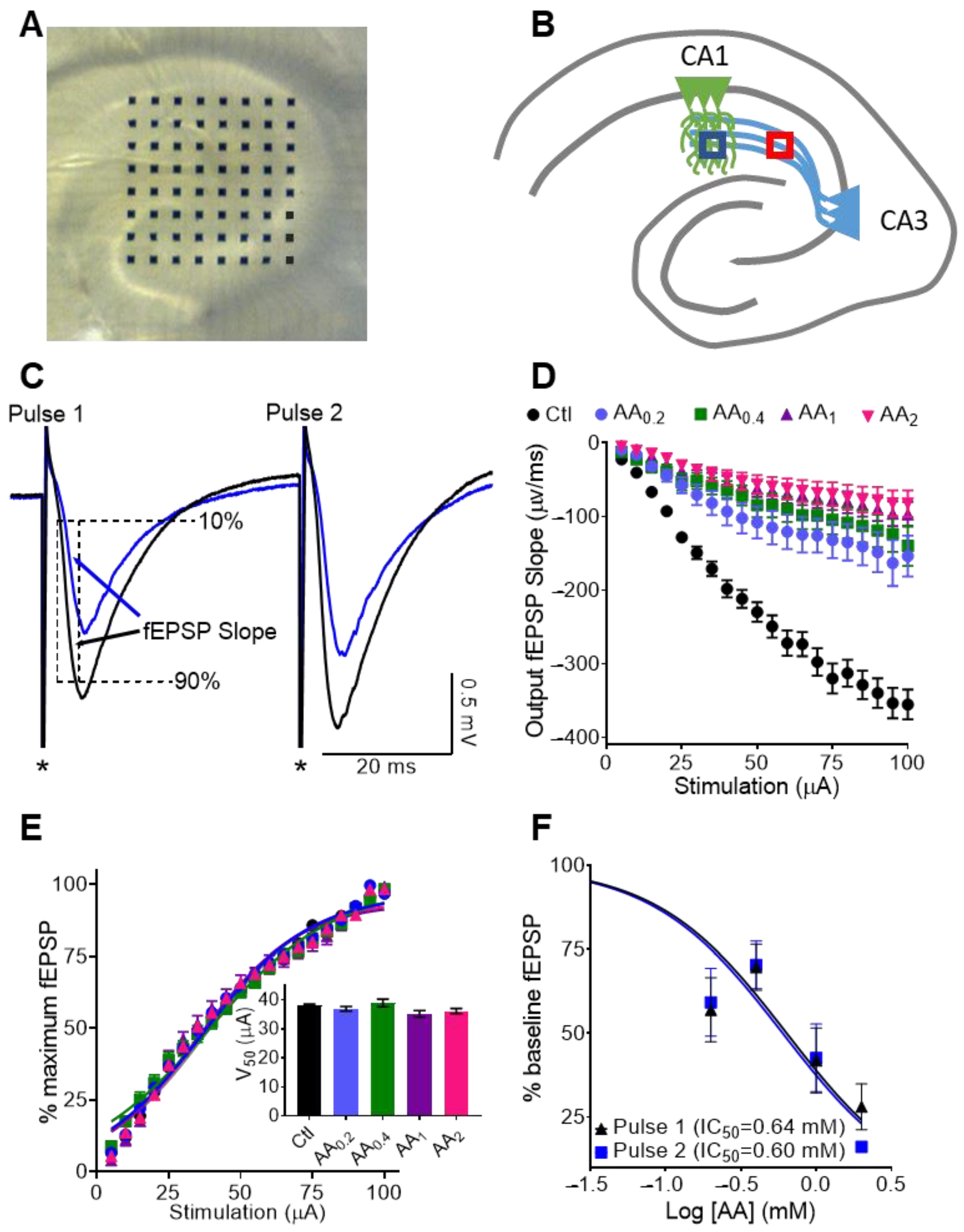
3.1. Physiological Concentrations of AA Reduce Dendritic fEPSPs at CA3-CA1 Synapses
3.2. AA Reduces Paired Pulse Slopes 1 and 2 Equally, thus Presynaptic Neurotransmitter Release Probablity Is Not Affected
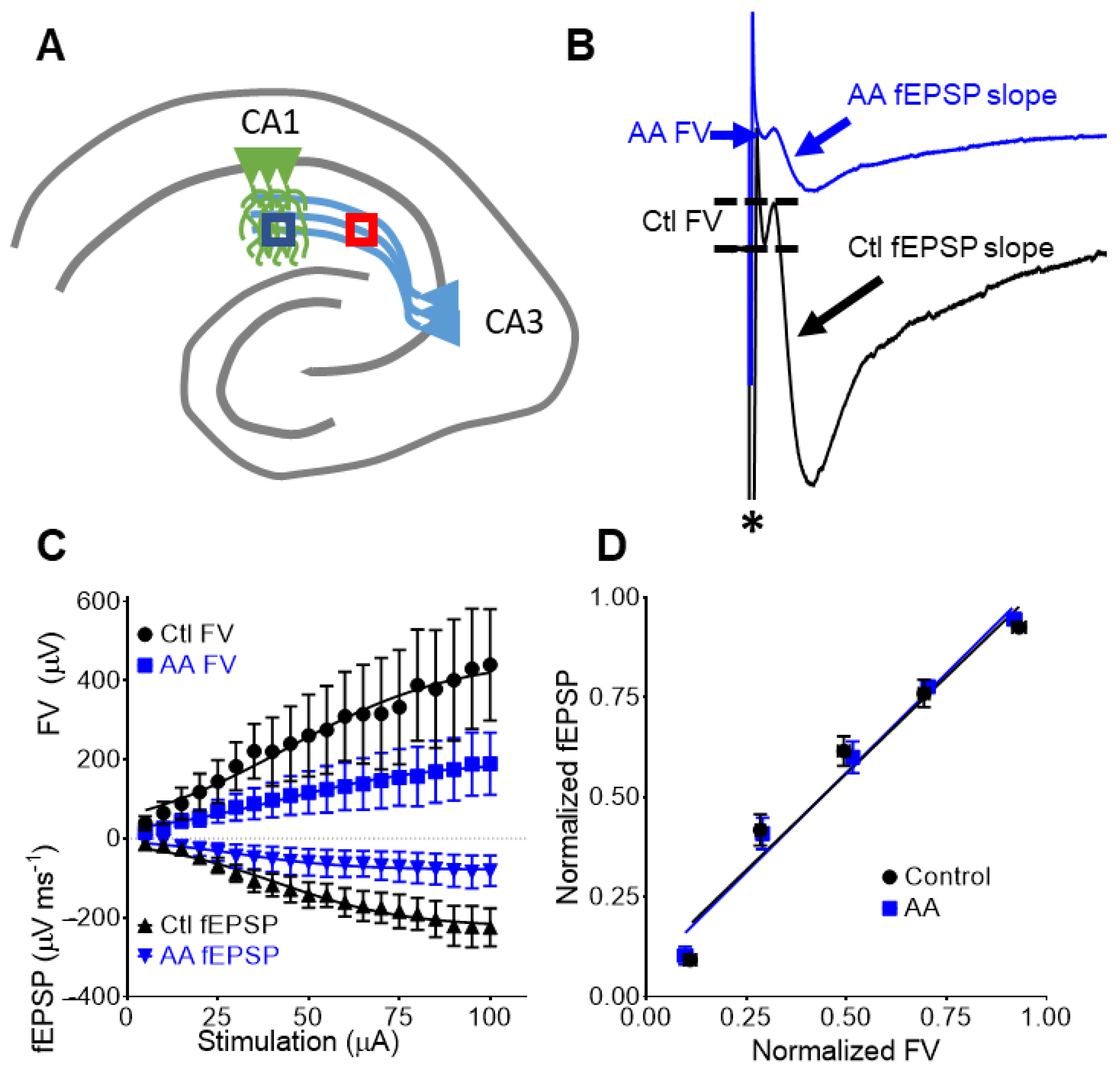
3.3. AA Reduces Presynaptic Fiber Volleys and Postsynaptic Dendritic fEPSPs
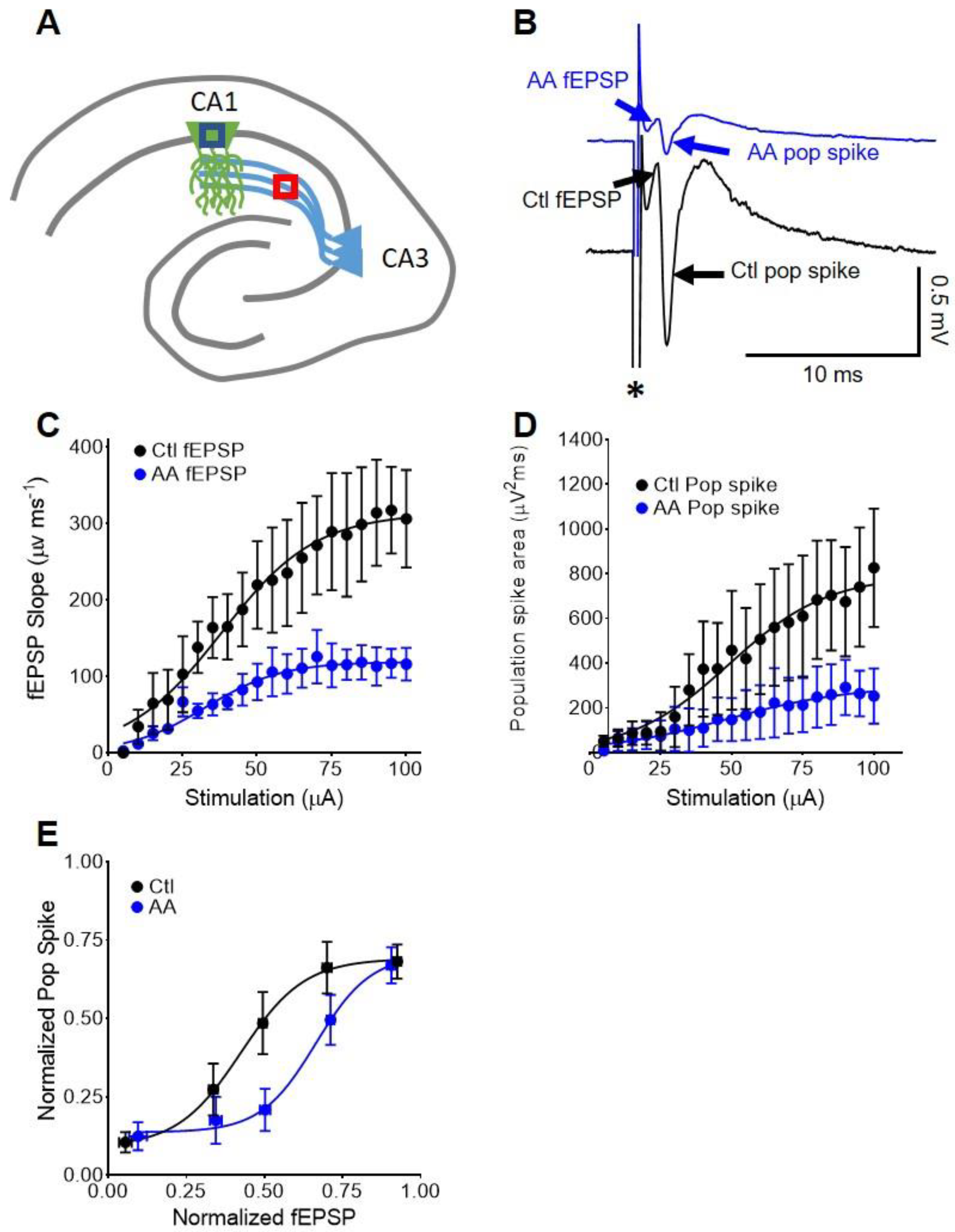
3.4. In CA1, AA Augments Somatic fEPSP and Population Spike Coupling
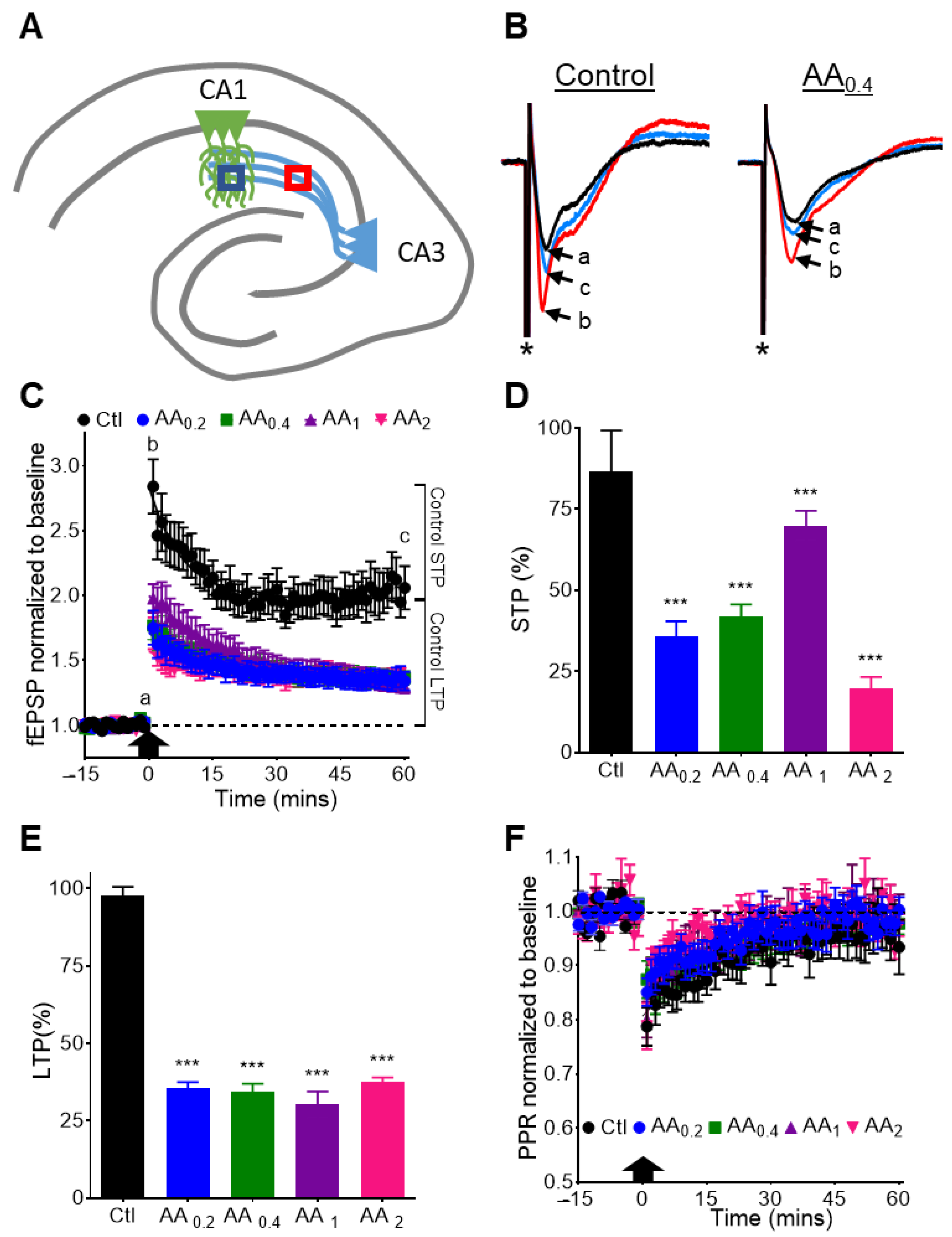
3.5. AA Reduces STP and LTP at the CA3-CA1 Synapse
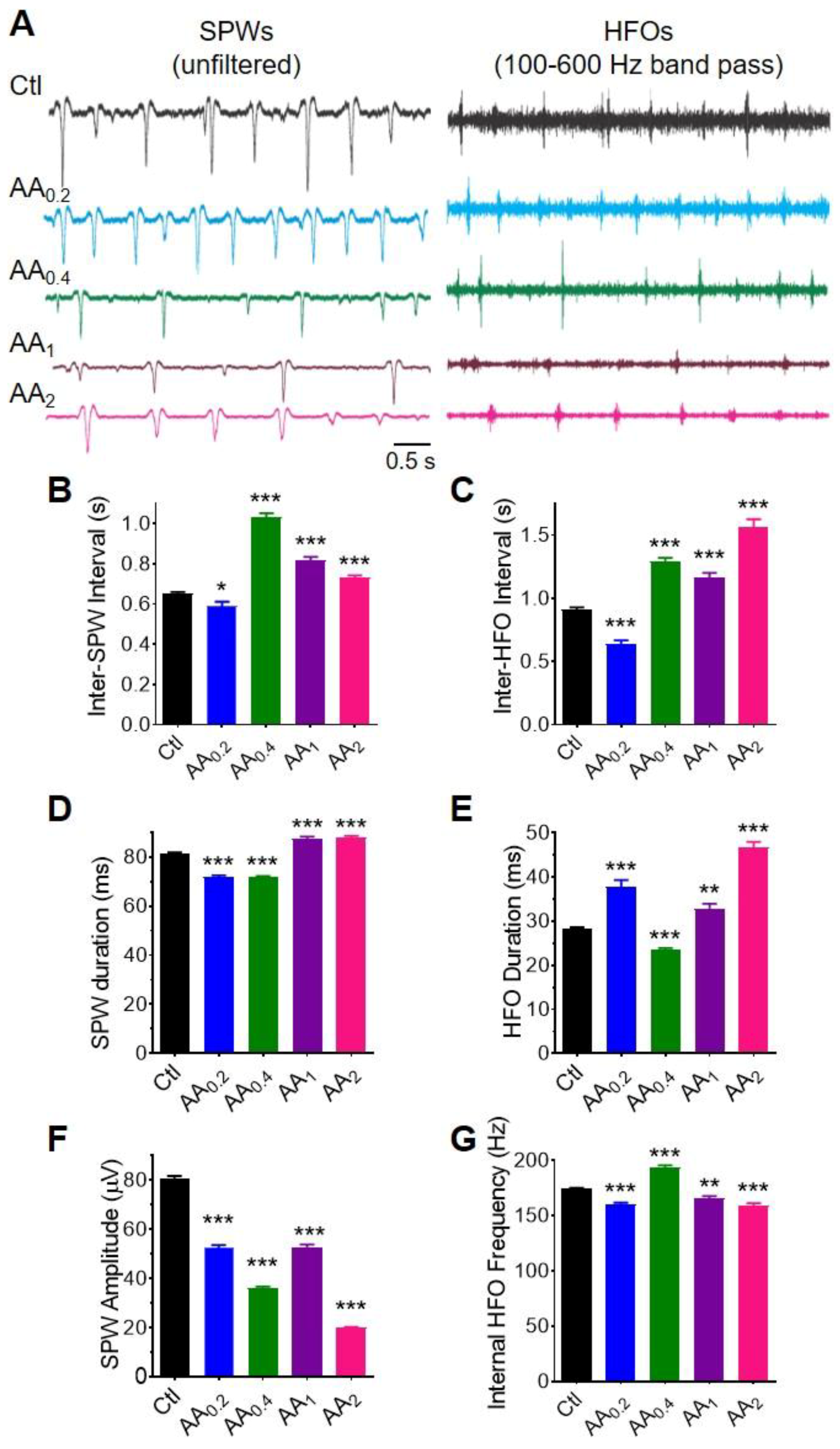
3.6. AA Reduces Spontaneous Sharp Waves and High Frequency Oscillations in Hippocampal Networks
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Doseděl, M.; Jirkovský, E.; Macáková, K.; Krčmová, L.K.; Javorská, L.; Pourová, J.; Mercolini, L.; Remião, F.; Nováková, L.; Mladěnka, P. On Behalf of The Oemonom. Vitamin C-Sources, Physiological Role, Kinetics, Deficiency, Use, Toxicity, and Determination. Nutrients 2021, 13, 615. [Google Scholar] [CrossRef] [PubMed]
- Padayatty, S.J.; Katz, A.; Wang, Y.; Eck, P.; Kwon, O.; Lee, J.-H.; Chen, S.; Corpe, C.P.; Dutta, A.; Dutta, S.K.; et al. Vitamin C as an Antioxidant: Evaluation of Its Role in Disease Prevention. J. Am. Coll. Nutr. 2003, 22, 18–35. [Google Scholar] [CrossRef] [PubMed]
- Evans, W.J. Vitamin E, vitamin C, and exercise. Am. J. Clin. Nutr. 2000, 72 (Suppl. 2), 647S–652S. [Google Scholar] [CrossRef]
