
Lipid Raft Integrity and Cellular Cholesterol Homeostasis Are Critical for SARS-CoV-2 Entry into Cells

 , , and
, , and 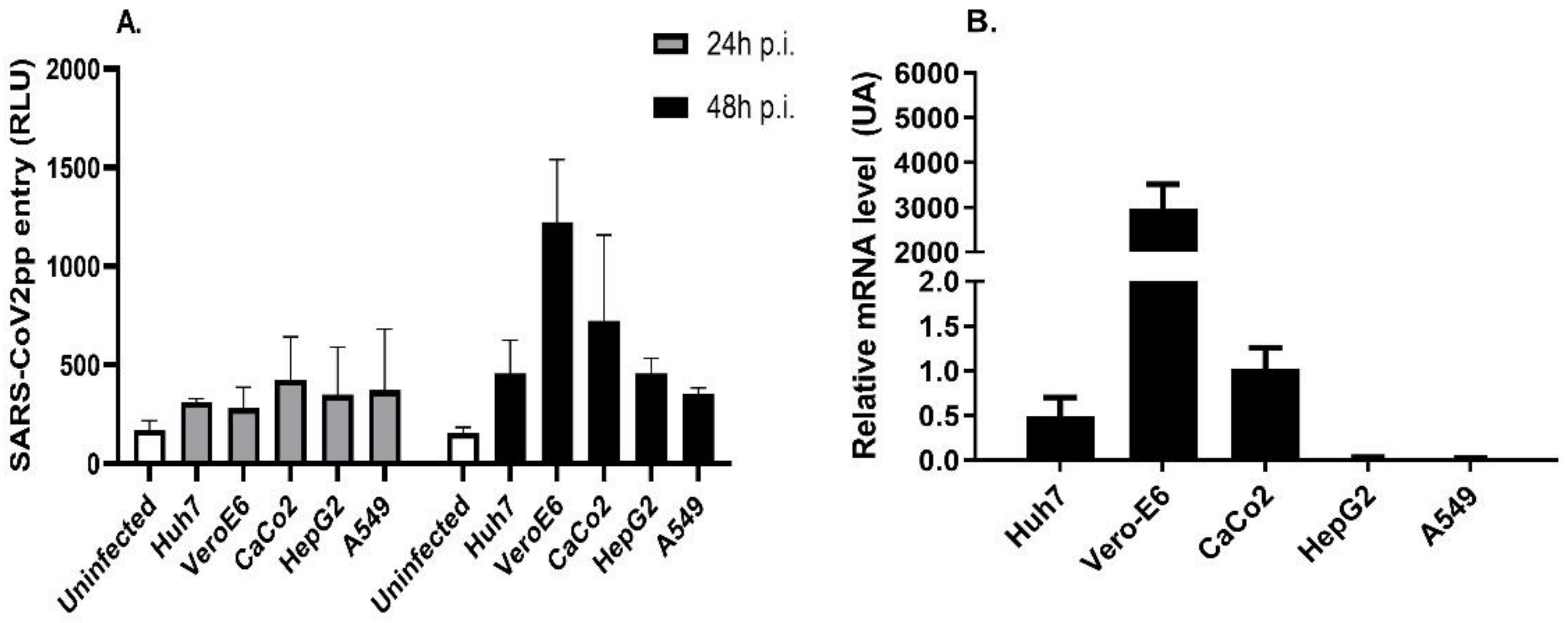{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Lines and Reagents
2.2. SARS-CoV-2pp Entry and Cell Viability Assays
2.3. Cell Treatment with Cholesterol-Distrupting Agents and Statins
2.4. Preparation of Wild-Type SARS-CoV-2
2.5. Cholesterol Depletion and Replenishment Using Wild-Type SARS-CoV-2
2.6. Intracellular Cholesterol Quantification
2.7. Western Blot Analysis
2.8. Immunofluorescence Staining Analysis
2.9. RNA Isolation and Quantitative Real-Time PCR (qRT-PCR)
2.10. SiRNA Transfection
2.11. Statistical Analysis
3. Results
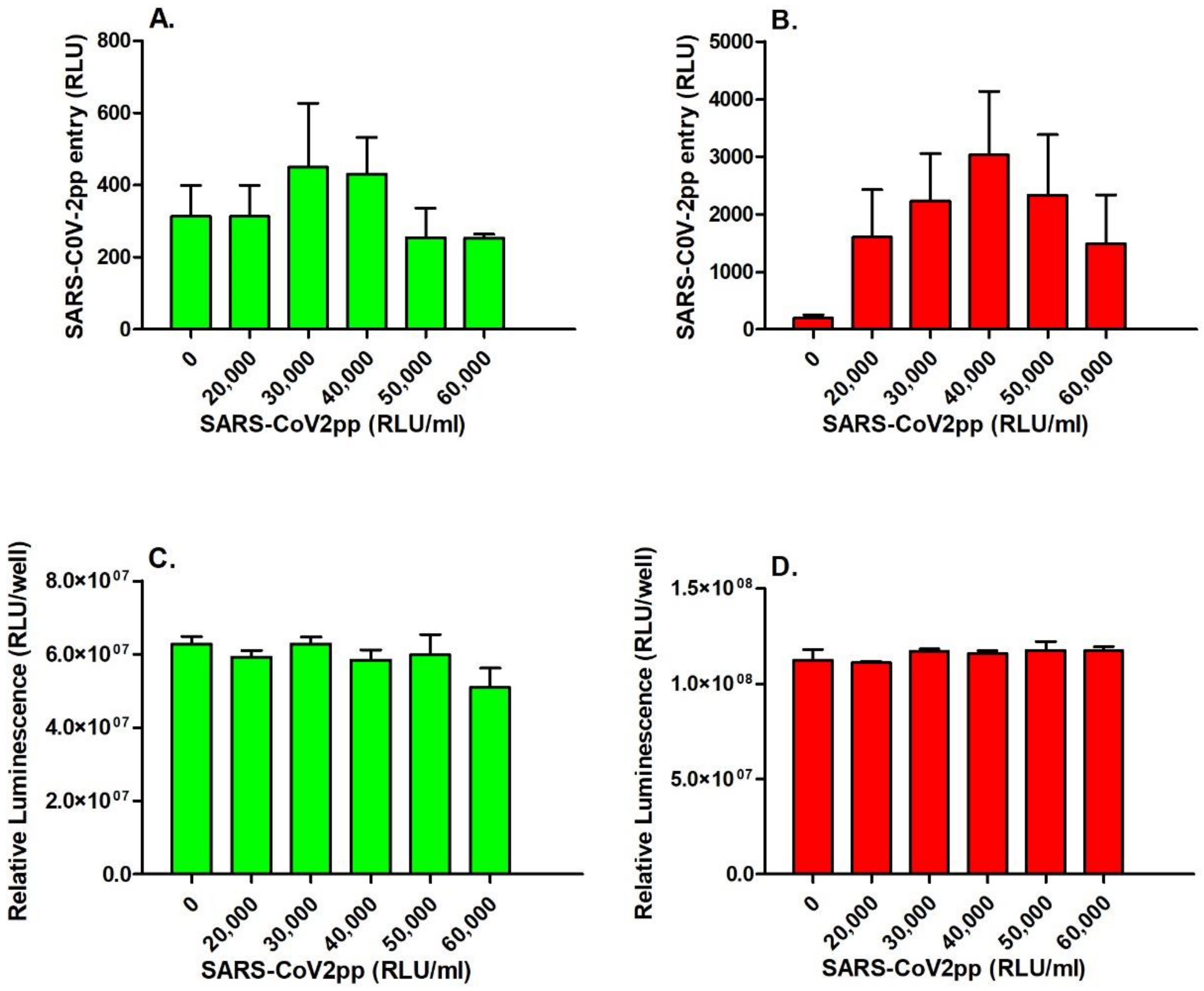
3.1. SARS-CoV-2pp Entry into Cells Is Dependent on ACE2 Expression Levels
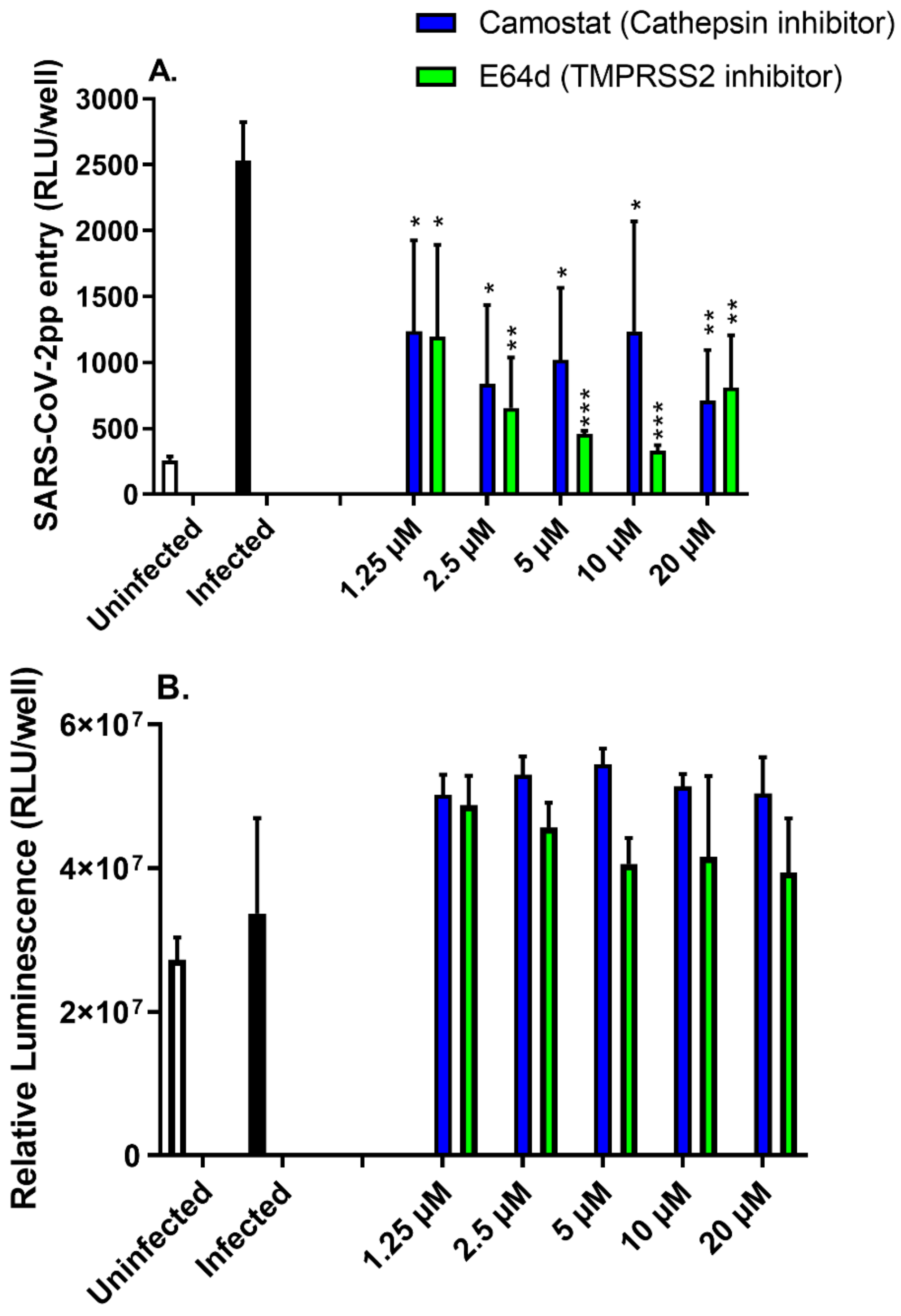
3.2. Protease Inhibitors Attenuated SARS-CoV-2pp Entry into Cells
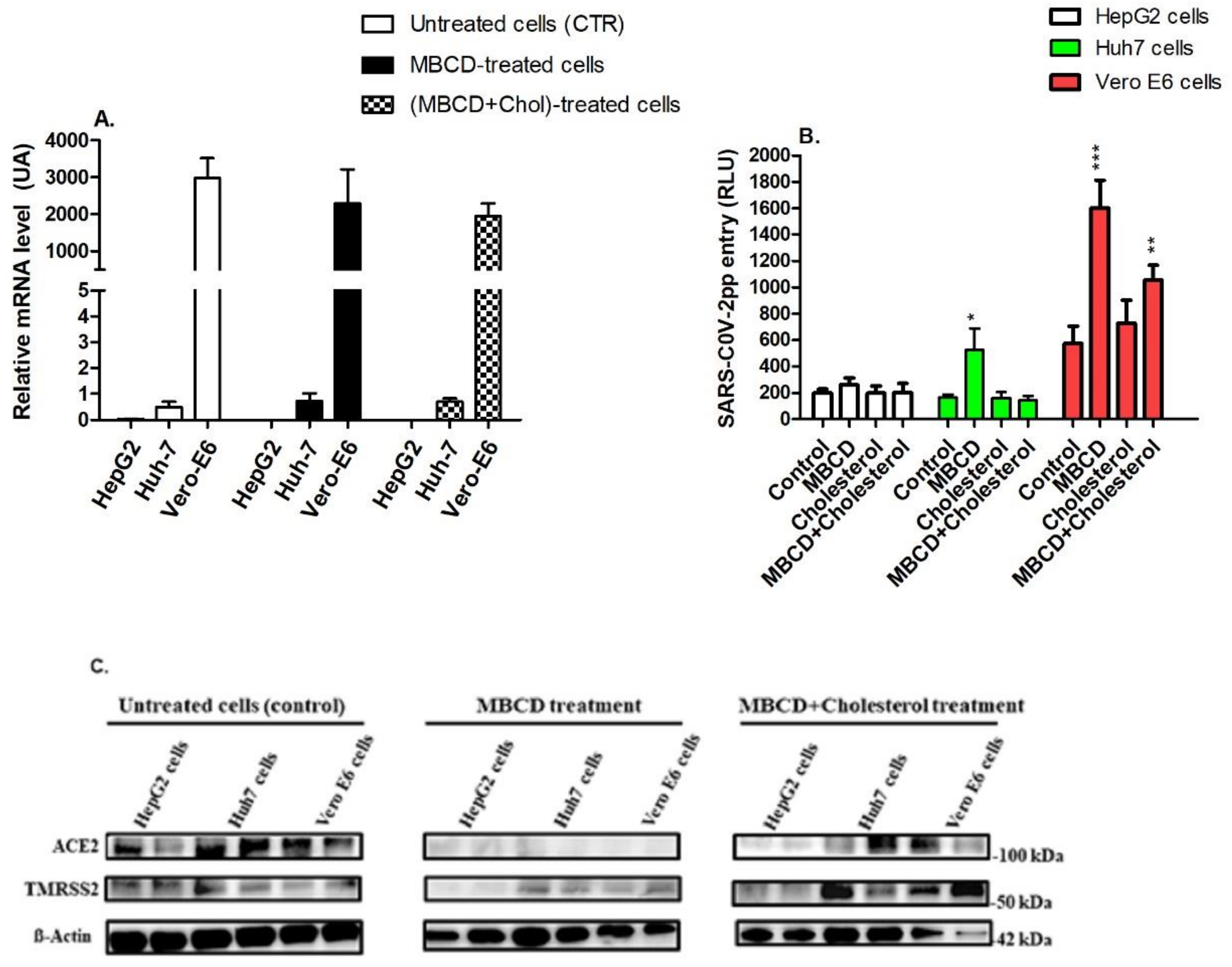
3.3. Depletion of Cholesterol Alters the Expression of Raft-Associated Receptors That Are Involved in Cell Entry of SARS-CoV-2pp
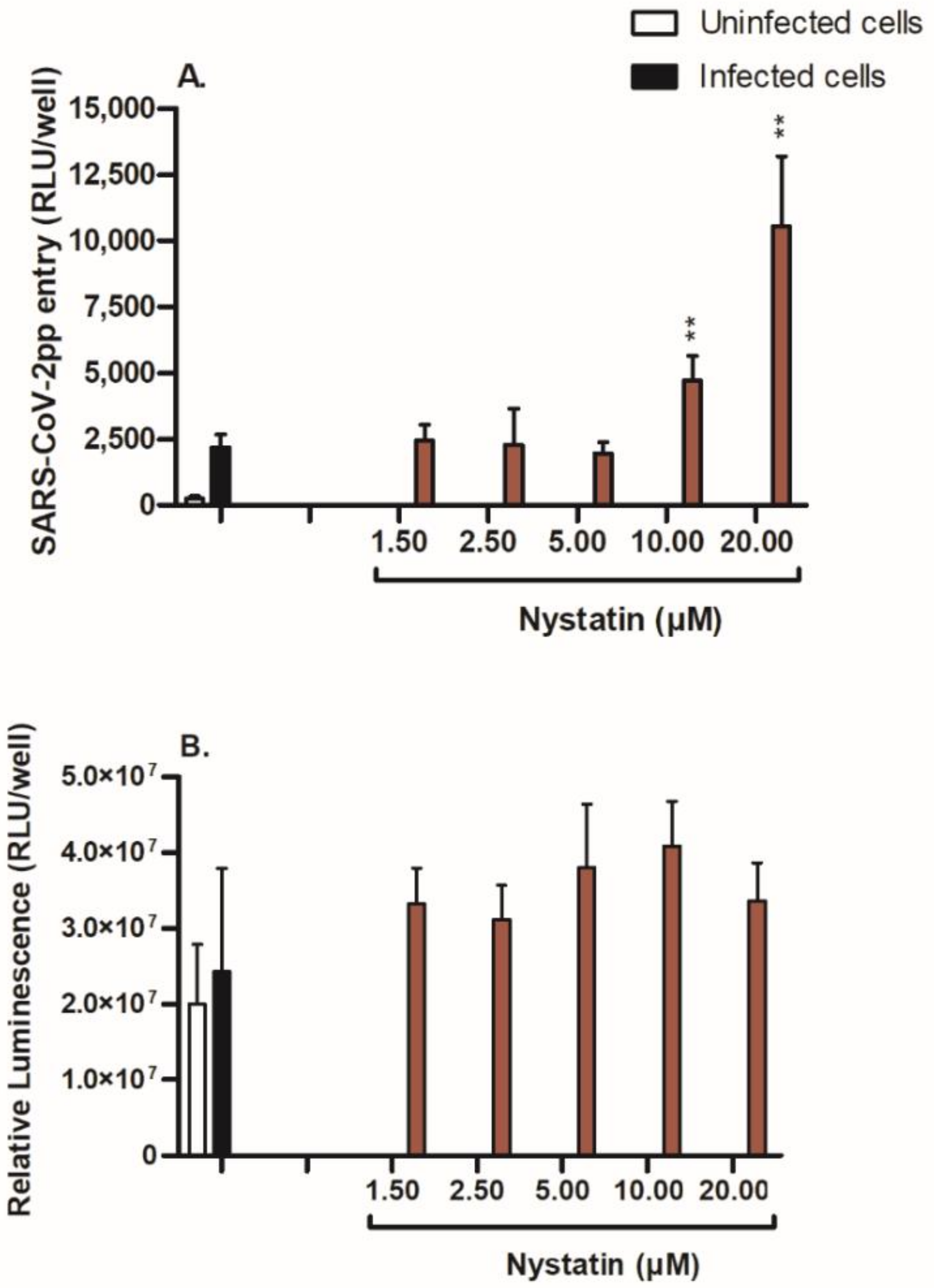
3.4. Nystatin Treatment Enhances Cell Entry of SARS-CoV-2pp
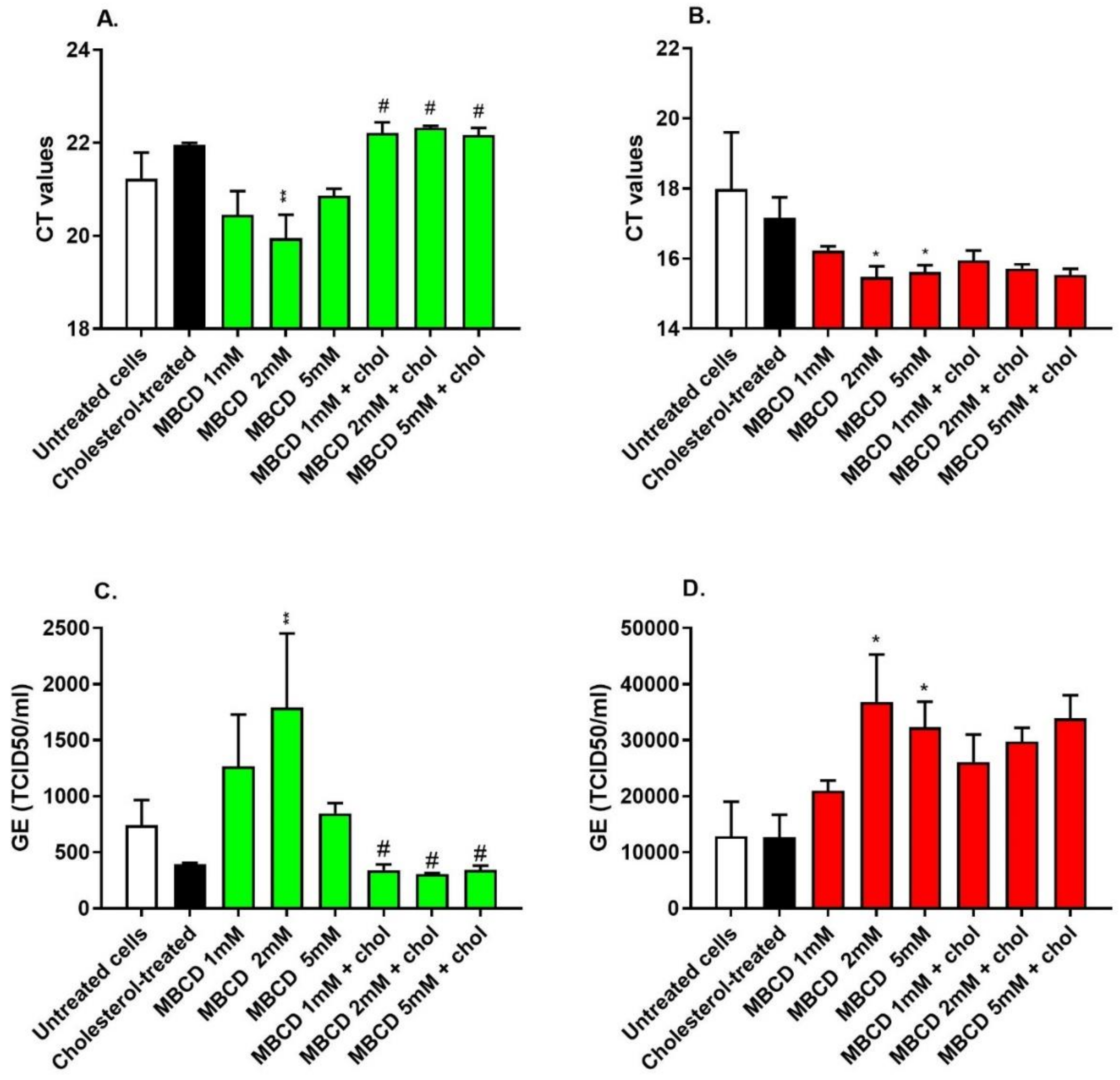
3.5. Depletion of Cholesterol-Rich Rafts Enhanced Cell Entry of the Wild-Type SARS-CoV-2 without Altering Cell Morphology
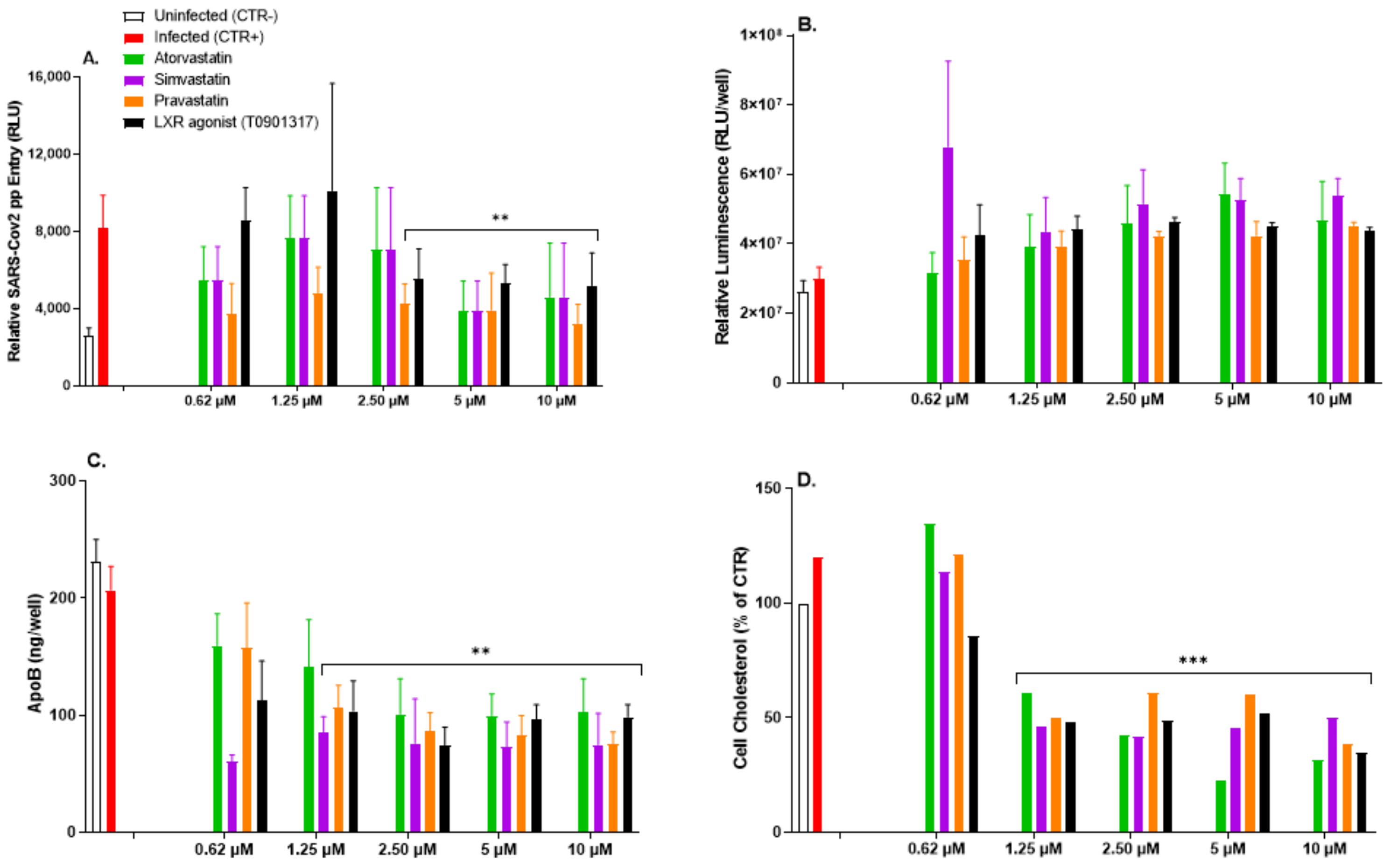
3.6. Impairment of Intracellular Cholesterol Homeostasis Mitigates SARS-CoV-2pp Infection
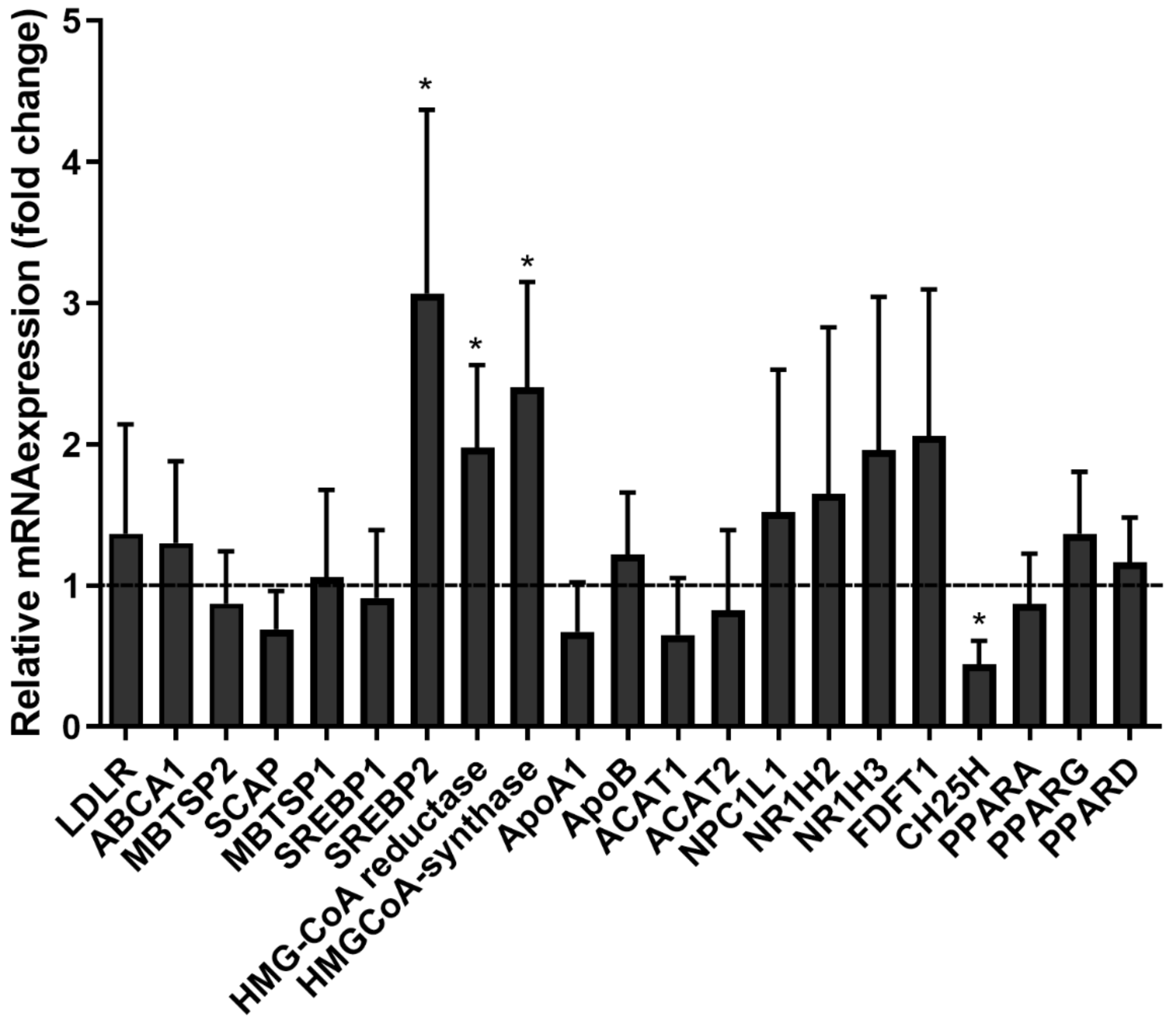
3.7. SARS-CoV-2pp Entry into Cells Alters Cholesterol- and Lipid-Modifying Gene Expression
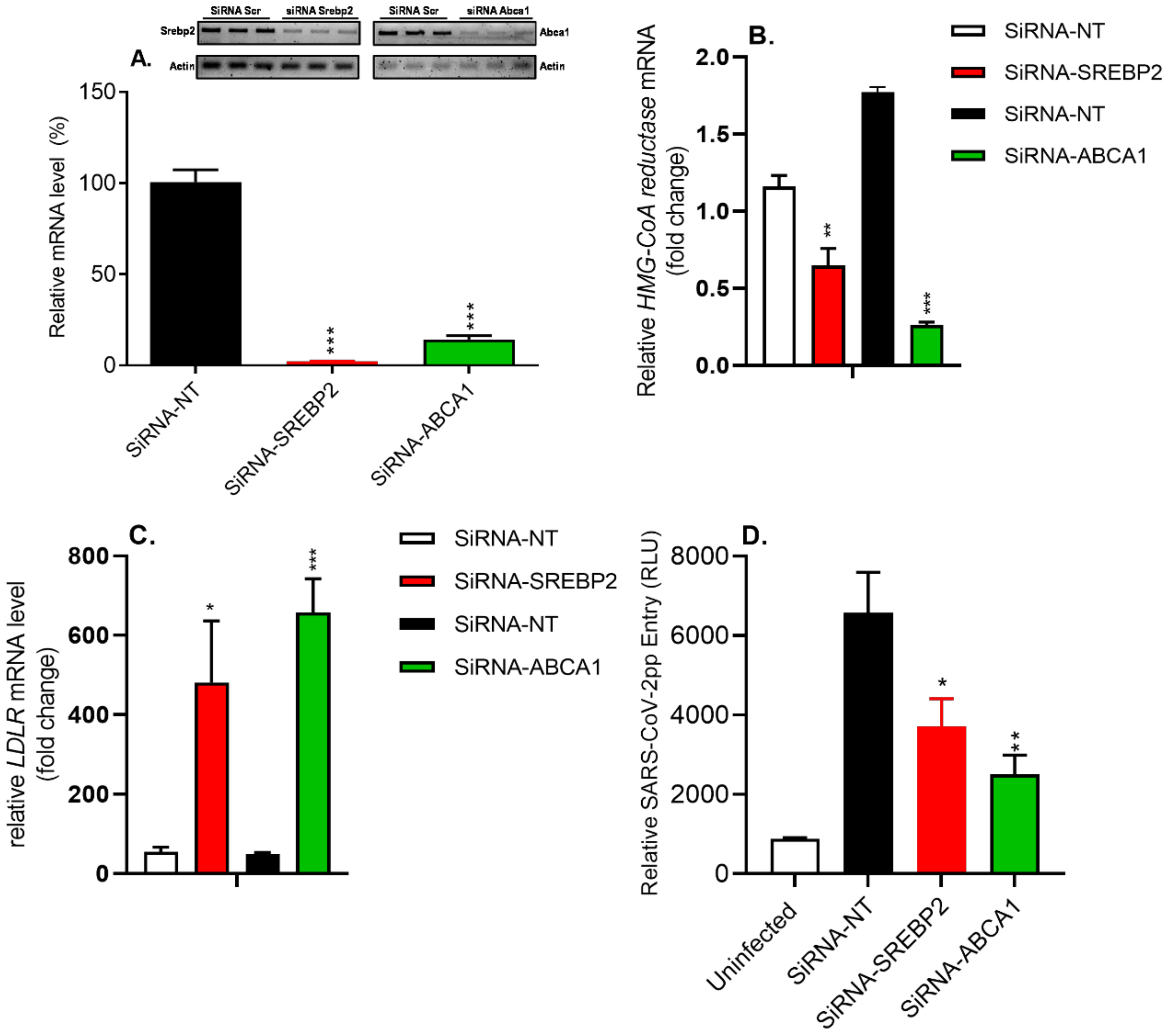
3.8. siRNA-Mediated Silencing of SREBP2 and ABCA1 Genes Reduced SARS-CoV-2pp Entry into Cells
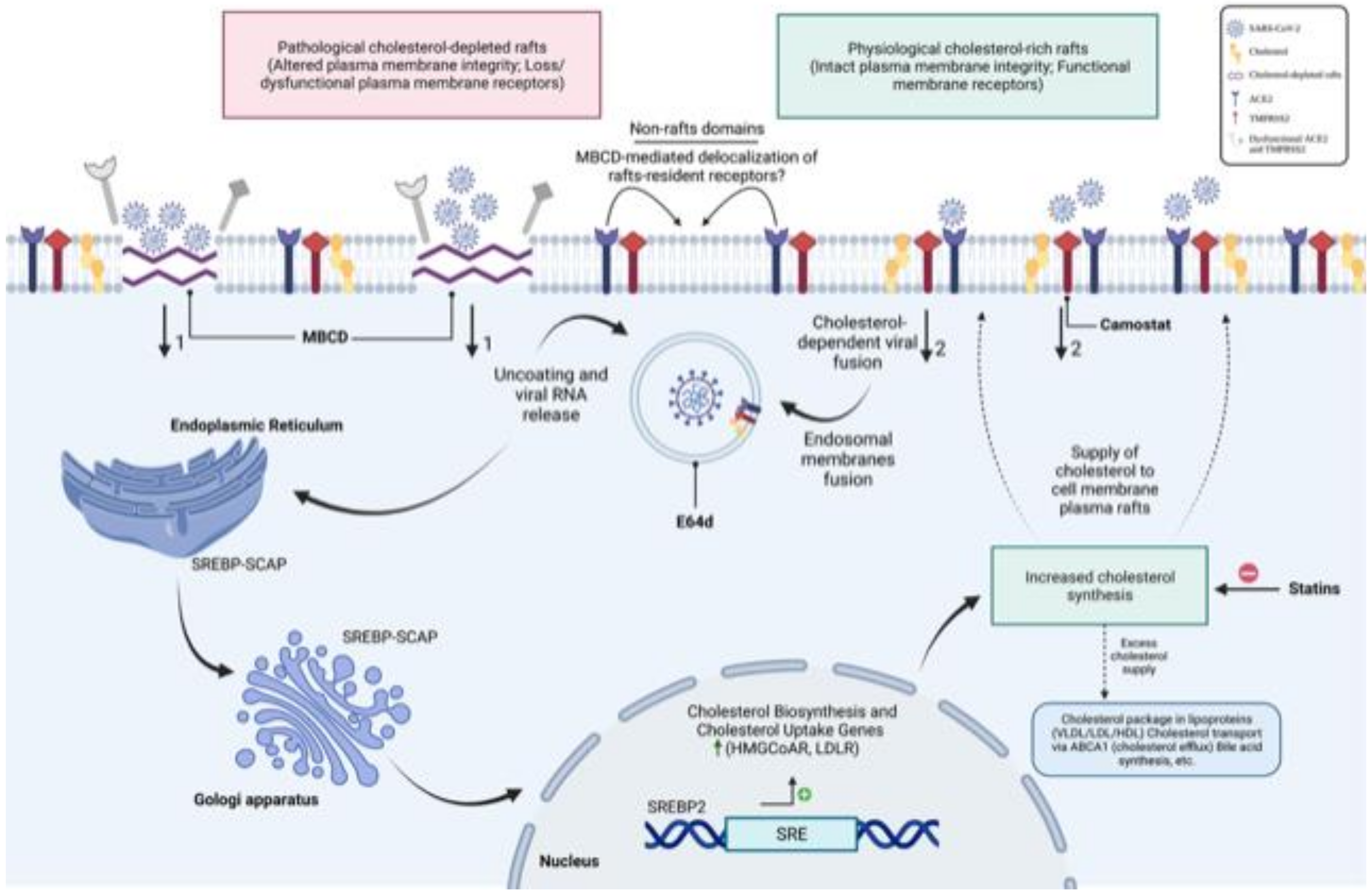
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mercer, J.; Schelhaas, M.; Helenius, A. Virus entry by endocytosis. Annu. Rev. Biochem. 2010, 79, 803–833. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Jia, H.; Tian, M.; Wu, N.; Yang, X.; Qi, J.; Ren, W.; Li, F.; Bian, H. SARS-CoV-2 and Emerging Variants: Unmasking Structure, Function, Infection, and Immune Escape Mechanisms. Front. Cell Infect. Microbiol. 2022, 12, 869832. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Petitjean, S.J.L.; Koehler, M.; Zhang, Q.; Dumitru, A.C.; Chen, W.; Derclaye, S.; Vincent, S.P.; Soumillion, P.; Alsteens, D. Molecular interaction and inhibition of SARS-CoV-2 binding to the ACE2 receptor. Nat. Commun. 2020, 11, 4541. [Google Scholar] [CrossRef] [PubMed]
- Yang, P.; Yang, Y.; Wu, Y.; Huang, C.; Ding, Y.; Wang, X.; Wang, S. An optimized and robust SARS-CoV-2 pseudovirus system for viral entry research. J. Virol. Methods. 2021, 295, 114221. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Kruger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280 e278. [Google Scholar] [CrossRef]
