
Combined Treatment of Dichloroacetic Acid and Pyruvate Increased Neuronal Survival after Seizure
 ,
,
Abstract
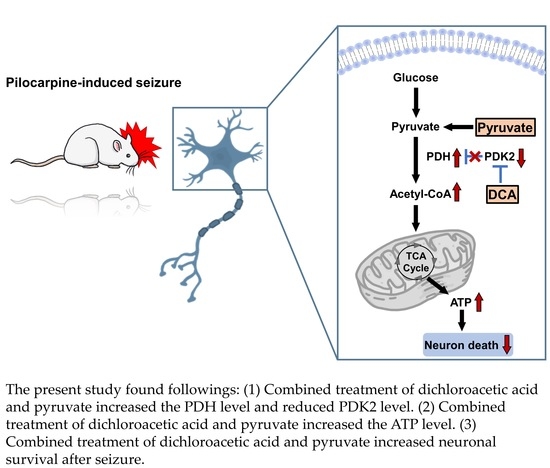
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Experimental Animals
2.3. Seizure Induction
2.4. Dichloroacetic Acid (DCA) and Pyruvate Injection
2.5. Brain Sample Preparation
2.6. Detection of PDH and PDK2
2.7. Detection of Oxidative Injury
2.8. Detection of Microglia and Astrocyte Activation
2.9. Detection of Neuronal Nuclei
2.10. Fluoro-Jade B (FJB) Staining
2.11. Western Blot
2.12. Tape Removal Test (TRT)
2.13. ATP Measurements
2.14. Data Analysis
3. Results
3.1. DCA and Pyruvate Co-Treatment Reduces Excessive PDK2 Levels after Seizures
3.2. DCA and Pyruvate Co-Treatment Increased PDH Levels after Seizure
3.3. DCA and Pyruvate Co-Treatment Decreases Oxidative Stress after Pilocarpine-Induced Seizure
3.4. DCA and Pyruvate Co-Treatment Prevents Inflammatory Reaction after Pilocarpine-Induced Seizure
3.5. DCA and Pyruvate Co-Treatment Increases Surviving Neurons after Pilocarpine-Induced Seizure
3.6. DCA and Pyruvate Co-Treatment Inhibitor Reduces Seizure-Induced Cognitive Impairment
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Chang, B.S.; Lowenstein, D.H. Epilepsy. N. Engl. J. Med. 2003, 349, 1257–1266. [Google Scholar] [CrossRef]
- Ashrafi, M.R.; Shams, S.; Nouri, M.; Mohseni, M.; Shabanian, R.; Yekaninejad, M.S.; Chegini, N.; Khodadad, A.; Safaralizadeh, R. A probable causative factor for an old problem: Selenium and glutathione peroxidase appear to play important roles in epilepsy pathogenesis. Epilepsia 2007, 48, 1750–1755. [Google Scholar] [CrossRef] [PubMed]
- Patel, M. Mitochondrial dysfunction and oxidative stress: Cause and consequence of epileptic seizures. Free Radic Biol. Med. 2004, 37, 1951–1962. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, J., II; Che Has, A.T. The evolution of the pilocarpine animal model of status epilepticus. Heliyon 2020, 6, e04557. [Google Scholar] [CrossRef]
- Martin, E.; Rosenthal, R.E.; Fiskum, G. Pyruvate dehydrogenase complex: Metabolic link to ischemic brain injury and target of oxidative stress. J. Neurosci. Res. 2005, 79, 240–247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, W.; Liu, Q.; Leng, J.; Zheng, Y.; Li, J. The role of Pyruvate Dehydrogenase Complex in cardiovascular diseases. Life Sci. 2015, 121, 97–103. [Google Scholar] [CrossRef] [PubMed]
- Holness, M.J.; Kraus, A.; Harris, R.A.; Sugden, M.C. Targeted upregulation of pyruvate dehydrogenase kinase (PDK)-4 in slow-twitch skeletal muscle underlies the stable modification of the regulatory characteristics of PDK induced by high-fat feeding. Diabetes 2000, 49, 775–781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holness, M.J.; Sugden, M.C. Regulation of pyruvate dehydrogenase complex activity by reversible phosphorylation. Biochem. Soc. Trans. 2003, 31, 1143–1151. [Google Scholar] [CrossRef]
- Peters, S.J.; Harris, R.A.; Heigenhauser, G.J.; Spriet, L.L. Muscle fiber type comparison of PDH kinase activity and isoform expression in fed and fasted rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2001, 280, R661–R668. [Google Scholar] [CrossRef] [Green Version]
