
ER-Stress and Senescence Coordinately Promote Endothelial Barrier Dysfunction in Diabetes-Induced Atherosclerosis

 , , , , and
, , , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods:
2.1. Reagents
2.2. Mice
2.3. Atherosclerotic Mouse Models
2.4. Cell Culture
2.5. Immunofluorescence Staining
2.6. Senescence-Associated Beta Galactosidase Staining
2.7. Immunoblotting
2.8. Fluorescein Isothiocyanate (FITC)-Dextran Vascular Endothelial Cells Permeability Assay
2.9. Transendothelial Electrical Resistance (TEER) Assay
2.10. Statistical Analysis
3. Results
3.1. High Glucose-Induced Senescence Promotes Barrier Disruption in Vascular Endothelial Cells
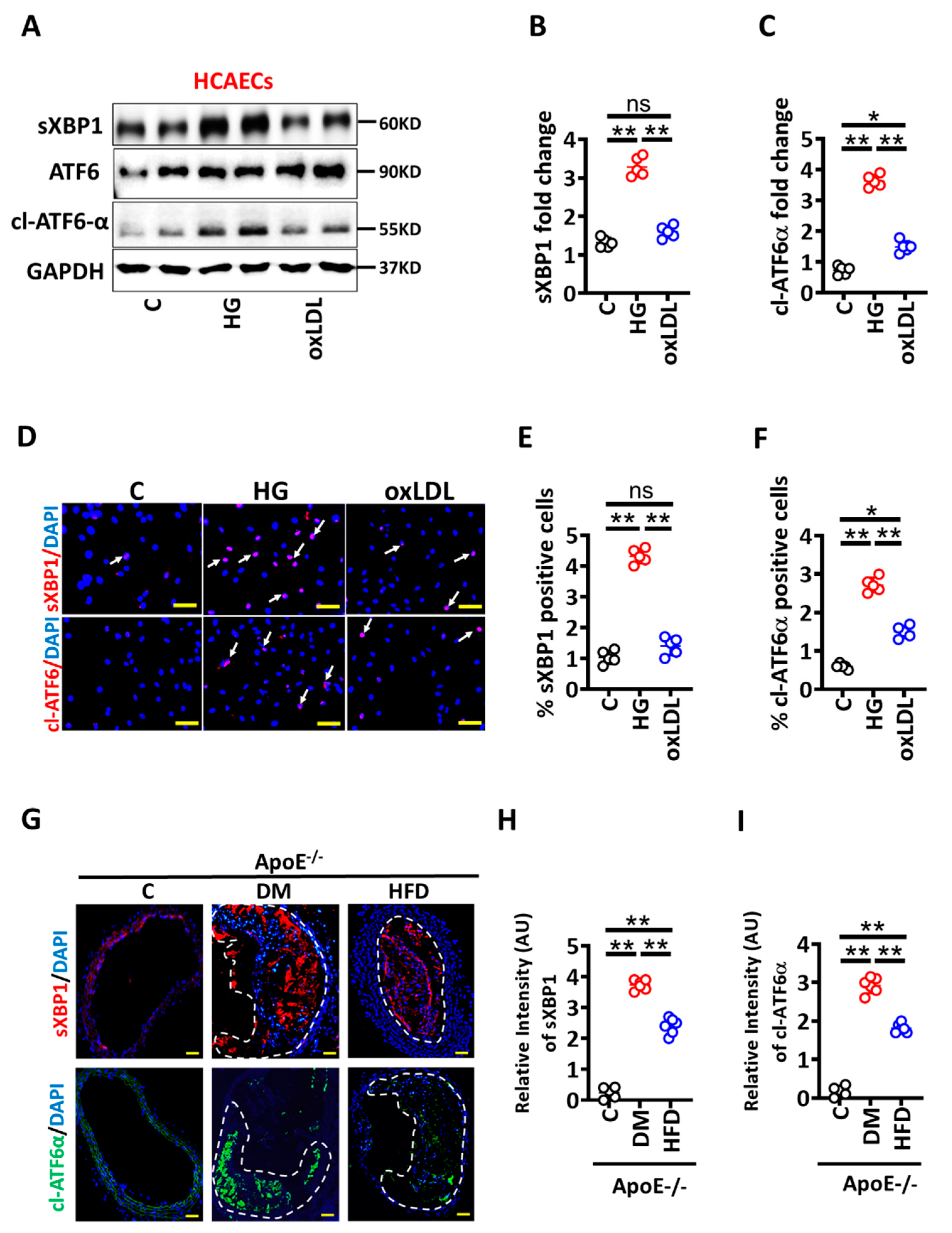
3.2. High Glucose Mediated Maladaptive UPR Signalling Impairs Vascular Endothelial Barrier Integrity
3.3. Activated Protein C Restricts the UPR and Prevents High Glucose-Induced Senescence in HCAECs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| aPC | activated protein C |
| ApoE | apolipoprotein E |
| ASCVD | atherosclerotic cardiovascular disease |
| ANOVA | analysis of variance |
| DM | diabetes mellitus |
| DAPI | 4′,6-diamidino-2-phenylindole |
| DEPC | diethyl pyrocarbonate |
| EPCR | endothelial protein C receptor |
| NF-κB | nuclear factor kappa-light-chain-enhancer of activated B cells |
| qPCR | quantitative polymerase chain reaction |
| ROS | reactive oxygen species |
| RT-PCR | reverse transcriptase polymerase chain reaction |
| SMC | smooth muscle cells |
| α-SMC | alpha-smooth muscle actin |
| SEM | standard error mean |
| STZ | streptozotocin |
| TM | thrombomodulin |
References
- Benjamin, E.J.; Muntner, P.; Alonso, A.; Bittencourt, M.S.; Callaway, C.W.; Carson, A.P.; Chamberlain, A.M.; Chang, A.R.; Cheng, S.; Das, S.R.; et al. Correction to: Heart Disease and Stroke Statistics-2019 Update: A Report From the American Heart Association. Circulation 2020, 141, e33. [Google Scholar] [CrossRef]
- Benjamin, E.J.; Muntner, P.; Alonso, A.; Bittencourt, M.S.; Callaway, C.W.; Carson, A.P.; Chamberlain, A.M.; Chang, A.R.; Cheng, S.; Das, S.R.; et al. Heart Disease and Stroke Statistics-2019 Update: A Report From the American Heart Association. Circulation 2019, 139, e56–e528. [Google Scholar] [CrossRef] [PubMed]
- Stitziel, N.O.; Kanter, J.E.; Bornfeldt, K.E. Emerging Targets for Cardiovascular Disease Prevention in Diabetes. Trends Mol. Med. 2020, 26, 744–757. [Google Scholar] [CrossRef] [PubMed]
- Maahs, D.M.; Daniels, S.R.; de Ferranti, S.D.; Dichek, H.L.; Flynn, J.; Goldstein, B.I.; Kelly, A.S.; Nadeau, K.J.; Martyn-Nemeth, P.; Osganian, S.K.; et al. Cardiovascular disease risk factors in youth with diabetes mellitus: A scientific statement from the American Heart Association. Circulation 2014, 130, 1532–1558. [Google Scholar] [CrossRef] [Green Version]