- Jackson, C.B.; Farzan, M.; Chen, B.; Choe, H. Mechanisms of SARS-CoV-2 entry into cells. Nat. Rev. Mol. Cell Biol. 2022, 23, 3–20. [Google Scholar] [CrossRef] [PubMed]
- Le Roy, C.; Wrana, J.L. Signaling and endocytosis: A team effort for cell migration. Dev. Cell 2005, 9, 167–168. [Google Scholar] [CrossRef]
- Varshney, P.; Yadav, V.; Saini, N. Lipid rafts in immune signalling: Current progress and future perspective. Immunology 2016, 149, 13–24. [Google Scholar] [CrossRef]
- Ouweneel, A.B.; Thomas, M.J.; Sorci-Thomas, M.G. The ins and outs of lipid rafts: Functions in intracellular cholesterol homeostasis, microparticles, and cell membranes: Thematic Review Series: Biology of Lipid Rafts. J. Lipid. Res. 2020, 61, 676–686. [Google Scholar] [CrossRef]
- Sviridov, D.; Mukhamedova, N.; Miller, Y.I. Lipid rafts as a therapeutic target. J. Lipid. Res. 2020, 61, 687–695. [Google Scholar] [CrossRef]
- Liao, Z.; Cimakasky, L.M.; Hampton, R.; Nguyen, D.H.; Hildreth, J.E. Lipid rafts and HIV pathogenesis: Host membrane cholesterol is required for infection by HIV type 1. AIDS Res. Hum. Retrovir. 2001, 17, 1009–1019. [Google Scholar] [CrossRef] [PubMed]
- Chung, C.S.; Huang, C.Y.; Chang, W. Vaccinia virus penetration requires cholesterol and results in specific viral envelope proteins associated with lipid rafts. J. Virol. 2005, 79, 1623–1634. [Google Scholar] [CrossRef] [PubMed]
- Li, G.M.; Li, Y.G.; Yamate, M.; Li, S.M.; Ikuta, K. Lipid rafts play an important role in the early stage of severe acute respiratory syndrome-coronavirus life cycle. Microbes. Infect. 2007, 9, 96–102. [Google Scholar] [CrossRef] [PubMed]
- Bremer, C.M.; Bung, C.; Kott, N.; Hardt, M.; Glebe, D. Hepatitis B virus infection is dependent on cholesterol in the viral envelope. Cell Microbiol. 2009, 11, 249–260. [Google Scholar] [CrossRef] [PubMed]
- Dou, X.; Li, Y.; Han, J.; Zarlenga, D.S.; Zhu, W.; Ren, X.; Dong, N.; Li, X.; Li, G. Cholesterol of lipid rafts is a key determinant for entry and post-entry control of porcine rotavirus infection. BMC Vet. Res. 2018, 14, 45. [Google Scholar] [CrossRef]
- Kulkarni, R.; Wiemer, E.A.C.; Chang, W. Role of Lipid Rafts in Pathogen-Host Interaction—A Mini Review. Front. Immunol. 2021, 12, 815020. [Google Scholar] [CrossRef] [PubMed]
- Pike, L.J.; Miller, J.M. Cholesterol depletion delocalizes phosphatidylinositol bisphosphate and inhibits hormone-stimulated phosphatidylinositol turnover. J. Biol. Chem. 1998, 273, 22298–22304. [Google Scholar] [CrossRef]
