
Adipose Tissue Dysfunction in Obesity: Role of Mineralocorticoid Receptor

 , ,
, , 
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
3. Adipose Tissue
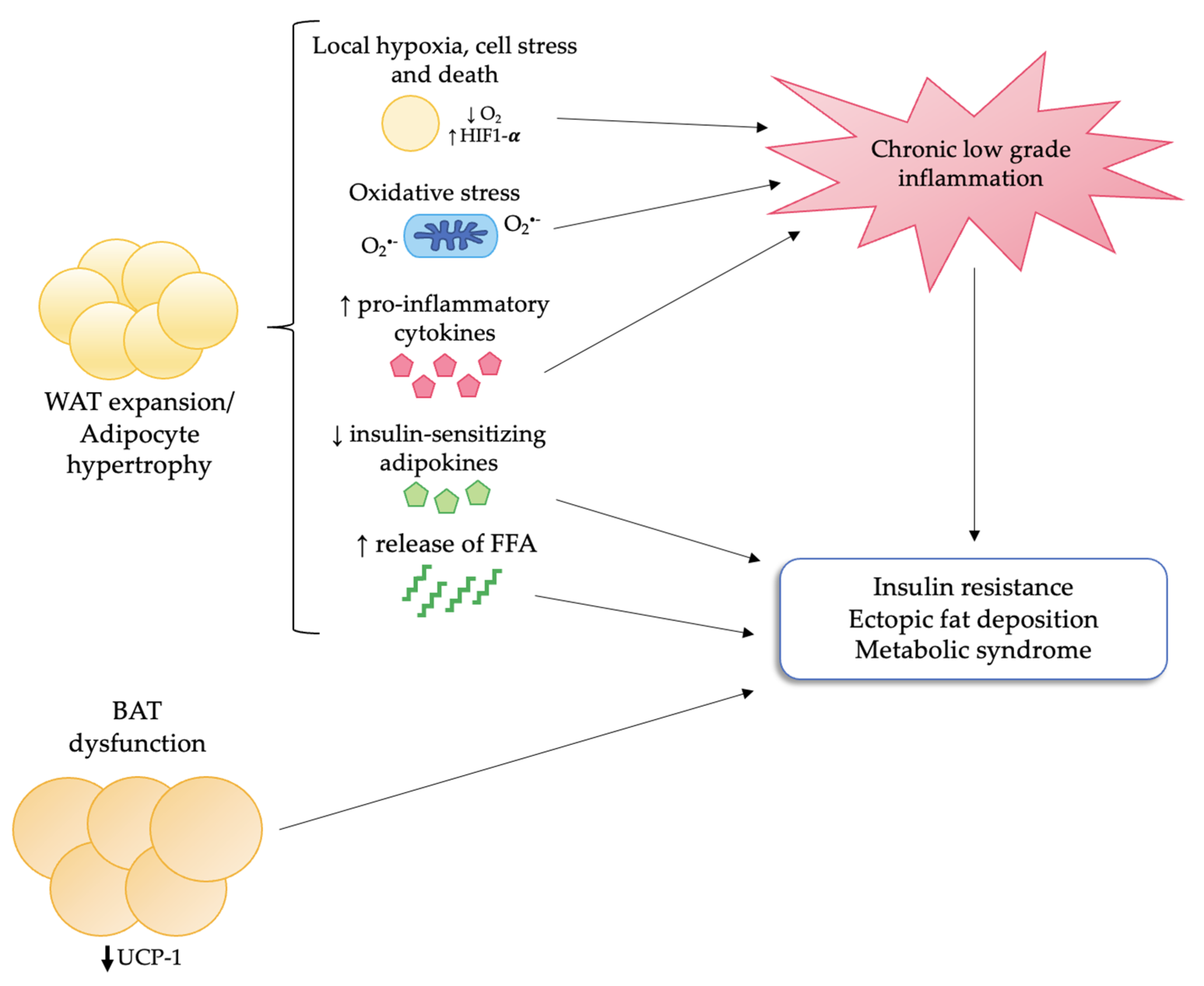
4. Adipose Tissue Dysfunction in Obesity
5. MR and Adipose Tissue Dysfunction
5.1. MR and Inflammation
5.2. MR and Oxidative Stress
5.3. MR Activation in BAT
5.4. MR, Insulin Resistance and Metabolic Syndrome
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- WHO. Obesity: Preventing and Managing the Global Epidemic: Report of a WHO Consultation. World Health Organ. Technol Rep. Ser. 2000, 894, i–xii, 1–253. [Google Scholar]
- Jastreboff, A.M.; Kotz, C.M.; Kahan, S.; Kelly, A.S.; Heymsfield, S.B. Obesity as a Disease: The Obesity Society 2018 Position Statement. Obesity 2018, 27, 7–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cawley, J.; Meyerhoefer, C. The medical care costs of obesity: An instrumental variables approach. J. Health Econ. 2012, 31, 219–230. [Google Scholar] [CrossRef] [PubMed]
- Cawley, J.; Biener, A.; Meyerhoefer, C.; Ding, Y.; Zvenyach, T.; Smolarz, B.G.; Ramasamy, A. Direct medical costs of obesity in the United States and the most populous states. J. Manag. Care Spéc. Pharm. 2021, 27, 354–366. [Google Scholar] [CrossRef]
- Finucane, M.M.; Stevens, G.A.; Cowan, M.J.; Danaei, G.; Lin, J.K.; Paciorek, C.J.; Singh, G.M.; Gutierrez, H.R.; Lu, Y.; Bahalim, A.N.; et al. National, regional, and global trends in body-mass index since 1980: Systematic analysis of health examination surveys and epidemiological studies with 960 country-years and 9.1 million participants. Lancet 2011, 377, 557–567. [Google Scholar] [CrossRef] [Green Version]
- Flegal, K.M.; Carroll, M.D.; Kit, B.K.; Ogden, C.L. Prevalence of Obesity and Trends in the Distribution of Body Mass Index among US Adults, 1999–2010. JAMA 2012, 307, 491–497. [Google Scholar] [CrossRef] [Green Version]
- Garvey, W.T.; Mechanick, J.I.; Brett, E.M.; Garber, A.J.; Hurley, D.L.; Jastreboff, A.M.; Nadolsky, K.; Pessah-Pollack, R.; Plodkowski, R.; Reviewers of the AACE/ACE Obesity Clinical Practice Guidelines. American Association of Clinical Endocrinologists and American College of Endocrinology Comprehensive Clinical Practice Guidelines for Medical Care of Patients with Obesity. Endocr. Pract. 2016, 22, 1–203. [Google Scholar] [CrossRef] [Green Version]
- Kawai, T.; Autieri, M.V.; Scalia, R. Adipose tissue inflammation and metabolic dysfunction in obesity. Am. J. Physiol. Physiol. 2021, 320, C375–C391. [Google Scholar] [CrossRef]
- Longo, M.; Zatterale, F.; Naderi, J.; Parrillo, L.; Formisano, P.; Raciti, G.A.; Beguinot, F.; Miele, C. Adipose Tissue Dysfunction as Determinant of Obesity-Associated Metabolic Complications. Int. J. Mol. Sci. 2019, 20, 2358. [Google Scholar] [CrossRef] [Green Version]
- Reyes-Farias, M.; Fos-Domenech, J.; Serra, D.; Herrero, L.; Sánchez-Infantes, D. White adipose tissue dysfunction in obesity and aging. Biochem. Pharmacol. 2021, 192, 114723. [Google Scholar] [CrossRef]
- Bo, S.; Ciccone, G.; Pearce, N.; Merletti, F.; Gentile, L.; Cassader, M.; Pagano, G. Prevalence of undiagnosed metabolic syndrome in a population of adult asymptomatic subjects. Diabetes Res. Clin. Pract. 2007, 75, 362–365. [Google Scholar] [CrossRef] [PubMed]
