
Maternal Low-Protein Diet during Puberty and Adulthood Aggravates Lipid Metabolism of Their Offspring Fed a High-Fat Diet in Mice
 , , , , ,
, , , , ,
Abstract
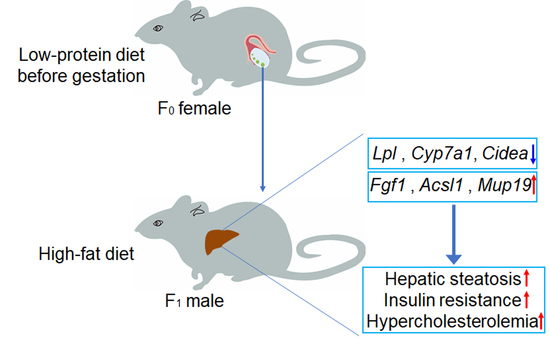
:
1. Introduction
2. Materials and Methods
2.1. Animal Care and Experimental Design
2.2. Serum Biochemical Analysis
2.3. Liver Histological Analysis
2.4. Insulin Tolerance Test
2.5. RNA Extraction and Real-Time PCR
2.6. Preparation of cDNA Libraries and Sequencing
2.7. Quality Control and Reads Mapping
2.8. Quantification and Analysis of Differentially Expressed Genes (DEGs)
2.9. Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) Enrichment Analysis of DEGs
2.10. Statistical Analysis
3. Results
3.1. LP Diet Did Not Change the Food Intake and Body Weight in Female Mice
3.2. Maternal LP Diet during Puberty and Adulthood Impaired Insulin Sensitivity in Offspring Fed High-Fat Diet
3.3. Maternal Low-Protein Diet during Puberty and Adulthood Aggravated Lipids Profiles in HFD-Fed Offspring
3.4. Transcriptome Profiling in Offspring’s Liver
3.5. Functional and Pathway Enrichment Analysis of DEGs
3.6. Validation of the DEGs Linked to Lipid and Glucose Metabolism
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 2016, 64, 73–84. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Cai, B.; Han, X.; Gao, Y.; Zhang, X.; Wang, R.; Zhang, Y.; Chen, Q. Vitamin D supplementation for nonalcoholic fatty liver disease in type 2 diabetes mellitus: A protocol for a systematic review and meta-analysis. Medicine 2020, 99, e20148. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.M.; Golabi, P.; De Avila, L.; Paik, J.M.; Srishord, M.; Fukui, N.; Qiu, Y.; Burns, L.; Afendy, A.; Nader, F. The global epidemiology of NAFLD and NASH in patients with type 2 diabetes: A systematic review and meta-analysis. J. Hepatol. 2019, 71, 793–801. [Google Scholar] [CrossRef]
- Pillon, N.J.; Loos, R.J.F.; Marshall, S.M.; Zierath, J.R. Metabolic consequences of obesity and type 2 diabetes: Balancing genes and environment for personalized care. Cell 2021, 184, 1530–1544. [Google Scholar] [CrossRef]
- Karbaschi, R.; Sadeghimahalli, F.; Zardooz, H. Maternal high-fat diet inversely affects insulin sensitivity in dams and young adult male rat offspring. J. Zhejiang Univ. Sci. B 2016, 17, 728–732. [Google Scholar] [CrossRef]
- Almeida, D.L.; Simoes, F.S.; Saavedra, L.P.J.; Praxedes Moraes, A.M.; Matiusso, C.C.I.; Malta, A.; Palma-Rigo, K.; Mathias, P.C.F. Maternal low-protein diet during lactation combined with early overfeeding impair male offspring’s long-term glucose homeostasis. Endocrine 2019, 63, 62–69. [Google Scholar] [CrossRef]
- Jahan-Mihan, A.; Luhovyy, B.L.; Khoury, D.E.; Anderson, G.H. Dietary Proteins as Determinants of Metabolic and Physiologic Functions of the Gastrointestinal Tract. Nutrients 2011, 3, 574–603. [Google Scholar] [CrossRef]
- Millward, D.J.; Jackson, A.A. Protein/energy ratios of current diets in developed and developing countries compared with a safe protein/energy ratio: Implications for recommended protein and amino acid intakes. Public Health Nutr. 2004, 7, 387–405. [Google Scholar] [CrossRef]