- Sanders, D.W.; Jumper, C.C.; Ackerman, P.J.; Bracha, D.; Donlic, A.; Kim, H.; Kenney, D.; Castello-Serrano, I.; Suzuki, S.; Tamura, T.; et al. SARS-CoV-2 requires cholesterol for viral entry and pathological syncytia formation. Elife 2021, 10, e65962. [Google Scholar] [CrossRef]
- Schmidt, N.M.; Wing, P.A.C.; McKeating, J.A.; Maini, M.K. Cholesterol-modifying drugs in COVID-19. Oxf Open Immunol. 2020, 1, iqaa001. [Google Scholar] [CrossRef]
- Wang, H.; Yuan, Z.; Pavel, M.A.; Jablonski, S.M.; Jablonski, J.; Hobson, R.; Valente, S.; Reddy, C.B.; Hansen, S.B. The role of high cholesterol in age-related COVID-19 lethality. Biorxiv 2021. [Google Scholar] [CrossRef]
- Kluck, G.E.G.; Yoo, J.A.; Sakarya, E.H.; Trigatti, B.L. Good Cholesterol Gone Bad? HDL and COVID-19. Int. J. Mol. Sci. 2021, 22, 10182. [Google Scholar] [CrossRef] [PubMed]
- Guyader, M.; Kiyokawa, E.; Abrami, L.; Turelli, P.; Trono, D. Role for human immunodeficiency virus type 1 membrane cholesterol in viral internalization. J. Virol. 2002, 76, 10356–10364. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Whittaker, G.R. Role for influenza virus envelope cholesterol in virus entry and infection. J. Virol. 2003, 77, 12543–12551. [Google Scholar] [CrossRef] [PubMed]
- Funk, A.; Mhamdi, M.; Hohenberg, H.; Heeren, J.; Reimer, R.; Lambert, C.; Prange, R.; Sirma, H. Duck hepatitis B virus requires cholesterol for endosomal escape during virus entry. J. Virol. 2008, 82, 10532–10542. [Google Scholar] [CrossRef]
- Umashankar, M.; Sanchez-San Martin, C.; Liao, M.; Reilly, B.; Guo, A.; Taylor, G.; Kielian, M. Differential cholesterol binding by class II fusion proteins determines membrane fusion properties. J. Virol. 2008, 82, 9245–9253. [Google Scholar] [CrossRef]
- Almasaud, A.; Alharbi, N.K.; Hashem, A.M. Generation of MERS-CoV Pseudotyped Viral Particles for the Evaluation of Neutralizing Antibodies in Mammalian Sera. Methods Mol. Biol. 2020, 2099, 117–126. [Google Scholar] [CrossRef]
- Alserehi, H.A.; Alqunaibet, A.M.; Al-Tawfiq, J.A.; Alharbi, N.K.; Alshukairi, A.N.; Alanazi, K.H.; Bin Saleh, G.M.; Alshehri, A.M.; Almasoud, A.; Hashem, A.M.; et al. Seroprevalence of SARS-CoV-2 (COVID-19) among healthcare workers in Saudi Arabia: Comparing case and control hospitals. Diagn. Microbiol. Infect. Dis. 2021, 99, 115273. [Google Scholar] [CrossRef]
- Mahammad, S.; Parmryd, I. Cholesterol depletion using methyl-beta-cyclodextrin. Methods Mol. Biol. 2015, 1232, 91–102. [Google Scholar] [CrossRef]
- Alharbi, N.K.; Qasim, I.; Almasoud, A.; Aljami, H.A.; Alenazi, M.W.; Alhafufi, A.; Aldibasi, O.S.; Hashem, A.M.; Kasem, S.; Albrahim, R.; et al. Humoral Immunogenicity and Efficacy of a Single Dose of ChAdOx1 MERS Vaccine Candidate in Dromedary Camels. Sci. Rep. 2019, 9, 16292. [Google Scholar] [CrossRef]