- Engelbertsen, D.; To, F.; Duner, P.; Kotova, O.; Soderberg, I.; Alm, R.; Gomez, M.F.; Nilsson, J.; Bengtsson, E. Increased inflammation in atherosclerotic lesions of diabetic Akita-LDLr(-)/(-) mice compared to nondiabetic LDLr(-)/(-) mice. Exp. Diabetes Res. 2012, 2012, 176162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Senatus, L.; Lopez-Diez, R.; Egana-Gorrono, L.; Liu, J.; Hu, J.; Daffu, G.; Li, Q.; Rahman, K.; Vengrenyuk, Y.; Barrett, T.J.; et al. RAGE impairs murine diabetic atherosclerosis regression and implicates IRF7 in macrophage inflammation and cholesterol metabolism. JCI Insight 2020, 5, e137289. [Google Scholar] [CrossRef]
- Nagareddy, P.R.; Murphy, A.J.; Stirzaker, R.A.; Hu, Y.; Yu, S.; Miller, R.G.; Ramkhelawon, B.; Distel, E.; Westerterp, M.; Huang, L.S.; et al. Hyperglycemia promotes myelopoiesis and impairs the resolution of atherosclerosis. Cell Metab. 2013, 17, 695–708. [Google Scholar] [CrossRef] [Green Version]
- Corti, R.; Fuster, V.; Fayad, Z.A.; Worthley, S.G.; Helft, G.; Chaplin, W.F.; Muntwyler, J.; Viles-Gonzalez, J.F.; Weinberger, J.; Smith, D.A.; et al. Effects of aggressive versus conventional lipid-lowering therapy by simvastatin on human atherosclerotic lesions: A prospective, randomized, double-blind trial with high-resolution magnetic resonance imaging. J. Am. Coll. Cardiol. 2005, 46, 106–112. [Google Scholar] [CrossRef]
- Hiro, T.; Kimura, T.; Morimoto, T.; Miyauchi, K.; Nakagawa, Y.; Yamagishi, M.; Ozaki, Y.; Kimura, K.; Saito, S.; Yamaguchi, T.; et al. Diabetes mellitus is a major negative determinant of coronary plaque regression during statin therapy in patients with acute coronary syndrome--serial intravascular ultrasound observations from the Japan Assessment of Pitavastatin and Atorvastatin in Acute Coronary Syndrome Trial (the JAPAN-ACS Trial). Circ. J. 2010, 74, 1165–1174. [Google Scholar] [CrossRef] [Green Version]
- Cholesterol Treatment Trialists’ (CTT) Collaboration; Baigent, C.; Blackwell, L.; Emberson, J.; Holland, L.E.; Reith, C.; Bhala, N.; Peto, R.; Barnes, E.H.; Keech, A.; et al. Efficacy and safety of more intensive lowering of LDL cholesterol: A meta-analysis of data from 170,000 participants in 26 randomised trials. Lancet 2010, 376, 1670–1681. [Google Scholar] [CrossRef] [Green Version]
- Beckman, J.A.; Creager, M.A. Vascular Complications of Diabetes. Circ. Res. 2016, 118, 1771–1785. [Google Scholar] [CrossRef] [Green Version]
- Madhusudhan, T.; Wang, H.; Ghosh, S.; Dong, W.; Kumar, V.; Al-Dabet, M.M.; Manoharan, J.; Nazir, S.; Elwakiel, A.; Bock, F.; et al. Signal integration at the PI3K-p85-XBP1 hub endows coagulation protease activated protein C with insulin-like function. Blood 2017, 130, 1445–1455. [Google Scholar] [CrossRef]
- Isermann, B.; Vinnikov, I.A.; Madhusudhan, T.; Herzog, S.; Kashif, M.; Blautzik, J.; Corat, M.A.; Zeier, M.; Blessing, E.; Oh, J.; et al. Activated protein C protects against diabetic nephropathy by inhibiting endothelial and podocyte apoptosis. Nat. Med. 2007, 13, 1349–1358. [Google Scholar] [CrossRef] [PubMed]