- Bo, S.; Musso, G.; Gambino, R.; Villois, P.; Gentile, L.; Durazzo, M.; Cavallo-Perin, P.; Cassader, M. Prognostic implications for insulin-sensitive and insulin-resistant normal-weight and obese individuals from a population-based cohort. Am. J. Clin. Nutr. 2012, 96, 962–969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Indulekha, K.; Anjana, R.M.; Surendar, J.; Mohan, V. Association of visceral and subcutaneous fat with glucose intolerance, insulin resistance, adipocytokines and inflammatory markers in Asian Indians (CURES-113). Clin. Biochem. 2011, 44, 281–287. [Google Scholar] [CrossRef] [PubMed]
- Lavagnino, L.; Amianto, F.; Parasiliti-Caprino, M.; Maccario, M.; Arvat, E.; Ghigo, E.; Abbate Daga, G.; Fassino, S. Urinary cortisol and psychopathology in obese binge eating subjects. Appetite. 2014, 83, 112–116. [Google Scholar] [CrossRef] [PubMed]
- Powell-Wiley, T.M.; Poirier, P.; Burke, L.E.; Després, J.-P.; Gordon-Larsen, P.; Lavie, C.J.; Lear, S.A.; Ndumele, C.E.; Neeland, I.J.; Sanders, P.; et al. Obesity and Cardiovascular Disease: A Scientific Statement From the American Heart Association. Circulation 2021, 143, e984–e1010. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Hu, T.; Zhang, S.; Zhou, L. Associations of Different Adipose Tissue Depots with Insulin Resistance: A Systematic Review and Meta-analysis of Observational Studies. Sci. Rep. 2015, 5, 18495. [Google Scholar] [CrossRef] [Green Version]
- Briet, M.; Schiffrin, E. Aldosterone: Effects on the kidney and cardiovascular system. Nat. Rev. Nephrol. 2010, 6, 261–273. [Google Scholar] [CrossRef]
- Gomez-Sanchez, E.; Gomez-Sanchez, C.E. The Multifaceted Mineralocorticoid Receptor. Compr. Physiol. 2014, 4, 965–994. [Google Scholar] [CrossRef] [Green Version]
- Brown, N.J. Contribution of aldosterone to cardiovascular and renal inflammation and fibrosis. Nat. Rev. Nephrol. 2013, 9, 459–469. [Google Scholar] [CrossRef]
- Bollati, M.; Lopez, C.; Bioletto, F.; Ponzetto, F.; Ghigo, E.; Maccario, M.; Parasiliti-Caprino, M. Atrial Fibrillation and Aortic Ectasia as Complications of Primary Aldosteronism: Focus on Pathophysiological Aspects. Int. J. Mol. Sci. 2022, 23, 2111. [Google Scholar] [CrossRef]
- Bioletto, F.; Bollati, M.; Lopez, C.; Arata, S.; Procopio, M.; Ponzetto, F.; Ghigo, E.; Maccario, M.; Parasiliti-Caprino, M. Primary Aldosteronism and Resistant Hypertension: A Pathophysiological Insight. Int. J. Mol. Sci. 2022, 23, 4803. [Google Scholar] [CrossRef] [PubMed]
- Urbanet, R.; Cat, A.N.D.; Feraco, A.; Venteclef, N.; El Mogrhabi, S.; Sierra-Ramos, C.; de la Rosa, D.A.; Adler, G.K.; Quilliot, D.; Rossignol, P.; et al. Adipocyte Mineralocorticoid Receptor Activation Leads to Metabolic Syndrome and Induction of Prostaglandin D2 Synthase. Hypertension 2015, 66, 149–157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirata, A.; Maeda, N.; Nakatsuji, H.; Hiuge-Shimizu, A.; Okada, T.; Funahashi, T.; Shimomura, I. Contribution of glucocorticoid–mineralocorticoid receptor pathway on the obesity-related adipocyte dysfunction. Biochem. Biophys. Res. Commun. 2012, 419, 182–187. [Google Scholar] [CrossRef] [PubMed]
- Marzolla, V.; Armani, A.; Feraco, A.; De Martino, M.U.; Fabbri, A.; Rosano, G.; Caprio, M. Mineralocorticoid receptor in adipocytes and macrophages: A promising target to fight metabolic syndrome. Steroids 2014, 91, 46–53. [Google Scholar] [CrossRef] [PubMed]