- Alluhaybi, K.A.; Alharbi, R.H.; Alhabbab, R.Y.; Aljehani, N.D.; Alamri, S.S.; Basabrain, M.; Alharbi, R.; Abdulaal, W.H.; Alfaleh, M.A.; Tamming, L.; et al. Cellular and Humoral Immunogenicity of a Candidate DNA Vaccine Expressing SARS-CoV-2 Spike Subunit 1. Vaccines 2021, 9, 852. [Google Scholar] [CrossRef]
- Guo, H.; Huang, M.; Yuan, Q.; Wei, Y.; Gao, Y.; Mao, L.; Gu, L.; Tan, Y.W.; Zhong, Y.; Liu, D.; et al. The Important Role of Lipid Raft-Mediated Attachment in the Infection of Cultured Cells by Coronavirus Infectious Bronchitis Virus Beaudette Strain. PLoS ONE 2017, 12, e0170123. [Google Scholar] [CrossRef]
- Iqbal, J.; Rudel, L.L.; Hussain, M.M. Microsomal triglyceride transfer protein enhances cellular cholesteryl esterification by relieving product inhibition. J. Biol. Chem. 2008, 283, 19967–19980. [Google Scholar] [CrossRef] [PubMed]
- Laporte, M.; Raeymaekers, V.; Van Berwaer, R.; Vandeput, J.; Marchand-Casas, I.; Thibaut, H.J.; Van Looveren, D.; Martens, K.; Hoffmann, M.; Maes, P.; et al. The SARS-CoV-2 and other human coronavirus spike proteins are fine-tuned towards temperature and proteases of the human airways. PLoS Pathog 2021, 17, e1009500. [Google Scholar] [CrossRef] [PubMed]
- Simons, P.; Rinaldi, D.A.; Bondu, V.; Kell, A.M.; Bradfute, S.; Lidke, D.; Buranda, T. Integrin activation is an essential component of SARS-CoV-2 infection. BioRxiv 2021, 11, 20398. [Google Scholar] [CrossRef] [PubMed]
- Simons, K.; Vaz, W.L. Model systems, lipid rafts, and cell membranes. Annu. Rev. Biophys. Biomol. Struct. 2004, 33, 269–295. [Google Scholar] [CrossRef] [PubMed]
- Radenkovic, D.; Chawla, S.; Pirro, M.; Sahebkar, A.; Banach, M. Cholesterol in Relation to COVID-19: Should We Care about It? J. Clin. Med. 2020, 9, 1909. [Google Scholar] [CrossRef]
- Wang, S.; Li, W.; Hui, H.; Tiwari, S.K.; Zhang, Q.; Croker, B.A.; Rawlings, S.; Smith, D.; Carlin, A.F.; Rana, T.M. Cholesterol 25-Hydroxylase inhibits SARS-CoV-2 and other coronaviruses by depleting membrane cholesterol. EMBO J. 2020, 39, e106057. [Google Scholar] [CrossRef] [PubMed]
- Cure, E.; Cumhur Cure, M. Strong relationship between cholesterol, low-density lipoprotein receptor, Na(+)/H(+) exchanger, and SARS-CoV-2: This association may be the cause of death in the patient with COVID-19. Lipids. Health Dis. 2021, 20, 179. [Google Scholar] [CrossRef]
- Zhao, M.; Luo, Z.; He, H.; Shen, B.; Liang, J.; Zhang, J.; Ye, J.; Xu, Y.; Wang, Z.; Ye, D.; et al. Decreased Low-Density Lipoprotein Cholesterol Level Indicates Poor Prognosis of Severe and Critical COVID-19 Patients: A Retrospective, Single-Center Study. Front. Med. (Lausanne) 2021, 8, 585851. [Google Scholar] [CrossRef]
- Masana, L.; Correig, E.; Ibarretxe, D.; Anoro, E.; Arroyo, J.A.; Jerico, C.; Guerrero, C.; Miret, M.; Naf, S.; Pardo, A.; et al. Low HDL and high triglycerides predict COVID-19 severity. Sci. Rep. 2021, 11, 7217. [Google Scholar] [CrossRef]