- Sugden, M.C.; Bulmer, K.; Augustine, D.; Holness, M.J. Selective modification of pyruvate dehydrogenase kinase isoform expression in rat pancreatic islets elicited by starvation and activation of peroxisome proliferator-activated receptor-alpha: Implications for glucose-stimulated insulin secretion. Diabetes 2001, 50, 2729–2736. [Google Scholar] [CrossRef]
- Di, R.M.; Feng, Q.T.; Chang, Z.; Luan, Q.; Zhang, Y.Y.; Huang, J.; Li, X.L.; Yang, Z.Z. PDK1 plays a critical role in regulating cardiac function in mice and human. Chin. Med. J. 2010, 123, 2358–2363. [Google Scholar] [PubMed]
- Bowker-Kinley, M.M.; Davis, W.I.; Wu, P.; Harris, R.A.; Popov, K.M. Evidence for existence of tissue-specific regulation of the mammalian pyruvate dehydrogenase complex. Biochem. J. 1998, 329 Pt 1, 191–196. [Google Scholar] [CrossRef] [PubMed]
- Wu, P.; Sato, J.; Zhao, Y.; Jaskiewicz, J.; Popov, K.M.; Harris, R.A. Starvation and diabetes increase the amount of pyruvate dehydrogenase kinase isoenzyme 4 in rat heart. Biochem. J. 1998, 329 Pt 1, 197–201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jha, M.K.; Jeon, S.; Suk, K. Pyruvate Dehydrogenase Kinases in the Nervous System: Their Principal Functions in Neuronal-glial Metabolic Interaction and Neuro-metabolic Disorders. Curr. Neuropharmacol. 2012, 10, 393–403. [Google Scholar] [CrossRef] [PubMed]
- Ruggieri, V.; Agriesti, F.; Scrima, R.; Laurenzana, I.; Perrone, D.; Tataranni, T.; Mazzoccoli, C.; Lo Muzio, L.; Capitanio, N.; Piccoli, C. Dichloroacetate, a selective mitochondria-targeting drug for oral squamous cell carcinoma: A metabolic perspective of treatment. Oncotarget 2015, 6, 1217–1230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barshop, B.A.; Naviaux, R.K.; McGowan, K.A.; Levine, F.; Nyhan, W.L.; Loupis-Geller, A.; Haas, R.H. Chronic treatment of mitochondrial disease patients with dichloroacetate. Mol. Genet. Metab. 2004, 83, 138–149. [Google Scholar] [CrossRef] [PubMed]
- Michelakis, E.D.; Webster, L.; Mackey, J.R. Dichloroacetate (DCA) as a potential metabolic-targeting therapy for cancer. Br. J. Cancer 2008, 99, 989–994. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Y.; Li, T.; Xie, C.; Zhang, Y.; Zhou, K.; Wang, X.; Blomgren, K.; Zhu, C. Dichloroacetate treatment improves mitochondrial metabolism and reduces brain injury in neonatal mice. Oncotarget 2016, 7, 31708–31722. [Google Scholar] [CrossRef] [Green Version]
- Stacpoole, P.W.; Kurtz, T.L.; Han, Z.; Langaee, T. Role of dichloroacetate in the treatment of genetic mitochondrial diseases. Adv. Drug Deliv. Rev. 2008, 60, 1478–1487. [Google Scholar] [CrossRef] [Green Version]
- Gray, L.R.; Tompkins, S.C.; Taylor, E.B. Regulation of pyruvate metabolism and human disease. Cell Mol. Life Sci. 2014, 71, 2577–2604. [Google Scholar] [CrossRef]
- Moro, N.; Ghavim, S.S.; Harris, N.G.; Hovda, D.A.; Sutton, R.L. Pyruvate treatment attenuates cerebral metabolic depression and neuronal loss after experimental traumatic brain injury. Brain Res. 2016, 1642, 270–277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.Y.; Kim, Y.H.; Koh, J.Y. Protection by pyruvate against transient forebrain ischemia in rats. J. Neurosci. 2001, 21, RC171. [Google Scholar] [CrossRef]
- Suh, S.W.; Aoyama, K.; Matsumori, Y.; Liu, J.; Swanson, R.A. Pyruvate administered after severe hypoglycemia reduces neuronal death and cognitive impairment. Diabetes 2005, 54, 1452–1458. [Google Scholar] [CrossRef] [Green Version]