- Harrison, F.E.; May, J.M. Vitamin C function in the brain: Vital role of the ascorbate transporter SVCT2. Free Radic. Biol. Med. 2009, 46, 719–730. [Google Scholar] [CrossRef] [Green Version]
- Lachapelle, M.Y.; Drouin, G. Inactivation dates of the human and guinea pig vitamin C genes. Genetica 2011, 139, 199–207. [Google Scholar] [CrossRef]
- Hyder, F.; Rothman, D.L.; Bennett, M.R. Cortical energy demands of signaling and nonsignaling components in brain are conserved across mammalian species and activity levels. Proc. Natl. Acad. Sci. USA 2013, 110, 3549–3554. [Google Scholar] [CrossRef] [Green Version]
- Harris, J.J.; Jolivet, R.; Attwell, D. Synaptic Energy Use and Supply. Neuron 2012, 75, 762–777. [Google Scholar] [CrossRef] [Green Version]
- Ballaz, S.J.; Rebec, G.V. Neurobiology of vitamin C: Expanding the focus from antioxidant to endogenous neuromodulator. Pharmacol. Res. 2019, 146, 104321. [Google Scholar] [CrossRef]
- Kumar, R.R.; Singh, L.; Thakur, A.; Singh, S.; Kumar, B. Role of Vitamins in Neurodegenerative Diseases: A Review. CNS Neurol. Disord. Drug Targets 2021, 20, 1. [Google Scholar] [CrossRef]
- Covarrubias-Pinto, A.; Acuña, A.I.; Beltrán, F.A.; Torres-Díaz, L.; Castro, M.A. Old Things New View: Ascorbic Acid Protects the Brain in Neurodegenerative Disorders. Int. J. Mol. Sci. 2015, 16, 28194–28217. [Google Scholar] [CrossRef]