- Daniels, L.B.; Sitapati, A.M.; Zhang, J.; Zou, J.; Bui, Q.M.; Ren, J.; Longhurst, C.A.; Criqui, M.H.; Messer, K. Relation of Statin Use Prior to Admission to Severity and Recovery Among COVID-19 Inpatients. Am. J. Cardiol. 2020, 136, 149–155. [Google Scholar] [CrossRef]
- Chazal, N.; Gerlier, D. Virus entry, assembly, budding, and membrane rafts. Microbiol. Mol. Biol. Rev. 2003, 67, 226–237, table of contents. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Suzuki, Y. Virus infection and lipid rafts. Biol. Pharm. Bull. 2006, 29, 1538–1541. [Google Scholar] [CrossRef] [PubMed]
- Zidovetzki, R.; Levitan, I. Use of cyclodextrins to manipulate plasma membrane cholesterol content: Evidence, misconceptions and control strategies. Biochim. Biophys. Acta 2007, 1768, 1311–1324. [Google Scholar] [CrossRef] [PubMed]
- Hotta, K.; Bazartseren, B.; Kaku, Y.; Noguchi, A.; Okutani, A.; Inoue, S.; Yamada, A. Effect of cellular cholesterol depletion on rabies virus infection. Virus. Res. 2009, 139, 85–90. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Li, C.; Fan, F.; Liu, N.; Boadi, F.; Shen, X.; Hao, B. Methyl-Beta-Cyclodextrin-Induced Macropinocytosis Results in Increased Infection of Sf21 Cells by Bombyx Mori Nucleopolyhedrovirus. Viruses 2019, 11, 937. [Google Scholar] [CrossRef]
- Barman, S.; Nayak, D.P. Lipid raft disruption by cholesterol depletion enhances influenza A virus budding from MDCK cells. J. Virol. 2007, 81, 12169–12178. [Google Scholar] [CrossRef] [PubMed]
- Fujita, H.; Tamai, K.; Kawachi, M.; Saga, K.; Shimbo, T.; Yamazaki, T.; Kaneda, Y. Methyl-beta cyclodextrin alters the production and infectivity of Sendai virus. Arch. Virol. 2011, 156, 995–1005. [Google Scholar] [CrossRef]
- Palacios-Rapalo, S.N.; De Jesus-Gonzalez, L.A.; Cordero-Rivera, C.D.; Farfan-Morales, C.N.; Osuna-Ramos, J.F.; Martinez-Mier, G.; Quistian-Galvan, J.; Munoz-Perez, A.; Bernal-Dolores, V.; Del Angel, R.M.; et al. Cholesterol-Rich Lipid Rafts as Platforms for SARS-CoV-2 Entry. Front. Immunol. 2021, 12, 796855. [Google Scholar] [CrossRef]
- Li, X.; Zhu, W.; Fan, M.; Zhang, J.; Peng, Y.; Huang, F.; Wang, N.; He, L.; Zhang, L.; Holmdahl, R.; et al. Dependence of SARS-CoV-2 infection on cholesterol-rich lipid raft and endosomal acidification. Comput. Struct. Biotechnol. J. 2021, 19, 1933–1943. [Google Scholar] [CrossRef]