- Marzolla, V.; Armani, A.; Zennaro, M.-C.; Cinti, F.; Mammi, C.; Fabbri, A.; Rosano, G.M.; Caprio, M. The role of the mineralocorticoid receptor in adipocyte biology and fat metabolism. Mol. Cell. Endocrinol. 2011, 350, 281–288. [Google Scholar] [CrossRef]
- Infante, M.; Armani, A.; Marzolla, V.; Fabbri, A.; Caprio, M. Adipocyte Mineralocorticoid Receptor. Vitam. Horm. 2018, 109, 189–209. [Google Scholar] [CrossRef]
- Hirsch, J.; Fried, S.K.; Edens, N.K.; Leibel, R.L. The Fat Cell. Med. Clin. North Am. 1989, 73, 83–96. [Google Scholar] [CrossRef]
- Kershaw, E.E.; Flier, J.S. Adipose Tissue as an Endocrine Organ. J. Clin. Endocrinol. Metab. 2004, 89, 2548–2556. [Google Scholar] [CrossRef]
- Coelho, M.; Oliveira, T.; Fernandes, R. Biochemistry of adipose tissue: An endocrine organ. Arch. Med. Sci. 2013, 9, 191–200. [Google Scholar] [CrossRef] [Green Version]
- Cinti, S. The adipose organ. Prostaglandins Leukot. Essent. Fat. Acids 2005, 73, 9–15. [Google Scholar] [CrossRef]
- Cinti, S. The adipose organ at a glance. Dis. Model. Mech. 2012, 5, 588–594. [Google Scholar] [CrossRef] [PubMed]
- Harms, M.; Seale, P. Brown and beige fat: Development, function and therapeutic potential. Nat. Med. 2013, 19, 1252–1263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fox, C.S.; Massaro, J.M.; Hoffmann, U.; Pou, K.M.; Maurovich-Horvat, P.; Liu, C.-Y.; Vasan, R.S.; Murabito, J.M.; Meigs, J.B.; Cupples, L.A.; et al. Abdominal Visceral and Subcutaneous Adipose Tissue Compartments: Association with metabolic risk factors in the Framingham Heart Study. Circulation 2007, 116, 39–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, P.; Abate, N. Body Fat Distribution and Insulin Resistance. Nutrients 2013, 5, 2019–2027. [Google Scholar] [CrossRef] [PubMed]
- Sam, S. Differential effect of subcutaneous abdominal and visceral adipose tissue on cardiometabolic risk. Horm. Mol. Biol. Clin. Investig. 2018, 33, 1–9. [Google Scholar] [CrossRef]
- McLaughlin, T.; Lamendola, C.; Liu, A.; Abbasi, F. Preferential Fat Deposition in Subcutaneous Versus Visceral Depots Is Associated with Insulin Sensitivity. J. Clin. Endocrinol. Metab. 2011, 96, E1756–E1760. [Google Scholar] [CrossRef] [Green Version]
- Farkhondeh, T.; Llorens, S.; Pourbagher-Shahri, A.M.; Ashrafizadeh, M.; Talebi, M.; Shakibaei, M.; Samarghandian, S. An Overview of the Role of Adipokines in Cardiometabolic Diseases. Molecules 2020, 25, 5218. [Google Scholar] [CrossRef]
- Zorena, K.; Jachimowicz-Duda, O.; Ślęzak, D.; Robakowska, M.; Mrugacz, M. Adipokines and Obesity. Potential Link to Metabolic Disorders and Chronic Complications. Int. J. Mol. Sci. 2020, 21, 3570. [Google Scholar] [CrossRef]
- Zhang, Y.; Proenca, R.; Maffei, M.; Barone, M.; Leopold, L.; Friedman, J.M. Positional cloning of the mouse obese gene and its human homologue. Nature 1994, 372, 425–432. [Google Scholar] [CrossRef]
- Cypess, A.M.; Lehman, S.; Williams, G.; Tal, I.; Rodman, D.; Goldfine, A.B.; Kuo, F.C.; Palmer, E.L.; Tseng, Y.-H.; Doria, A.; et al. Identification and Importance of Brown Adipose Tissue in Adult Humans. N. Engl. J. Med. 2009, 360, 1509–1517. [Google Scholar] [CrossRef] [Green Version]
- Nedergaard, J.; Bengtsson, T.; Cannon, B. Unexpected evidence for active brown adipose tissue in adult humans. Am. J. Physiol. Metab. 2007, 293, E444–E452. [Google Scholar] [CrossRef] [PubMed]