- Pereira de Vasconcelos, A.; Ferrandon, A.; Nehlig, A. Local cerebral blood flow during lithium-pilocarpine seizures in the developing and adult rat: Role of coupling between blood flow and metabolism in the genesis of neuronal damage. J. Cereb. Blood Flow Metab. 2002, 22, 196–205. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Lee, D.W.; Choi, B.Y.; Sohn, M.; Lee, S.H.; Choi, H.C.; Song, H.K.; Suh, S.W. Cytidine 5′-diphosphocholine (CDP-choline) adversely effects on pilocarpine seizure-induced hippocampal neuronal death. Brain Res. 2015, 1595, 156–165. [Google Scholar] [CrossRef]
- Persinger, M.A.; Makarec, K.; Bradley, J.C. Characteristics of limbic seizures evoked by peripheral injections of lithium and pilocarpine. Physiol. Behav. 1988, 44, 27–37. [Google Scholar] [CrossRef]
- Racine, R.J.; Gartner, J.G.; Burnham, W.M. Epileptiform activity and neural plasticity in limbic structures. Brain Res. 1972, 47, 262–268. [Google Scholar] [CrossRef]
- Jeong, J.H.; Lee, S.H.; Kho, A.R.; Hong, D.K.; Kang, D.H.; Kang, B.S.; Park, M.K.; Choi, B.Y.; Choi, H.C.; Lim, M.S.; et al. The Transient Receptor Potential Melastatin 7 (TRPM7) Inhibitors Suppress Seizure-Induced Neuron Death by Inhibiting Zinc Neurotoxicity. Int. J. Mol. Sci. 2020, 21, 7897. [Google Scholar] [CrossRef]
- Hong, D.K.; Kho, A.R.; Choi, B.Y.; Lee, S.H.; Jeong, J.H.; Lee, S.H.; Park, K.H.; Park, J.B.; Suh, S.W. Combined Treatment With Dichloroacetic Acid and Pyruvate Reduces Hippocampal Neuronal Death After Transient Cerebral Ischemia. Front. Neurol. 2018, 9, 137. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.H.; Jang, B.G.; Choi, B.Y.; Kim, H.S.; Sohn, M.; Chung, T.N.; Choi, H.C.; Song, H.K.; Suh, S.W. Post-treatment of an NADPH oxidase inhibitor prevents seizure-induced neuronal death. Brain Res. 2013, 1499, 163–172. [Google Scholar] [CrossRef] [PubMed]
- Suh, S.W.; Gum, E.T.; Hamby, A.M.; Chan, P.H.; Swanson, R.A. Hypoglycemic neuronal death is triggered by glucose reperfusion and activation of neuronal NADPH oxidase. J. Clin. Investig. 2007, 117, 910–918. [Google Scholar] [CrossRef] [PubMed]
- Choi, B.Y.; Jang, B.G.; Kim, J.H.; Seo, J.N.; Wu, G.; Sohn, M.; Chung, T.N.; Suh, S.W. Copper/zinc chelation by clioquinol reduces spinal cord white matter damage and behavioral deficits in a murine MOG-induced multiple sclerosis model. Neurobiol. Dis. 2013, 54, 382–391. [Google Scholar] [CrossRef] [PubMed]
- Huang, G.B.; Zhao, T.; Muna, S.S.; Jin, H.M.; Park, J.I.; Jo, K.S.; Lee, B.H.; Chae, S.W.; Kim, S.Y.; Park, S.H.; et al. Therapeutic potential of Gastrodia elata Blume for the treatment of Alzheimer’s disease. Neural Regen. Res. 2013, 8, 1061–1070. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.J.; Lee, H.M.; Choi, S.W.; Oh, S.J.; Kong, S.K.; Lee, I.W. Comparative study of endoscopic and microscopic tympanoplasty performed by a single experienced surgeon. Am. J. Otolaryngol. 2021, 42, 102788. [Google Scholar] [CrossRef] [PubMed]
- McCarty, M.F.; Lerner, A. Nutraceuticals Targeting Generation and Oxidant Activity of Peroxynitrite May Aid Prevention and Control of Parkinson’s Disease. Int. J. Mol. Sci. 2020, 21, 3624. [Google Scholar] [CrossRef] [PubMed]