- Roncato, R.; Angelini, J.; Pani, A.; Talotta, R. Lipid rafts as viral entry routes and immune platforms: A double-edged sword in SARS-CoV-2 infection? Biochim. Biophys. Acta Mol. Cell Biol. Lipids. 2022, 1867, 159140. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Liu, S.; Shen, S.; Guo, H.; Huang, H.; Wei, W. Methyl-beta-cyclodextrin inhibits EV-D68 virus entry by perturbing the accumulation of virus particles and ICAM-5 in lipid rafts. Antiviral. Res. 2020, 176, 104752. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhang, Y.; Zhang, C.; Hu, M.; Yan, Q.; Zhao, H.; Zhang, X.; Wu, Y. Cholesterol-Rich Lipid Rafts in the Cellular Membrane Play an Essential Role in Avian Reovirus Replication. Front. Microbiol. 2020, 11, 597794. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Perry, J.W.; Lauring, A.S.; Neddermann, P.; De Francesco, R.; Tai, A.W. Oxysterol-binding protein is a phosphatidylinositol 4-kinase effector required for HCV replication membrane integrity and cholesterol trafficking. Gastroenterology 2014, 146, 1373–1385.e11. [Google Scholar] [CrossRef] [PubMed]
- Subczynski, W.K.; Pasenkiewicz-Gierula, M.; Widomska, J.; Mainali, L.; Raguz, M. High Cholesterol/Low Cholesterol: Effects in Biological Membranes: A Review. Cell Biochem. Biophys. 2017, 75, 369–385. [Google Scholar] [CrossRef] [PubMed]
- Ammendolia, D.A.; Bement, W.M.; Brumell, J.H. Plasma membrane integrity: Implications for health and disease. BMC Biol. 2021, 19, 71. [Google Scholar] [CrossRef]
- Silva, L.; Coutinho, A.; Fedorov, A.; Prieto, M. Nystatin-induced lipid vesicles permeabilization is strongly dependent on sterol structure. Biochim. Biophys. Acta 2006, 1758, 452–459. [Google Scholar] [CrossRef]
- Bieberich, E. Sphingolipids and lipid rafts: Novel concepts and methods of analysis. Chem. Phys. Lipids. 2018, 216, 114–131. [Google Scholar] [CrossRef]
- Carter, G.C.; Bernstone, L.; Sangani, D.; Bee, J.W.; Harder, T.; James, W. HIV entry in macrophages is dependent on intact lipid rafts. Virology 2009, 386, 192–202. [Google Scholar] [CrossRef]
- Zhang, X.J.; Qin, J.J.; Cheng, X.; Shen, L.; Zhao, Y.C.; Yuan, Y.; Lei, F.; Chen, M.M.; Yang, H.; Bai, L.; et al. In-Hospital Use of Statins Is Associated with a Reduced Risk of Mortality among Individuals with COVID-19. Cell Metab. 2020, 32, 176–187.e174. [Google Scholar] [CrossRef]
- Peymani, P.; Dehesh, T.; Aligolighasemabadi, F.; Sadeghdoust, M.; Kotfis, K.; Ahmadi, M.; Mehrbod, P.; Iranpour, P.; Dastghaib, S.; Nasimian, A.; et al. Statins in patients with COVID-19: A retrospective cohort study in Iranian COVID-19 patients. Transl. Med. Commun. 2021, 6, 3. [Google Scholar] [CrossRef] [PubMed]
- Ayeh, S.K.; Abbey, E.J.; Khalifa, B.A.A.; Nudotor, R.D.; Osei, A.D.; Chidambaram, V.; Osuji, N.; Khan, S.; Salia, E.L.; Oduwole, M.O.; et al. Statins use and COVID-19 outcomes in hospitalized patients. PLoS ONE 2021, 16, e0256899. [Google Scholar] [CrossRef]
- Diaz-Arocutipa, C.; Melgar-Talavera, B.; Alvarado-Yarasca, A.; Saravia-Bartra, M.M.; Cazorla, P.; Belzusarri, I.; Hernandez, A.V. Statins reduce mortality in patients with COVID-19: An updated meta-analysis of 147 824 patients. Int. J. Infect. Dis. 2021, 110, 374–381. [Google Scholar] [CrossRef] [PubMed]