- Ghosh, S.; Suri, D.; Uauy, R. Assessment of protein adequacy in developing countries: Quality matters. Br. J. Nutr. 2012, 108 (Suppl. S2), S77–S87. [Google Scholar] [CrossRef]
- Maslova, E.; Rytter, D.; Bech, B.H.; Henriksen, T.B.; Rasmussen, M.A.; Olsen, S.F.; Halldorsson, T.I. Maternal protein intake during pregnancy and offspring overweight 20 y later. Am. J. Clin. Nutr. 2014, 100, 1139–1148. [Google Scholar] [CrossRef] [Green Version]
- Geraghty, A.A.; O’Brien, E.C.; Alberdi, G.; Horan, M.K.; Donnelly, J.; Larkin, E.; Segurado, R.; Mehegan, J.; Molloy, E.J.; McAuliffe, F.M. Maternal protein intake during pregnancy is associated with child growth up to 5 years of age, but not through insulin-like growth factor-1: Findings from the ROLO study. Br. J. Nutr. 2018, 120, 1252–1261. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Choi, A.; Kwon, Y.H. Maternal Protein Restriction Altered Insulin Resistance and Inflammation-Associated Gene Expression in Adipose Tissue of Young Adult Mouse Offspring in Response to a High-Fat Diet. Nutrients 2020, 12, 1103. [Google Scholar] [CrossRef] [PubMed]
- Han, R.; Li, A.; Li, L.; Kitlinska, J.B.; Zukowska, Z. Maternal low-protein diet up-regulates the neuropeptide Y system in visceral fat and leads to abdominal obesity and glucose intolerance in a sex- and time-specific manner. FASEB J. 2012, 26, 3528–3536. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Zhang, L.; Liu, J.; Li, Y.; Zhang, J. Long-Term Effects of Maternal Low-Protein Diet and Post-weaning High-Fat Feeding on Glucose Metabolism and Hypothalamic POMC Promoter Methylation in Offspring Mice. Front. Nutr. 2021, 8, 657848. [Google Scholar] [CrossRef] [PubMed]
- Sato, N.; Sudo, K.; Mori, M.; Imai, C.; Muramatsu, M.; Sugimoto, M. Early gestational maternal low-protein diet diminishes hepatic response to fasting in young adult male mice. Sci. Rep. 2017, 7, 9812. [Google Scholar] [CrossRef]
- Ozanne, S.E.; Hales, C.N. The long-term consequences of intra-uterine protein malnutrition for glucose metabolism. Proc. Nutr. Soc. 1999, 58, 615–619. [Google Scholar] [CrossRef]
- Fleming, T.P.; Velazquez, M.A.; Eckert, J.J.; Lucas, E.S.; Watkins, A.J. Nutrition of females during the peri-conceptional period and effects on foetal programming and health of offspring. Anim. Reprod. Sci. 2012, 130, 193–197. [Google Scholar] [CrossRef]
- Watkins, A.J.; Wilkins, A.; Cunningham, C.; Perry, V.H.; Seet, M.J.; Osmond, C.; Eckert, J.J.; Torrens, C.; Cagampang, F.R.; Cleal, J.; et al. Low protein diet fed exclusively during mouse oocyte maturation leads to behavioural and cardiovascular abnormalities in offspring. J. Physiol. 2008, 586, 2231–2244. [Google Scholar] [CrossRef]
- Dudele, A.; Lund, S.; Jessen, N.; Wegener, G.; Winther, G.; Elnif, J.; Frische, S.; Wang, T.; Mayntz, D. Maternal protein restriction before pregnancy reduces offspring early body mass and affects glucose metabolism in C57BL/6JBom mice. J. Dev. Orig. Health Dis. 2012, 3, 364–374. [Google Scholar] [CrossRef]