- Moretti, M.; Rodrigues, A.L.S. Functional role of ascorbic acid in the central nervous system: A focus on neurogenic and synaptogenic processes. Nutr. Neurosci. 2021, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Nelson, M.T.; Joksovic, P.M.; Su, P.; Kang, H.-W.; Van Deusen, A.; Baumgart, J.; David, L.S.; Snutch, T.P.; Barrett, P.Q.; Lee, J.-H.; et al. Molecular Mechanisms of Subtype-Specific Inhibition of Neuronal T-Type Calcium Channels by Ascorbate. J. Neurosci. 2007, 27, 12577–12583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Domith, I.; Socodato, R.; Portugal, C.C.; Munis, A.F.; Duarte-Silva, A.T.; Paes-De-Carvalho, R. Vitamin C modulates glutamate transport and NMDA receptor function in the retina. J. Neurochem. 2018, 144, 408–420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sutor, B.; ten Bruggencate, G. Ascorbic acid: A useful reductant to avoid oxidation of catecholamines in electrophysiological experiments in vitro? Neurosci. Lett. 1990, 116, 287–292. [Google Scholar] [CrossRef] [Green Version]
- Calero, C.I.; Vickers, E.; Cid, G.M.; Aguayo, L.G.; Von Gersdorff, H.; Calvo, D.J. Allosteric Modulation of Retinal GABA Receptors by Ascorbic Acid. J. Neurosci. 2011, 31, 9672–9682. [Google Scholar] [CrossRef]
- Simeone, T.A.; Simeone, K.A.; Samson, K.K.; Kim, D.Y.; Rho, J.M. Loss of the Kv1.1 potassium channel promotes pathologic sharp waves and high frequency oscillations in in vitro hippocampal slices. Neurobiol. Dis. 2013, 54, 68–81. [Google Scholar] [CrossRef] [Green Version]