- Saito, M.; Okamatsu-Ogura, Y.; Matsushita, M.; Watanabe, K.; Yoneshiro, T.; Nio-Kobayashi, J.; Iwanaga, T.; Miyagawa, M.; Kameya, T.; Nakada, K.; et al. High incidence of metabolically active brown adipose tissue in healthy adult humans: Effects of cold exposure and adiposity. Diabetes 2009, 58, 1526–1531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Marken Lichtenbelt, W.D.; Vanhommerig, J.W.; Smulders, N.M.; Drossaerts, J.M.A.F.L.; Kemerink, G.J.; Bouvy, N.D.; Schrauwen, P.; Teule, G.J.J. Cold-Activated Brown Adipose Tissue in Healthy Men. N. Engl. J. Med. 2009, 360, 1500–1508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Virtanen, K.A.; Lidell, M.E.; Orava, J.; Heglind, M.; Westergren, R.; Niemi, T.; Taittonen, M.; Laine, J.; Savisto, N.-J.; Enerbäck, S.; et al. Functional Brown Adipose Tissue in Healthy Adults. N. Engl. J. Med. 2009, 360, 1518–1525. [Google Scholar] [CrossRef] [PubMed]
- Marlatt, K.L.; Ravussin, E. Brown Adipose Tissue: An Update on Recent Findings. Curr. Obes. Rep. 2017, 6, 389–396. [Google Scholar] [CrossRef]
- Catrysse, L.; van Loo, G. Adipose tissue macrophages and their polarization in health and obesity. Cell. Immunol. 2018, 330, 114–119. [Google Scholar] [CrossRef]
- Ahmed, B.; Sultana, R.; Greene, M.W. Adipose tissue and insulin resistance in obese. Biomed. Pharmacother. 2021, 137, 111315. [Google Scholar] [CrossRef]
- Ferrara, D.; Montecucco, F.; Dallegri, F.; Carbone, F. Impact of different ectopic fat depots on cardiovascular and metabolic diseases. J. Cell. Physiol. 2019, 234, 21630–21641. [Google Scholar] [CrossRef]
- Briones, A.M.; Cat, A.N.D.; Callera, G.E.; Yogi, A.; Burger, D.; He, Y.; Corrêa, J.W.; Gagnon, A.M.; Gomez-Sanchez, C.E.; Gomez-Sanchez, E.P.; et al. Adipocytes Produce Aldosterone Through Calcineurin-Dependent Signaling Pathways. Hypertension 2012, 59, 1069–1078. [Google Scholar] [CrossRef] [Green Version]
- Lefranc, C.; Friederich-Persson, M.; Foufelle, F.; Cat, A.N.D.; Jaisser, F. Adipocyte-Mineralocorticoid Receptor Alters Mitochondrial Quality Control Leading to Mitochondrial Dysfunction and Senescence of Visceral Adipose Tissue. Int. J. Mol. Sci. 2021, 22, 2881. [Google Scholar] [CrossRef]
- Caprio, M.; Fève, B.; Claës, A.; Viengchareun, S.; Lombès, M.; Zennaro, M.-C. Pivotal role of the mineralocorticoid receptor in corticosteroid-induced adipogenesis. FASEB J. 2007, 21, 2185–2194. [Google Scholar] [CrossRef] [PubMed]
- Hoppmann, J.; Perwitz, N.; Meier, B.; Fasshauer, M.; Hadaschik, D.; Lehnert, H.; Klein, J. The balance between gluco- and mineralo-corticoid action critically determines inflammatory adipocyte responses. J. Endocrinol. 2009, 204, 153–164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirata, A.; Maeda, N.; Hiuge, A.; Hibuse, T.; Fujita, K.; Okada, T.; Kihara, S.; Funahashi, T.; Shimomura, I. Blockade of mineralocorticoid receptor reverses adipocyte dysfunction and insulin resistance in obese mice. Cardiovasc. Res. 2009, 84, 164–172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pradhan, A.D.; Manson, J.E.; Rifai, N.; Buring, J.E.; Ridker, P.M. C-Reactive Protein, Interleukin 6, and Risk of Developing Type 2 Diabetes Mellitus. JAMA 2001, 286, 327–334. [Google Scholar] [CrossRef] [PubMed]