- Zhuo, Y.; Hua, L.; Feng, B.; Jiang, X.; Li, J.; Jiang, D.; Huang, X.; Zhu, Y.; Li, Z.; Yan, L.; et al. Fibroblast growth factor 21 coordinates adiponectin to mediate the beneficial effects of low-protein diet on primordial follicle reserve. EBioMedicine 2019, 41, 623–635. [Google Scholar] [CrossRef] [Green Version]
- Feng, B.; Huang, X.; Jiang, D.; Hua, L.; Zhuo, Y.; Wu, D. Endoplasmic Reticulum Stress Inducer Tunicamycin Alters Hepatic Energy Homeostasis in Mice. Int. J. Mol. Sci. 2017, 18, 1710. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Jiang, D.; Zhu, Y.; Fang, Z.; Che, L.; Lin, Y.; Xu, S.; Li, J.; Huang, C.; Zou, Y.; et al. Chronic high dose zinc supplementation induces visceral adipose tissue hypertrophy without altering body weight in mice. Nutrients 2017, 9, 1138. [Google Scholar] [CrossRef] [PubMed]
- Feng, B.; Jiao, P.; Helou, Y.; Li, Y.; He, Q.; Walters, M.S.; Salomon, A.; Xu, H. Mitogen-activated protein kinase phosphatase 3 (MKP-3)-deficient mice are resistant to diet-induced obesity. Diabetes 2014, 63, 2924–2934. [Google Scholar] [CrossRef]
- Joshi, S.; Garole, V.; Daware, M.; Girigosavi, S.; Rao, S. Maternal protein restriction before pregnancy affects vital organs of offspring in Wistar rats. Metabolism 2003, 52, 13–18. [Google Scholar] [CrossRef]
- Mortensen, E.L.; Wang, T.; Malte, H.; Raubenheimer, D.; Mayntz, D. Maternal preconceptional nutrition leads to variable fat deposition and gut dimensions of adult offspring mice (C57BL/6JBom). Int. J. Obes. 2010, 34, 1618–1624. [Google Scholar] [CrossRef] [PubMed]
- Gastaldelli, A.; Baldi, S.; Pettiti, M.; Toschi, E.; Camastra, S.; Natali, A.; Landau, B.R.; Ferrannini, E. Influence of obesity and type 2 diabetes on gluconeogenesis and glucose output in humans: A quantitative study. Diabetes 2000, 49, 1367–1373. [Google Scholar] [CrossRef]
- Sargeant, J.A.; Gray, L.J.; Bodicoat, D.H.; Willis, S.A.; Stensel, D.J.; Nimmo, M.A.; Aithal, G.P.; King, J.A. The effect of exercise training on intrahepatic triglyceride and hepatic insulin sensitivity: A systematic review and meta-analysis. Obes. Rev. 2018, 19, 1446–1459. [Google Scholar] [CrossRef]
- Jonker, J.W.; Suh, J.M.; Atkins, A.R.; Ahmadian, M.; Li, P.; Whyte, J.; He, M.; Juguilon, H.; Yin, Y.Q.; Phillips, C.T.; et al. A PPARgamma-FGF1 axis is required for adaptive adipose remodelling and metabolic homeostasis. Nature 2012, 485, 391–394. [Google Scholar] [CrossRef]
- Fan, L.; Ding, L.; Lan, J.; Niu, J.; He, Y.; Song, L. Fibroblast Growth Factor-1 Improves Insulin Resistance via Repression of JNK-Mediated Inflammation. Front. Pharmacol. 2019, 10, 1478. [Google Scholar] [CrossRef]
- Suh, J.M.; Jonker, J.W.; Ahmadian, M.; Goetz, R.; Lackey, D.; Osborn, O.; Huang, Z.; Liu, W.; Yoshihara, E.; Van Dijk, T.H.; et al. Corrigendum: Endocrinization of FGF1 produces a neomorphic and potent insulin sensitizer. Nature 2015, 520, 388. [Google Scholar] [CrossRef] [Green Version]
- Rahmadi, M.; Nurhan, A.D.; Pratiwi, E.D.; Prameswari, D.A.; Panggono, S.M.; Nisak, K.; Khotib, J. The effect of various high-fat diet on liver histology in the development of NAFLD models in mice. J. Basic Clin. Physiol. Pharmacol. 2021, 32, 547–553. [Google Scholar] [CrossRef] [PubMed]