- Lohia, P.; Kapur, S.; Benjaram, S.; Cantor, Z.; Mahabadi, N.; Mir, T.; Badr, M.S. Statins and clinical outcomes in hospitalized COVID-19 patients with and without Diabetes Mellitus: A retrospective cohort study with propensity score matching. Cardiovasc. Diabetol. 2021, 20, 140. [Google Scholar] [CrossRef]
- Bergqvist, R.; Ahlqvist, V.H.; Lundberg, M.; Hergens, M.P.; Sundstrom, J.; Bell, M.; Magnusson, C. HMG-CoA reductase inhibitors and COVID-19 mortality in Stockholm, Sweden: A registry-based cohort study. PLoS Med. 2021, 18, e1003820. [Google Scholar] [CrossRef] [PubMed]
- Hariyanto, T.I.; Kurniawan, A. Statin and outcomes of coronavirus disease 2019 (COVID-19): A systematic review, meta-analysis, and meta-regression. Nutr. Metab. Cardiovasc. Dis. 2021, 31, 1662–1670. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.; Ahn, J.H.; Park, H.H.; Kim, H.N.; Kim, H.; Yoo, Y.; Shin, H.; Hong, K.S.; Jang, J.G.; Park, C.G.; et al. COVID-19-activated SREBP2 disturbs cholesterol biosynthesis and leads to cytokine storm. Signal Transduct. Target. Ther. 2020, 5, 186. [Google Scholar] [CrossRef]
- Taylor, F.R.; Kandutsch, A.A. Oxysterol binding protein. Chem. Phys. Lipids. 1985, 38, 187–194. [Google Scholar] [CrossRef]
- Brown, M.S.; Goldstein, J.L. The SREBP pathway: Regulation of cholesterol metabolism by proteolysis of a membrane-bound transcription factor. Cell 1997, 89, 331–340. [Google Scholar] [CrossRef]
- Goldstein, J.L.; Brown, M.S. The LDL receptor. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 431–438. [Google Scholar] [CrossRef]
- Zhang, L.; Reue, K.; Fong, L.G.; Young, S.G.; Tontonoz, P. Feedback regulation of cholesterol uptake by the LXR-IDOL-LDLR axis. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 2541–2546. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.S.; Goldstein, J.L. A proteolytic pathway that controls the cholesterol content of membranes, cells, and blood. Proc. Natl. Acad. Sci. USA 1999, 96, 11041–11048. [Google Scholar] [CrossRef] [PubMed]
- Bashore, A.C.; Liu, M.; Key, C.C.; Boudyguina, E.; Wang, X.; Carroll, C.M.; Sawyer, J.K.; Mullick, A.E.; Lee, R.G.; Macauley, S.L.; et al. Targeted Deletion of Hepatocyte Abca1 Increases Plasma HDL (High-Density Lipoprotein) Reverse Cholesterol Transport via the LDL (Low-Density Lipoprotein) Receptor. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 1747–1761. [Google Scholar] [CrossRef]
- Joyce, C.W.; Wagner, E.M.; Basso, F.; Amar, M.J.; Freeman, L.A.; Shamburek, R.D.; Knapper, C.L.; Syed, J.; Wu, J.; Vaisman, B.L.; et al. ABCA1 overexpression in the liver of LDLr-KO mice leads to accumulation of pro-atherogenic lipoproteins and enhanced atherosclerosis. J. Biol. Chem. 2006, 281, 33053–33065. [Google Scholar] [CrossRef] [PubMed]