- Hardy, O.T.; Perugini, R.A.; Nicoloro, S.M.; Gallagher-Dorval, K.; Puri, V.; Straubhaar, J.; Czech, M.P. Body mass index-independent inflammation in omental adipose tissue associated with insulin resistance in morbid obesity. Surg. Obes. Relat. Dis. 2011, 7, 60–67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bo, S.; Gentile, L.; Ciccone, G.; Baldi, C.; Benini, L.; Dusio, F.; Lucia, C.; Forastiere, G.; Nuti, C.; Cassader, M.; et al. The metabolic syndrome and high C-reactive protein: Prevalence and differences by sex in a southern-European population-based cohort. Diabetes/Metabolism Res. Rev. 2005, 21, 515–524. [Google Scholar] [CrossRef] [PubMed]
- Skurk, T.; Alberti-Huber, C.; Herder, C.; Hauner, H. Relationship between Adipocyte Size and Adipokine Expression and Secretion. J. Clin. Endocrinol. Metab. 2007, 92, 1023–1033. [Google Scholar] [CrossRef]
- Murano, I.; Barbatelli, G.; Parisani, V.; Latini, C.; Muzzonigro, G.; Castellucci, M.; Cinti, S. Dead adipocytes, detected as crown-like structures, are prevalent in visceral fat depots of genetically obese mice. J. Lipid Res. 2008, 49, 1562–1568. [Google Scholar] [CrossRef] [Green Version]
- Russo, L.; Lumeng, C.N. Properties and functions of adipose tissue macrophages in obesity. Immunology 2018, 155, 407–417. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.; Wood, I.S.; Trayhurn, P. Dysregulation of the expression and secretion of inflammation-related adipokines by hypoxia in human adipocytes. 2007, 455, 479–492. Pflügers Arch.-Eur. J. Physiol. [CrossRef] [Green Version]
- Xu, L.; Yan, X.; Zhao, Y.; Wang, J.; Liu, B.; Yu, S.; Fu, J.; Liu, Y.; Su, J. Macrophage Polarization Mediated by Mitochondrial Dysfunction Induces Adipose Tissue Inflammation in Obesity. Int. J. Mol. Sci. 2022, 23, 9252. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Ricchiuti, V.; Lian, B.Q.; Yao, T.M.; Coutinho, P.; Romero, J.R.; Li, J.; Williams, G.H.; Adler, G.K. Mineralocorticoid Receptor Blockade Reverses Obesity-Related Changes in Expression of Adiponectin, Peroxisome Proliferator-Activated Receptor-γ, and Proinflammatory Adipokines. Circulation 2008, 117, 2253–2261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Łabuzek, K.; Liber, S.; Buldak, L.; Machnik, G.; Liber, J.; Okopień, B. Eplerenone promotes alternative activation in human monocyte-derived macrophages. Pharmacol. Rep. 2013, 65, 226–234. [Google Scholar] [CrossRef]
- Wada, T.; Ishikawa, A.; Watanabe, E.; Nakamura, Y.; Aruga, Y.; Hasegawa, H.; Onogi, Y.; Honda, H.; Nagai, Y.; Takatsu, K.; et al. Eplerenone prevented obesity-induced inflammasome activation and glucose intolerance. J. Endocrinol. 2017, 235, 179–191. [Google Scholar] [CrossRef] [PubMed]
- Ray, P.D.; Huang, B.-W.; Tsuji, Y. Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell. Signal. 2012, 24, 981–990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finkel, T.; Holbrook, N.J. Oxidants, oxidative stress and the biology of ageing. Nature 2000, 408, 239–247. [Google Scholar] [CrossRef]
- Förstermann, U.; Xia, N.; Li, H. Roles of Vascular Oxidative Stress and Nitric Oxide in the Pathogenesis of Atherosclerosis. Circ. Res. 2017, 120, 713–735. [Google Scholar] [CrossRef]
- Piconi, L.; Quagliaro, L.; Ceriello, A. Oxidative Stress in Diabetes. Clin. Chem. Lab. Med. (CCLM) 2003, 41, 1144–1149. [Google Scholar] [CrossRef]