- Mashek, D.G.; Li, L.O.; Coleman, R.A. Rat long-chain acyl-CoA synthetase mRNA, protein, and activity vary in tissue distribution and in response to diet. J. Lipid Res. 2006, 47, 2004–2010. [Google Scholar] [CrossRef] [PubMed]
- Li, L.O.; Mashek, D.G.; An, J.; Doughman, S.D.; Newgard, C.B.; Coleman, R.A. Overexpression of rat long chain acyl-coa synthetase 1 alters fatty acid metabolism in rat primary hepatocytes. J. Biol. Chem. 2006, 281, 37246–37255. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.B.; Dong, B.; Xu, Y.; Zhang, Y.; Liu, J. Identification of a novel function of hepatic long-chain acyl-CoA synthetase-1 (ACSL1) in bile acid synthesis and its regulation by bile acid-activated farnesoid X receptor. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2019, 1864, 358–371. [Google Scholar] [CrossRef] [PubMed]
- Han, P.; Wei, G.; Cai, K.; Xiang, X.; Deng, W.P.; Li, Y.B.; Kuang, S.; Dong, Z.; Zheng, T.; Luo, Y.; et al. Identification and functional characterization of mutations in LPL gene causing severe hypertriglyceridaemia and acute pancreatitis. J. Cell. Mol. Med. 2020, 24, 1286–1299. [Google Scholar] [CrossRef]
- Liu, G.; Xu, J.N.; Liu, D.; Ding, Q.; Liu, M.N.; Chen, R.; Fan, M.; Zhang, Y.; Zheng, C.; Zou, D.J.; et al. Regulation of plasma lipid homeostasis by hepatic lipoprotein lipase in adult mice. J. Lipid Res. 2016, 57, 1155–1161. [Google Scholar] [CrossRef]
- Kim, J.K.; Fillmore, J.J.; Chen, Y.; Yu, C.; Moore, I.K.; Pypaert, M.; Lutz, E.P.; Kako, Y.; Velez-Carrasco, W.; Goldberg, I.J.; et al. Tissue-specific overexpression of lipoprotein lipase causes tissue-specific insulin resistance. Proc. Natl. Acad. Sci. USA 2001, 98, 7522–7527. [Google Scholar] [CrossRef]
- Bol, V.V.; Delattre, A.I.; Reusens, B.; Raes, M.; Remacle, C. Forced catch-up growth after fetal protein restriction alters the adipose tissue gene expression program leading to obesity in adult mice. Am. J. Physiol.-Regul. Integr. Comp. Physiol. 2009, 297, R291–R299. [Google Scholar] [CrossRef]
- Chambers, K.F.; Day, P.E.; Aboufarrag, H.T.; Kroon, P.A. Polyphenol Effects on Cholesterol Metabolism via Bile Acid Biosynthesis, CYP7A1: A Review. Nutrients 2019, 11, 2588. [Google Scholar] [CrossRef]
- Zhou, L.; Xu, L.; Ye, J.; Li, D.; Wang, W.; Li, X.; Wu, L.; Wang, H.; Guan, F.; Li, P. Cidea promotes hepatic steatosis by sensing dietary fatty acids. Hepatology 2012, 56, 95–107. [Google Scholar] [CrossRef]
- Zhou, Z.; Yon Toh, S.; Chen, Z.; Guo, K.; Ng, C.P.; Ponniah, S.; Lin, S.C.; Hong, W.; Li, P. Cidea-deficient mice have lean phenotype and are resistant to obesity. Nat. Genet. 2003, 35, 49–56. [Google Scholar] [CrossRef] [PubMed]
- He, Q.; Diao, Y.; Zhao, T.; Hou, B.; Ngokana, L.D.; Liang, H.; Nie, J.; Tan, P.; Huang, H.; Li, Y.; et al. SREBP1c mediates the effect of acetaldehyde on Cidea expression in Alcoholic fatty liver Mice. Sci. Rep. 2018, 8, 1200. [Google Scholar] [CrossRef] [PubMed]