- Bocchetta, S.; Maillard, P.; Yamamoto, M.; Gondeau, C.; Douam, F.; Lebreton, S.; Lagaye, S.; Pol, S.; Helle, F.; Plengpanich, W.; et al. Up-regulation of the ATP-binding cassette transporter A1 inhibits hepatitis C virus infection. PLoS ONE 2014, 9, e92140. [Google Scholar] [CrossRef]
- Cui, H.L.; Grant, A.; Mukhamedova, N.; Pushkarsky, T.; Jennelle, L.; Dubrovsky, L.; Gaus, K.; Fitzgerald, M.L.; Sviridov, D.; Bukrinsky, M. HIV-1 Nef mobilizes lipid rafts in macrophages through a pathway that competes with ABCA1-dependent cholesterol efflux. J. Lipid. Res. 2012, 53, 696–708. [Google Scholar] [CrossRef] [PubMed]
- Morrow, M.P.; Grant, A.; Mujawar, Z.; Dubrovsky, L.; Pushkarsky, T.; Kiselyeva, Y.; Jennelle, L.; Mukhamedova, N.; Remaley, A.T.; Kashanchi, F.; et al. Stimulation of the liver X receptor pathway inhibits HIV-1 replication via induction of ATP-binding cassette transporter A1. Mol. Pharmacol. 2010, 78, 215–225. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, A.V.; Valuev-Elliston, V.T.; Ivanova, O.N.; Kochetkov, S.N.; Starodubova, E.S.; Bartosch, B.; Isaguliants, M.G. Oxidative Stress during HIV Infection: Mechanisms and Consequences. Oxid. Med. Cell. Longev. 2016, 2016, 8910396. [Google Scholar] [CrossRef] [PubMed]
- Cloherty, A.P.M.; Olmstead, A.D.; Ribeiro, C.M.S.; Jean, F. Hijacking of Lipid Droplets by Hepatitis C, Dengue and Zika Viruses-From Viral Protein Moonlighting to Extracellular Release. Int. J. Mol. Sci. 2020, 21, 7901. [Google Scholar] [CrossRef]
- Tornquist, K.; Asghar, M.Y.; Srinivasan, V.; Korhonen, L.; Lindholm, D. Sphingolipids as Modulators of SARS-CoV-2 Infection. Front. Cell Dev. Biol. 2021, 9, 689854. [Google Scholar] [CrossRef]










Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bakillah, A.; Hejji, F.A.; Almasaud, A.; Jami, H.A.; Hawwari, A.; Qarni, A.A.; Iqbal, J.; Alharbi, N.K. Lipid Raft Integrity and Cellular Cholesterol Homeostasis Are Critical for SARS-CoV-2 Entry into Cells. Nutrients 2022, 14, 3417. https://doi.org/10.3390/nu14163417
Bakillah A, Hejji FA, Almasaud A, Jami HA, Hawwari A, Qarni AA, Iqbal J, Alharbi NK. Lipid Raft Integrity and Cellular Cholesterol Homeostasis Are Critical for SARS-CoV-2 Entry into Cells. Nutrients. 2022; 14(16):3417. https://doi.org/10.3390/nu14163417
Chicago/Turabian StyleBakillah, Ahmed, Fatimah Al Hejji, Abdulrahman Almasaud, Haya Al Jami, Abbas Hawwari, Ali Al Qarni, Jahangir Iqbal, and Naif Khalaf Alharbi. 2022. "Lipid Raft Integrity and Cellular Cholesterol Homeostasis Are Critical for SARS-CoV-2 Entry into Cells" Nutrients 14, no. 16: 3417. https://doi.org/10.3390/nu14163417