- Kho, A.R.; Choi, B.Y.; Lee, S.H.; Hong, D.K.; Lee, S.H.; Jeong, J.H.; Park, K.H.; Song, H.K.; Choi, H.C.; Suh, S.W. Effects of Protocatechuic Acid (PCA) on Global Cerebral Ischemia-Induced Hippocampal Neuronal Death. Int. J. Mol. Sci. 2018, 19, 1420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Čakar, U.; Čolović, M.; Milenković, D.; Medić, B.; Krstić, D.; Petrović, A.; Đorđević, B. Protective effects of fruit wines against hydrogen peroxide—Induced oxidative stress in rat synaptosomes. Agronomy 2021, 11, 1414. [Google Scholar] [CrossRef]
- Kirkley, K.S.; Popichak, K.A.; Afzali, M.F.; Legare, M.E.; Tjalkens, R.B. Microglia amplify inflammatory activation of astrocytes in manganese neurotoxicity. J. Neuroinflamm. 2017, 14, 99. [Google Scholar] [CrossRef] [Green Version]
- Melchior, B.; Puntambekar, S.S.; Carson, M.J. Microglia and the control of autoreactive T cell responses. Neurochem. Int. 2006, 49, 145–153. [Google Scholar] [CrossRef] [Green Version]
- Hanisch, U.K.; Kettenmann, H. Microglia: Active sensor and versatile effector cells in the normal and pathologic brain. Nat. Neurosci. 2007, 10, 1387–1394. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, E.C.; Hunot, S. Neuroinflammation in Parkinson’s disease: A target for neuroprotection? Lancet Neurol. 2009, 8, 382–397. [Google Scholar] [CrossRef]
- Calcutt, N.A.; Lopez, V.L.; Bautista, A.D.; Mizisin, L.M.; Torres, B.R.; Shroads, A.L.; Mizisin, A.P.; Stacpoole, P.W. Peripheral neuropathy in rats exposed to dichloroacetate. J. Neuropathol. Exp. Neurol. 2009, 68, 985–993. [Google Scholar] [CrossRef] [PubMed]
- Moser, V.C.; Phillips, P.M.; McDaniel, K.L.; MacPhail, R.C. Behavioral evaluation of the neurotoxicity produced by dichloroacetic acid in rats. Neurotoxicol. Teratol. 1999, 21, 719–731. [Google Scholar] [CrossRef]
- Sun, X.Q.; Zhang, R.; Zhang, H.D.; Yuan, P.; Wang, X.J.; Zhao, Q.H.; Wang, L.; Jiang, R.; Jan Bogaard, H.; Jing, Z.C. Reversal of right ventricular remodeling by dichloroacetate is related to inhibition of mitochondria-dependent apoptosis. Hypertens Res. 2016, 39, 302–311. [Google Scholar] [CrossRef]
- Staneviciute, J.; Jukneviciene, M.; Palubinskiene, J.; Balnyte, I.; Valanciute, A.; Vosyliute, R.; Suziedelis, K.; Lesauskaite, V.; Stakisaitis, D. Sodium Dichloroacetate Pharmacological Effect as Related to Na-K-2Cl Cotransporter Inhibition in Rats. Dose Response 2018, 16, 1559325818811522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Liu, J.; Hu, H.; Lu, S.; Lu, Q.; Quan, N.; Rousselle, T.; Patel, M.S.; Li, J. Dichloroacetate Ameliorates Cardiac Dysfunction Caused by Ischemic Insults Through AMPK Signal Pathway-Not Only Shifts Metabolism. Toxicol. Sci. 2019, 167, 604–617. [Google Scholar] [CrossRef] [PubMed]
- Kho, A.R.; Choi, B.Y.; Lee, S.H.; Hong, D.K.; Jeong, J.H.; Kang, B.S.; Kang, D.H.; Park, K.H.; Park, J.B.; Suh, S.W. The Effects of Sodium Dichloroacetate on Mitochondrial Dysfunction and Neuronal Death Following Hypoglycemia-Induced Injury. Cells 2019, 8, 405. [Google Scholar] [CrossRef] [Green Version]
- Stacpoole, P.W.; Henderson, G.N.; Yan, Z.; James, M.O. Clinical pharmacology and toxicology of dichloroacetate. Environ. Health Perspect. 1998, 106 (Suppl. 4), 989–994. [Google Scholar] [CrossRef]
- Dingledine, R.; Varvel, N.H.; Dudek, F.E. When and how do seizures kill neurons, and is cell death relevant to epileptogenesis? Adv. Exp. Med. Biol. 2014, 813, 109–122. [Google Scholar] [CrossRef] [Green Version]
- Yaari, Y.; Selzer, M.E.; Pincus, J.H. Phenytoin: Mechanisms of its anticonvulsant action. Ann. Neurol. 1986, 20, 171–184. [Google Scholar] [CrossRef]