- Shaw, P.H.; Held, W.A.; Hastie, N.D. The gene family for major urinary proteins: Expression in several secretory tissues of the mouse. Cell 1983, 32, 755–761. [Google Scholar] [CrossRef]
- Flowers, M.T.; Groen, A.K.; Oler, A.T.; Keller, M.P.; Choi, Y.; Schueler, K.L.; Richards, O.C.; Lan, H.; Miyazaki, M.; Kuipers, F.; et al. Cholestasis and hypercholesterolemia in SCD1-deficient mice fed a low-fat, high-carbohydrate diet. J. Lipid Res. 2006, 47, 2668–2680. [Google Scholar] [CrossRef]
- Zhou, Y.; Jiang, L.; Rui, L. Identification of MUP1 as a regulator for glucose and lipid metabolism in mice. J. Biol. Chem. 2009, 284, 11152–11159. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ensembl ID | Gene | Description | Log2FC | p Value |
|---|---|---|---|---|
| ENSMUSG00000078673 | Mup19 | major urinary protein 19 | −4.46 | 0.0001 |
| ENSMUSG00000036585 | Fgf1 | fibroblast growth factor 1 | −0.35 | 0.0068 |
| ENSMUSG00000028240 | Cyp7a1 | cytochrome P450, family 7, subfamily a, polypeptide 1 | 0.55 | 0.0131 |
| ENSMUSG00000024378 | Stard4 | StAR-related lipid transfer (START) domain containing 4 | −0.32 | 0.0157 |
| ENSMUSG00000028195 | Cyr61 | cysteine rich protein 61 | 0.36 | 0.0265 |
| ENSMUSG00000032083 | Apoa1 | apolipoprotein A-I | −0.24 | 0.0373 |
| ENSMUSG00000005514 | Por | P450 (cytochrome) oxidoreductase | −0.28 | 0.0426 |
| ENSMUSG00000095320 | Ccl21a | chemokine (C-C motif) ligand 21A (serine) | 1.53 | 0.0059 |
| ENSMUSG00000029648 | Flt1 | FMS-like tyrosine kinase 1 | −0.30 | 0.0295 |
| ENSMUSG00000024526 | Cidea | cell death-inducing DNA fragmentation factor, alpha subunit-like effector A | 0.91 | 0.000 |
| ENSMUSG00000020644 | Id2 | inhibitor of DNA binding 2 | −0.38 | 0.0305 |
| ENSMUSG00000015568 | Lpl | lipoprotein lipase | 0.47 | 0.0201 |
| ENSMUSG00000031767 | Nudt7 | nudix (nucleoside diphosphate linked moiety X)-type motif 7 | −0.53 | 0.0119 |
| ENSMUSG00000006517 | Mvd | mevalonate (diphospho) decarboxylase | −0.46 | 0.0135 |
| ENSMUSG00000057228 | Aadat | aminoadipate aminotransferase | −0.29 | 0.0359 |
| ENSMUSG00000028011 | Tdo2 | tryptophan 2,3-dioxygenase | −0.24 | 0.0407 |
| ENSMUSG00000034593 | Myo5a | myosin VA | 0.76 | 0.0092 |
| ENSMUSG00000043461 | Sptssb | serine palmitoyltransferase, small subunit B | 3.34 | 0.0122 |
| ENSMUSG00000032231 | Anxa2 | annexin A2 | −0.41 | 0.0134 |
| ENSMUSG00000021135 | Slc10a1 | solute carrier family 10 (sodium/bile acid cotransporter family), member 1 | −0.25 | 0.0162 |
| ENSMUSG00000030382 | Slc27a5 | solute carrier family 27 (fatty acid transporter), member 5 | −0.21 | 0.0252 |
| ENSMUSG00000041828 | Abca8a | ATP-binding cassette, sub-family A (ABC1), member 8a | −0.34 | 0.0343 |
| ENSMUSG00000018796 | Acsl1 | acyl-CoA synthetase long-chain family member 1 | −0.21 | 0.0366 |
| ENSMUSG00000079608 | Stard6 | StAR-related lipid transfer (START) domain containing 6 | −2.86 | 0.0378 |
| ENSMUSG00000020484 | Xbp1 | X-box binding protein 1 | −0.41 | 0.0120 |
| ENSMUSG00000024292 | Cyp4f14 | cytochrome P450, family 4, subfamily f, polypeptide 14 | −0.30 | 0.0039 |
| ENSMUSG00000042248 | Cyp2c37 | cytochrome P450, family 2. Subfamily c, polypeptide 37 | −0.41 | 0.0073 |