- Darenskaya, M.A.; Kolesnikova, L.I. Oxidative Stress: Pathogenetic Role in Diabetes Mellitus and Its Complications and Therapeutic Approaches to Correction. Bull. Exp. Biol. Med. 2021, 171, 179–189. [Google Scholar] [CrossRef]
- Cheung, E.C.; Vousden, K.H. The role of ROS in tumour development and progression. Nat. Cancer 2022, 22, 280–297. [Google Scholar] [CrossRef]
- Furukawa, S.; Fujita, T.; Shimabukuro, M.; Iwaki, M.; Yamada, Y.; Nakajima, Y.; Nakayama, O.; Makishima, M.; Matsuda, M.; Shimomura, I. Increased oxidative stress in obesity and its impact on metabolic syndrome. J. Clin. Investig. 2004, 114, 1752–1761. [Google Scholar] [CrossRef] [PubMed]
- Chattopadhyay, M.; Khemka, V.K.; Chatterjee, G.; Ganguly, A.; Mukhopadhyay, S.; Chakrabarti, S. Enhanced ROS production and oxidative damage in subcutaneous white adipose tissue mitochondria in obese and type 2 diabetes subjects. Mol. Cell. Biochem. 2014, 399, 95–103. [Google Scholar] [CrossRef] [PubMed]
- Nagata, D.; Takahashi, M.; Sawai, K.; Tagami, T.; Usui, T.; Shimatsu, A.; Hirata, Y.; Naruse, M. Molecular Mechanism of the Inhibitory Effect of Aldosterone on Endothelial NO Synthase Activity. Hypertension 2006, 48, 165–171. [Google Scholar] [CrossRef]
- Leopold, J.A.; Dam, A.; Maron, B.A.; Scribner, A.W.; Liao, R.; Handy, D.E.; Stanton, R.C.; Pitt, B.; Loscalzo, J. Aldosterone impairs vascular reactivity by decreasing glucose-6-phosphate dehydrogenase activity. Nat. Med. 2007, 13, 189–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, C.; Huang, S.; Yuan, Y.; Ding, G.; Chen, R.; Liu, B.; Yang, T.; Zhang, A. Mitochondrial Dysfunction Mediates Aldosterone-Induced Podocyte Damage: A Therapeutic Target of PPARγ. Am. J. Pathol. 2011, 178, 2020–2031. [Google Scholar] [CrossRef] [Green Version]
- Su, M.; Dhoopun, A.-R.; Yuan, Y.; Huang, S.; Zhu, C.; Ding, G.; Liu, B.; Yang, T.; Zhang, A. Mitochondrial dysfunction is an early event in aldosterone-induced podocyte injury. Am. J. Physiol. Physiol. 2013, 305, F520–F531. [Google Scholar] [CrossRef] [Green Version]
- Lefranc, C.; Friederich-Persson, M.; Braud, L.; Palacios-Ramirez, R.; Karlsson, S.; Boujardine, N.; Motterlini, R.; Jaisser, F.; Cat, A.N.D. MR (Mineralocorticoid Receptor) Induces Adipose Tissue Senescence and Mitochondrial Dysfunction Leading to Vascular Dysfunction in Obesity. Hypertension 2019, 73, 458–468. [Google Scholar] [CrossRef]
- Yoneshiro, T.; Aita, S.; Matsushita, M.; Kayahara, T.; Kameya, T.; Kawai, Y.; Iwanaga, T.; Saito, M. Recruited brown adipose tissue as an antiobesity agent in humans. J. Clin. Investig. 2013, 123, 3404–3408. [Google Scholar] [CrossRef] [Green Version]
- Lee, P.; Smith, S.; Linderman, J.; Courville, A.B.; Brychta, R.J.; Dieckmann, W.; Werner, C.D.; Chen, K.Y.; Celi, F.S. Temperature-Acclimated Brown Adipose Tissue Modulates Insulin Sensitivity in Humans. Diabetes 2014, 63, 3686–3698. [Google Scholar] [CrossRef] [Green Version]
- Zennaro, M.-C.; Le Menuet, D.; Viengchareun, S.; Walker, F.; Ricquier, D.; Lombès, M. Hibernoma development in transgenic mice identifies brown adipose tissue as a novel target of aldosterone action. J. Clin. Investig. 1998, 101, 1254–1260. [Google Scholar] [CrossRef] [Green Version]