- Simeone, K.A.; Matthews, S.A.; Samson, K.K.; Simeone, T.A. Targeting deficiencies in mitochondrial respiratory complex I and functional uncoupling exerts anti-seizure effects in a genetic model of temporal lobe epilepsy and in a model of acute temporal lobe seizures. Exp. Neurol. 2014, 251, 84–90. [Google Scholar] [CrossRef] [Green Version]
- Simeone, T.; Samson, K.K.; Matthews, S.A.; Simeone, K.A. In vivo ketogenic diet treatment attenuates pathologic sharp waves and high frequency oscillations in in vitro hippocampal slices from epileptic Kv1.1α knockout mice. Epilepsia 2014, 55, e44–e49. [Google Scholar] [CrossRef] [Green Version]
- Simeone, T.A.; Heruye, S.H.; Kostansek, J.A., IV; Yeh, M.Y.; Matthews, S.A.; Samson, K.K.; Simeone, K.A. Carbamazepine Reduces Sharp Wave-Ripple Complexes and Exerts Synapse-Specific Inhibition of Neurotransmission in Ex Vivo Hippocampal Slices. Brain Sci. 2021, 11, 787. [Google Scholar] [CrossRef]
- Sheridan, G.K.; Wdowicz, A.; Pickering, M.; Watters, O.; Halley, P.G.; O’Sullivan, N.C.; Mooney, C.; O’Connell, D.J.; O’Connor, J.J.; Murphy, K.J. CX3CL1 is up-regulated in the rat hippocampus during memory-associated synaptic plasticity. Front. Cell. Neurosci. 2014, 8, 233. [Google Scholar] [CrossRef]
- Rice, M.E. Ascorbate regulation and its neuroprotective role in the brain. Trends Neurosci. 2000, 23, 209–216. [Google Scholar] [CrossRef]