- Balagura, G.; Iapadre, G.; Verrotti, A.; Striano, P. Moving beyond sodium valproate: Choosing the right anti-epileptic drug in children. Expert Opin. Pharm. 2019, 20, 1449–1456. [Google Scholar] [CrossRef]
- Kelly, K.M. Gabapentin. Antiepileptic mechanism of action. Neuropsychobiology 1998, 38, 139–144. [Google Scholar] [CrossRef] [PubMed]
- Schachter, S.C. A review of the antiepileptic drug tiagabine. Clin. Neuropharmacol. 1999, 22, 312–317. [Google Scholar]
- Genton, P.; Van Vleymen, B. Piracetam and levetiracetam: Close structural similarities but different pharmacological and clinical profiles. Epileptic Disord. 2000, 2, 99–105. [Google Scholar] [PubMed]
- Fischer, W.; Kittner, H.; Regenthal, R.; Russo, E.; De Sarro, G. Effects of piracetam alone and in combination with antiepileptic drugs in rodent seizure models. J. Neural. Transm 2004, 111, 1121–1139. [Google Scholar] [CrossRef] [PubMed]
- Hovinga, C.A. Levetiracetam: A novel antiepileptic drug. Pharmacotherapy 2001, 21, 1375–1388. [Google Scholar] [CrossRef] [PubMed]
- Rapport, R.L., 2nd; Harris, A.B.; Friel, P.N.; Ojemann, G.A. Human epileptic brain Na, K ATPase activity and phenytoin concentrations. Arch. Neurol. 1975, 32, 549–554. [Google Scholar] [CrossRef]
- Nagy, A.K.; Houser, C.R.; Delgado-Escueta, A.V. Synaptosomal ATPase activities in temporal cortex and hippocampal formation of humans with focal epilepsy. Brain Res. 1990, 529, 192–201. [Google Scholar] [CrossRef]
- Fei, Y.; Shi, R.; Song, Z.; Wu, J. Metabolic Control of Epilepsy: A Promising Therapeutic Target for Epilepsy. Front. Neurol. 2020, 11, 592514. [Google Scholar] [CrossRef]
- Kovac, S.; Dinkova Kostova, A.T.; Herrmann, A.M.; Melzer, N.; Meuth, S.G.; Gorji, A. Metabolic and Homeostatic Changes in Seizures and Acquired Epilepsy-Mitochondria, Calcium Dynamics and Reactive Oxygen Species. Int. J. Mol. Sci. 2017, 18, 1935. [Google Scholar] [CrossRef] [Green Version]
- Kovac, S.; Domijan, A.M.; Walker, M.C.; Abramov, A.Y. Prolonged seizure activity impairs mitochondrial bioenergetics and induces cell death. J. Cell Sci. 2012, 125, 1796–1806. [Google Scholar] [CrossRef]
- Kovac, S.; Abramov, A.Y.; Walker, M.C. Energy depletion in seizures: Anaplerosis as a strategy for future therapies. Neuropharmacology 2013, 69, 96–104. [Google Scholar] [CrossRef] [PubMed]
- Campbell, I.; Campbell, H. A pyruvate dehydrogenase complex disorder hypothesis for bipolar disorder. Med. Hypotheses 2019, 130, 109263. [Google Scholar] [CrossRef] [PubMed]
- Lucchi, C.; Marchio, M.; Caramaschi, E.; Giordano, C.; Giordano, R.; Guerra, A.; Biagini, G. Electrographic Changes Accompanying Recurrent Seizures under Ketogenic Diet Treatment. Pharmaceuticals 2017, 10, 82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yokobori, S.; Mazzeo, A.T.; Gajavelli, S.; Bullock, M.R. Mitochondrial neuroprotection in traumatic brain injury: Rationale and therapeutic strategies. CNS Neurol. Disord Drug Targets 2014, 13, 606–619. [Google Scholar] [CrossRef]
- Fiskum, G.; Rosenthal, R.E.; Vereczki, V.; Martin, E.; Hoffman, G.E.; Chinopoulos, C.; Kowaltowski, A. Protection against ischemic brain injury by inhibition of mitochondrial oxidative stress. J. Bioenerg. Biomembr. 2004, 36, 347–352. [Google Scholar] [CrossRef]
- Zsurka, G.; Kunz, W.S. Mitochondrial dysfunction and seizures: The neuronal energy crisis. Lancet Neurol. 2015, 14, 956–966. [Google Scholar] [CrossRef]