- Penfornis, P.; Viengchareun, S.; Le Menuet, D.; Cluzeaud, F.; Zennaro, M.-C.; Lombès, M. The mineralocorticoid receptor mediates aldosterone-induced differentiation of T37i cells into brown adipocytes. Am. J. Physiol. Metab. 2000, 279, E386–E394. [Google Scholar] [CrossRef] [PubMed]
- Viengchareun, S.; Penfornis, P.; Zennaro, M.-C.; Lombès, M. Mineralocorticoid and glucocorticoid receptors inhibit UCP expression and function in brown adipocytes. Am. J. Physiol. Metab. 2001, 280, E640–E649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kraus, D.; Jäger, J.; Meier, B.; Fasshauer, M.; Klein, J. Aldosterone Inhibits Uncoupling Protein-1, Induces Insulin Resistance, and Stimulates Proinflammatory Adipokines in Adipocytes. Horm. Metab. Res. 2005, 37, 455–459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pisani, D.F.; Beranger, G.E.; Corinus, A.; Giroud, M.; Ghandour, R.A.; Altirriba, J.; Chambard, J.; Mazure, N.M.; Bendahhou, S.; Duranton, C.; et al. The K+channel TASK1 modulates β-adrenergic response in brown adipose tissue through the mineralocorticoid receptor pathway. FASEB J. 2015, 30, 909–922. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuhn, E.; Lamribet, K.; Viengchareun, S.; Le Menuet, D.; Fève, B.; Lombès, M. UCP1 transrepression in Brown Fat in vivo and mineralocorticoid receptor anti-thermogenic effects. Ann. d’Endocrinologie 2018, 80, 1–9. [Google Scholar] [CrossRef]
- Armani, A.; Cinti, F.; Marzolla, V.; Morgan, J.; Cranston, G.A.; Antelmi, A.; Carpinelli, G.; Canese, R.; Pagotto, U.; Quarta, C.; et al. Mineralocorticoid receptor antagonism induces browning of white adipose tissue through impairment of autophagy and prevents adipocyte dysfunction in high-fat-diet-fed mice. FASEB J. 2014, 28, 3745–3757. [Google Scholar] [CrossRef]
- Marzolla, V.; Feraco, A.; Gorini, S.; Mammi, C.; Marrese, C.; Mularoni, V.; Boitani, C.; Lombès, M.; Kolkhof, P.; Ciriolo, M.R.; et al. The novel non-steroidal MR antagonist finerenone improves metabolic parameters in high-fat diet-fed mice and activates brown adipose tissue via AMPK-ATGL pathway. FASEB J. 2020, 34, 12450–12465. [Google Scholar] [CrossRef]
- Marzolla, V.; Feraco, A.; Limana, F.; Kolkhof, P.; Armani, A.; Caprio, M. Class-specific responses of brown adipose tissue to steroidal and nonsteroidal mineralocorticoid receptor antagonists. J. Endocrinol. Investig. 2021, 45, 215–220. [Google Scholar] [CrossRef]
- Thuzar, M.; Law, W.P.; Dimeski, G.; Stowasser, M.; Ho, K.K.Y. Mineralocorticoid antagonism enhances brown adipose tissue function in humans: A randomized placebo-controlled cross-over study. Diabetes, Obes. Metab. 2018, 21, 509–516. [Google Scholar] [CrossRef]
- Kahn, C.R.; Wang, G.; Lee, K.Y. Altered adipose tissue and adipocyte function in the pathogenesis of metabolic syndrome. J. Clin. Investig. 2019, 129, 3990–4000. [Google Scholar] [CrossRef]
- Zhang, Y.-Y.; Li, C.; Yao, G.-F.; Du, L.-J.; Liu, Y.; Zheng, X.-J.; Yan, S.; Sun, J.-Y.; Liu, M.-Z.; Zhang, X.; et al. Deletion of Macrophage Mineralocorticoid Receptor Protects Hepatic Steatosis and Insulin Resistance Through ERα/HGF/Met Pathway. Diabetes 2017, 66, 1535–1547. [Google Scholar] [CrossRef] [PubMed]
- Bavuu, O.; Fukuda, D.; Ganbaatar, B.; Matsuura, T.; Ise, T.; Kusunose, K.; Yamaguchi, K.; Yagi, S.; Yamada, H.; Soeki, T.; et al. Esaxerenone, a selective mineralocorticoid receptor blocker, improves insulin sensitivity in mice consuming high-fat diet. Eur. J. Pharmacol. 2022, 931, 172190. [Google Scholar] [CrossRef] [PubMed]