- Petersen, C.C.; Malenka, R.C.; Nicoll, R.A.; Hopfield, J.J. All-or-none potentiation at CA3-CA1 synapses. Proc. Natl. Acad. Sci. USA 1998, 95, 4732–4737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Enoki, R.; Hu, Y.-L.; Hamilton, D.; Fine, A. Expression of Long-Term Plasticity at Individual Synapses in Hippocampus Is Graded, Bidirectional, and Mainly Presynaptic: Optical Quantal Analysis. Neuron 2009, 62, 242–253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Majewska, M.D.; Bell, J.A.; London, E.D. Regulation of the NMDA receptor by redox phenomena: Inhibitory role of ascorbate. Brain Res. 1990, 537, 328–332. [Google Scholar] [CrossRef]
- Kazmierska, P.; Grebowski, J.; Konopacki, J. Does an anti-oxidant ascorbic acid improve the condition of hippocampal formation slice preparations? A micro-EEG approach. Int. J. Exp. Pathol. 2012, 93, 406–413. [Google Scholar] [CrossRef] [PubMed]
- Buzsáki, G. Hippocampal sharp wave-ripple: A cognitive biomarker for episodic memory and planning. Hippocampus 2015, 25, 1073–1188. [Google Scholar] [CrossRef]
- Papatheodoropoulos, C. NMDA receptor-dependent high-frequency network oscillations (100–300 Hz) in rat hippocampal slices. Neurosci. Lett. 2007, 414, 197–202. [Google Scholar] [CrossRef]
- Howe, T.; Blockeel, A.J.; Taylor, H.; Jones, M.W.; Bazhenov, M.; Malerba, P. NMDA receptors promote hippocampal sharp-wave ripples and the associated coactivity of CA1 pyramidal cells. Hippocampus 2020, 30, 1356–1370. [Google Scholar] [CrossRef]