| ENSMUSG00000092008 | Cyp2c69 | cytochrome P450, family 2, subfamily c, polypeptide 69 | −2.07 | 0.0075 |
| ENSMUSG00000019768 | Esr1 | estrogen receptor 1 (alpha) | −0.48 | 0.0276 |
| ENSMUSG00000054827 | Cyp2c50 | cytochrome P450, family 2, subfamily c, polypeptide 50 | −0.41 | 0.0179 |
| ENSMUSG00000020258 | Glyctk | glycerate kinase | −0.20 | 0.0437 |
| ENSMUSG00000002831 | Plin4 | perilipin 4 | 0.42 | 0.0030 |
| ENSMUSG00000028427 | Aqp7 | aquaporin 7 | 1.16 | 0.0238 |
| ENSMUSG00000025006 | Sorbs1 | sorbin and SH3 domain containing 1 | 0.27 | 0.0285 |
| ENSMUSG00000016194 | Hsd11b1 | hydroxysteroid 11-beta dehydrogenase 1 | −0.28 | 0.0073 |
| ENSMUSG00000035780 | Ugt2a3 | UDP glucuronosyltransferase 2 family, polypeptide A3 | −0.31 | 0.0139 |
| ENSMUSG00000039648 | Kyat1 | kynurenine aminotransferase 1 | −0.29 | 0.0244 |
| ENSMUSG00000030711 | Sult1a1 | sulfotransferase family 1A, phenol-preferring, member 1 | −0.30 | 0.0262 |
| ENSMUSG00000057425 | Ugt2b37 | UDP glucuronosyltransferase 2 family, polypeptide B37 | −0.57 | 0.0362 |
| ENSMUSG00000039519 | Cyp7b1 | cytochrome P450, family 7, subfamily b, polypeptide 1 | −0.80 | 0.0422 |
| ENSMUSG00000067144 | Slc22a7 | solute carrier family 22 (organic anion transporter), member 7 | −0.91 | 0.0230 |
| ENSMUSG00000041698 | Slco1a1 | solute carrier organic anion transporter family, member 1a1 | −1.64 | 0.0419 |
| ENSMUSG00000027938 | Creb3l4 | cAMP responsive element binding protein 3-like 4 | 1.58 | 0.0128 |
| ENSMUSG00000038648 | Creb3l2 | cAMP responsive element binding protein 3-like 2 | −0.36 | 0.0444 |
| ENSMUSG00000078683 | Mup1 | major urinary protein 1 | −1.96 | 0.0414 |
| ENSMUSG00000078687 | Mup8 | major urinary protein 8 | −2.80 | 0.0039 |
| ENSMUSG00000096674 | Mup15 | major urinary protein 15 | −2.03 | 0.0087 |
| ENSMUSG00000082644 | Mup-ps19 | major urinary protein, pseudogene 19 | −1.24 | 0.0280 |
| ENSMUSG00000020053 | Igf1 | insulin-like growth factor 1 | −0.29 | 0.0461 |
| ENSMUSG00000024378 | Stard4 | StAR-related lipid transfer (START) domain containing 4 | −0.32 | 0.0157 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, X.; Zhuo, Y.; Jiang, D.; Zhu, Y.; Fang, Z.; Che, L.; Lin, Y.; Xu, S.; Hua, L.; Zou, Y.; et al. Maternal Low-Protein Diet during Puberty and Adulthood Aggravates Lipid Metabolism of Their Offspring Fed a High-Fat Diet in Mice. Nutrients 2022, 14, 4057. https://doi.org/10.3390/nu14194057
Huang X, Zhuo Y, Jiang D, Zhu Y, Fang Z, Che L, Lin Y, Xu S, Hua L, Zou Y, et al. Maternal Low-Protein Diet during Puberty and Adulthood Aggravates Lipid Metabolism of Their Offspring Fed a High-Fat Diet in Mice. Nutrients. 2022; 14(19):4057. https://doi.org/10.3390/nu14194057
Chicago/Turabian StyleHuang, Xiaohua, Yong Zhuo, Dandan Jiang, Yingguo Zhu, Zhengfeng Fang, Lianqiang Che, Yan Lin, Shengyu Xu, Lun Hua, Yuanfeng Zou, and et al. 2022. "Maternal Low-Protein Diet during Puberty and Adulthood Aggravates Lipid Metabolism of Their Offspring Fed a High-Fat Diet in Mice" Nutrients 14, no. 19: 4057. https://doi.org/10.3390/nu14194057