- Lazzarino, G.; Amorini, A.M.; Signoretti, S.; Musumeci, G.; Lazzarino, G.; Caruso, G.; Pastore, F.S.; Di Pietro, V.; Tavazzi, B.; Belli, A. Pyruvate Dehydrogenase and Tricarboxylic Acid Cycle Enzymes Are Sensitive Targets of Traumatic Brain Injury Induced Metabolic Derangement. Int. J. Mol. Sci. 2019, 20, 5774. [Google Scholar] [CrossRef] [Green Version]
- Roche, T.E.; Hiromasa, Y. Pyruvate dehydrogenase kinase regulatory mechanisms and inhibition in treating diabetes, heart ischemia, and cancer. Cell Mol. Life Sci. 2007, 64, 830–849. [Google Scholar] [CrossRef]
- Kovac, S.; Domijan, A.M.; Walker, M.C.; Abramov, A.Y. Seizure activity results in calcium- and mitochondria-independent ROS production via NADPH and xanthine oxidase activation. Cell Death Dis. 2014, 5, e1442. [Google Scholar] [CrossRef] [Green Version]
- Puttachary, S.; Sharma, S.; Stark, S.; Thippeswamy, T. Seizure-induced oxidative stress in temporal lobe epilepsy. Biomed. Res. Int. 2015, 2015, 745613. [Google Scholar] [CrossRef] [PubMed]
- Olowe, R.; Sandouka, S.; Saadi, A.; Shekh-Ahmad, T. Approaches for Reactive Oxygen Species and Oxidative Stress Quantification in Epilepsy. Antioxidants 2020, 9, 990. [Google Scholar] [CrossRef]
- Lai, A.Y.; Todd, K.G. Microglia in cerebral ischemia: Molecular actions and interactions. Can. J. Physiol. Pharmacol. 2006, 84, 49–59. [Google Scholar] [CrossRef]
- Garabadu, D.; Agrawal, N.; Sharma, A.; Sharma, S. Mitochondrial metabolism: A common link between neuroinflammation and neurodegeneration. Behav. Pharmacol. 2019, 30, 642–652. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, L.A.; Hardie, D.G. Metabolism of inflammation limited by AMPK and pseudo-starvation. Nature 2013, 493, 346–355. [Google Scholar] [CrossRef] [PubMed]
- Gelderblom, M.; Leypoldt, F.; Steinbach, K.; Behrens, D.; Choe, C.U.; Siler, D.A.; Arumugam, T.V.; Orthey, E.; Gerloff, C.; Tolosa, E.; et al. Temporal and spatial dynamics of cerebral immune cell accumulation in stroke. Stroke 2009, 40, 1849–1857. [Google Scholar] [CrossRef] [Green Version]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Munch, A.E.; Chung, W.S.; Peterson, T.C.; et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stevens, S.L.; Bao, J.; Hollis, J.; Lessov, N.S.; Clark, W.M.; Stenzel-Poore, M.P. The use of flow cytometry to evaluate temporal changes in inflammatory cells following focal cerebral ischemia in mice. Brain Res. 2002, 932, 110–119. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, S.H.; Choi, B.Y.; Kho, A.R.; Hong, D.K.; Kang, B.S.; Park, M.K.; Lee, S.H.; Choi, H.C.; Song, H.K.; Suh, S.W. Combined Treatment of Dichloroacetic Acid and Pyruvate Increased Neuronal Survival after Seizure. Nutrients 2022, 14, 4804. https://doi.org/10.3390/nu14224804
Lee SH, Choi BY, Kho AR, Hong DK, Kang BS, Park MK, Lee SH, Choi HC, Song HK, Suh SW. Combined Treatment of Dichloroacetic Acid and Pyruvate Increased Neuronal Survival after Seizure. Nutrients. 2022; 14(22):4804. https://doi.org/10.3390/nu14224804
Chicago/Turabian StyleLee, Song Hee, Bo Young Choi, A Ra Kho, Dae Ki Hong, Beom Seok Kang, Min Kyu Park, Si Hyun Lee, Hui Chul Choi, Hong Ki Song, and Sang Won Suh. 2022. "Combined Treatment of Dichloroacetic Acid and Pyruvate Increased Neuronal Survival after Seizure" Nutrients 14, no. 22: 4804. https://doi.org/10.3390/nu14224804