- Papatheodoropoulos, C.; Sotiriou, E.; Kotzadimitriou, D.; Drimala, P. At clinically relevant concentrations the anaesthetic/amnesic thiopental but not the anticonvulsant phenobarbital interferes with hippocampal sharp wave-ripple complexes. BMC Neurosci. 2007, 8, 60. [Google Scholar] [CrossRef] [Green Version]
- Koniaris, E.; Drimala, P.; Sotiriou, E.; Papatheodoropoulos, C. Different effects of zolpidem and diazepam on hippocampal sharp wave—Ripple activity in vitro. Neuroscience 2011, 175, 224–234. [Google Scholar] [CrossRef]
- Kranig, S.A.; Duhme, N.; Waldeck, C.; Draguhn, A.; Reichinnek, S.; Both, M. Different functions of hyperpolarization-activated cation channels for hippocampal sharp waves and ripples in vitro. Neuroscience 2013, 228, 325–333. [Google Scholar] [CrossRef] [PubMed]
- Miyawaki, T.; Norimoto, H.; Ishikawa, T.; Watanabe, Y.; Matsuki, N.; Ikegaya, Y. Dopamine Receptor Activation Reorganizes Neuronal Ensembles during Hippocampal Sharp Waves In Vitro. PLoS ONE 2014, 9, e104438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haq, R.U.; Liotta, A.; Kovacs, R.; Rösler, A.; Jarosch, M.; Heinemann, U.; Behrens, C. Adrenergic modulation of sharp wave-ripple activity in rat hippocampal slices. Hippocampus 2012, 22, 516–533. [Google Scholar] [CrossRef] [PubMed]