- Homma, T.; Fujisawa, M.; Arai, K.; Ishii, M.; Sada, T.; Ikeda, M. Spironolactone, but not Eplerenone, Impairs Glucose Tolerance in a Rat Model of Metabolic Syndrome. J. Veter Med. Sci. 2012, 74, 1015–1022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ingelsson, E.; Pencina, M.J.; Tofler, G.H.; Benjamin, E.; Lanier, K.J.; Jacques, P.F.; Fox, C.S.; Meigs, J.B.; Levy, D.; Larson, M.; et al. Multimarker Approach to Evaluate the Incidence of the Metabolic Syndrome and Longitudinal Changes in Metabolic Risk Factors. Circulation 2007, 116, 984–992. [Google Scholar] [CrossRef] [Green Version]
- Monticone, S.; D’Ascenzo, F.; Moretti, C.; Williams, T.A.; Veglio, F.; Gaita, F.; Mulatero, P. Cardiovascular events and target organ damage in primary aldosteronism compared with essential hypertension: A systematic review and meta-analysis. Lancet Diabetes Endocrinol. 2018, 6, 41–50. [Google Scholar] [CrossRef]
- Šindelka, G.; Widimský, J.; Haas, T.; Prázný, M.; Hilgertová, J.; Škrha, J. Insulin action in primary hyperaldosteronism before and after surgical or pharmacological treatment. Exp. Clin. Endocrinol. Diabetes 2000, 108, 21–25. [Google Scholar] [CrossRef]
- Catena, C.; Lapenna, R.; Baroselli, S.; Nadalini, E.; Colussi, G.; Novello, M.; Favret, G.; Melis, A.; Cavarape, A.; Sechi, L.A. Insulin Sensitivity in Patients with Primary Aldosteronism: A Follow-Up Study. J. Clin. Endocrinol. Metab. 2006, 91, 3457–3463. [Google Scholar] [CrossRef]
- Strauch, B.; Widimský, J.; Sindelka, G.; Skrha, J. Does the Treatment of Primary Hyperaldosteronism Influence Glucose Tol-erance? Physiol. Res. 2003, 52, 503–506. [Google Scholar]
- Lucatello, B.; Benso, A.; Tabaro, I.; Capello, E.; Caprino, M.P.; Marafetti, L.; Rossato, D.; Oleandri, S.E.; Ghigo, E.; Maccario, M. Long-term re-evaluation of primary aldosteronism after medical treatment reveals high proportion of normal mineralocorticoid secretion. Eur. J. Endocrinol. 2013, 168, 525–532. [Google Scholar] [CrossRef] [Green Version]
- DeRosa, G.; Bonaventura, A.; Bianchi, L.; Romano, D.; D’Angelo, A.; Fogari, E.; Maffioli, P. Effects of canrenone in patients with metabolic syndrome. Expert Opin. Pharmacother. 2013, 14, 2161–2169. [Google Scholar] [CrossRef]
- Garg, R.; Kneen, L.; Williams, G.H.; Adler, G.K. Effect of mineralocorticoid receptor antagonist on insulin resistance and endothelial function in obese subjects. Diabetes, Obes. Metab. 2013, 16, 268–272. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Parasiliti-Caprino, M.; Bollati, M.; Merlo, F.D.; Ghigo, E.; Maccario, M.; Bo, S. Adipose Tissue Dysfunction in Obesity: Role of Mineralocorticoid Receptor. Nutrients 2022, 14, 4735. https://doi.org/10.3390/nu14224735
Parasiliti-Caprino M, Bollati M, Merlo FD, Ghigo E, Maccario M, Bo S. Adipose Tissue Dysfunction in Obesity: Role of Mineralocorticoid Receptor. Nutrients. 2022; 14(22):4735. https://doi.org/10.3390/nu14224735
Chicago/Turabian StyleParasiliti-Caprino, Mirko, Martina Bollati, Fabio Dario Merlo, Ezio Ghigo, Mauro Maccario, and Simona Bo. 2022. "Adipose Tissue Dysfunction in Obesity: Role of Mineralocorticoid Receptor" Nutrients 14, no. 22: 4735. https://doi.org/10.3390/nu14224735