- Kangisser, L.; Tan, E.; Bellomo, R.; Deane, A.M.; Plummer, M.P. Neuroprotective Properties of Vitamin C: A Scoping Review of Pre-Clinical and Clinical Studies. J. Neurotrauma 2021, 38, 2194–2205. [Google Scholar] [CrossRef]
- Hájos, N.; Mody, I. Establishing a physiological environment for visualized in vitro brain slice recordings by increasing oxygen supply and modifying aCSF content. J. Neurosci. Methods 2009, 183, 107–113. [Google Scholar] [CrossRef] [Green Version]
- González-Ramírez, M.; Razo-Juárez, L.I.; Sauer-Ramírez, J.L.; González-Trujano, M.E.; Salgado-Ceballos, H.; Orozco-Suarez, S. Anticonvulsive effect of vitamin C on pentylenetetrazol-induced seizures in immature rats. Pharmacol. Biochem. Behav. 2010, 97, 267–272. [Google Scholar] [CrossRef]
- Dong, Y.; Wang, S.; Zhang, T.; Zhao, X.; Liu, X.; Cao, L.; Chi, Z. Ascorbic acid ameliorates seizures and brain damage in rats through inhibiting autophagy. Brain Res. 2013, 1535, 115–123. [Google Scholar] [CrossRef]
- Klepper, J.; Vera, J.C.; De Vivo, D.C. Deficient transport of dehydroascorbic acid in the glucose transporter protein syndrome. Ann. Neurol. 1998, 44, 286–287. [Google Scholar] [CrossRef]
- Mi, D.J.; Dixit, S.; Warner, T.A.; Kennard, J.A.; Scharf, D.A.; Kessler, E.S.; Moore, L.M.; Consoli, D.C.; Bown, C.W.; Eugene, A.J.; et al. Altered glutamate clearance in ascorbate deficient mice increases seizure susceptibility and contributes to cognitive impairment in APP/PSEN1 mice. Neurobiol. Aging 2018, 71, 241–254. [Google Scholar] [CrossRef]
- Warner, T.A.; Kang, J.Q.; Kennard, J.A.; Harrison, F.E. Low brain ascorbic acid increases susceptibility to seizures in mouse models of decreased brain ascorbic acid transport and Alzheimer’s disease. Epilepsy Res. 2015, 110, 20–25. [Google Scholar] [CrossRef] [Green Version]
- Harrison, F.E. A critical review of vitamin C for the prevention of age-related cognitive decline and Alzheimer’s disease. J. Alzheimers Dis. 2012, 29, 711–726. [Google Scholar] [CrossRef] [PubMed] [Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Heruye, S.H.; Warren, T.J.; Kostansek IV, J.A.; Draves, S.B.; Matthews, S.A.; West, P.J.; Simeone, K.A.; Simeone, T.A. Ascorbic Acid Reduces Neurotransmission, Synaptic Plasticity, and Spontaneous Hippocampal Rhythms in In Vitro Slices. Nutrients 2022, 14, 613. https://doi.org/10.3390/nu14030613
Heruye SH, Warren TJ, Kostansek IV JA, Draves SB, Matthews SA, West PJ, Simeone KA, Simeone TA. Ascorbic Acid Reduces Neurotransmission, Synaptic Plasticity, and Spontaneous Hippocampal Rhythms in In Vitro Slices. Nutrients. 2022; 14(3):613. https://doi.org/10.3390/nu14030613
Chicago/Turabian StyleHeruye, Segewkal H., Ted J. Warren, Joseph A. Kostansek IV, Samantha B. Draves, Stephanie A. Matthews, Peter J. West, Kristina A. Simeone, and Timothy A. Simeone. 2022. "Ascorbic Acid Reduces Neurotransmission, Synaptic Plasticity, and Spontaneous Hippocampal Rhythms in In Vitro Slices" Nutrients 14, no. 3: 613. https://doi.org/10.3390/nu14030613