- Vukovich, T.C.; Schernthaner, G. Decreased protein C levels in patients with insulin-dependent type I diabetes mellitus. Diabetes 1986, 35, 617–619. [Google Scholar] [CrossRef] [PubMed]
- Laszik, Z.G.; Zhou, X.J.; Ferrell, G.L.; Silva, F.G.; Esmon, C.T. Down-regulation of endothelial expression of endothelial cell protein C receptor and thrombomodulin in coronary atherosclerosis. Am. J. Pathol. 2001, 159, 797–802. [Google Scholar] [CrossRef] [Green Version]
- Shahzad, K.; Gadi, I.; Nazir, S.; Al-Dabet, M.M.; Kohli, S.; Bock, F.; Breitenstein, L.; Ranjan, S.; Fuchs, T.; Halloul, Z.; et al. Activated protein C reverses epigenetically sustained p66(Shc) expression in plaque-associated macrophages in diabetes. Commun. Biol. 2018, 1, 104. [Google Scholar] [CrossRef]
- Gimbrone, M.A., Jr.; Garcia-Cardena, G. Endothelial Cell Dysfunction and the Pathobiology of Atherosclerosis. Circ. Res. 2016, 118, 620–636. [Google Scholar] [CrossRef] [Green Version]
- Walter, P.; Ron, D. The unfolded protein response: From stress pathway to homeostatic regulation. Science 2011, 334, 1081–1086. [Google Scholar] [CrossRef] [Green Version]
- Prasad, M.K.; Mohandas, S.; Ramkumar, K.M. Role of ER stress inhibitors in the management of diabetes. Eur. J. Pharmacol. 2022, 922, 174893. [Google Scholar] [CrossRef]
- Tabas, I. The role of endoplasmic reticulum stress in the progression of atherosclerosis. Circ. Res. 2010, 107, 839–850. [Google Scholar] [CrossRef]
- Seehaus, S.; Shahzad, K.; Kashif, M.; Vinnikov, I.A.; Schiller, M.; Wang, H.; Madhusudhan, T.; Eckstein, V.; Bierhaus, A.; Bea, F.; et al. Hypercoagulability inhibits monocyte transendothelial migration through protease-activated receptor-1-, phospholipase-Cbeta-, phosphoinositide 3-kinase-, and nitric oxide-dependent signaling in monocytes and promotes plaque stability. Circulation 2009, 120, 774–784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bock, F.; Shahzad, K.; Wang, H.; Stoyanov, S.; Wolter, J.; Dong, W.; Pelicci, P.G.; Kashif, M.; Ranjan, S.; Schmidt, S.; et al. Activated protein C ameliorates diabetic nephropathy by epigenetically inhibiting the redox enzyme p66Shc. Proc. Natl. Acad. Sci. USA 2013, 110, 648–653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shahzad, K.; Bock, F.; Al-Dabet, M.M.; Gadi, I.; Nazir, S.; Wang, H.; Kohli, S.; Ranjan, S.; Mertens, P.R.; Nawroth, P.P.; et al. Stabilization of endogenous Nrf2 by minocycline protects against Nlrp3-inflammasome induced diabetic nephropathy. Sci. Rep. 2016, 6, 34228. [Google Scholar] [CrossRef] [PubMed]
- Childs, B.G.; Baker, D.J.; Wijshake, T.; Conover, C.A.; Campisi, J.; van Deursen, J.M. Senescent intimal foam cells are deleterious at all stages of atherosclerosis. Science 2016, 354, 472–477. [Google Scholar] [CrossRef]
- Machado-Oliveira, G.; Ramos, C.; Marques, A.R.A.; Vieira, O.V. Cell Senescence, Multiple Organelle Dysfunction and Atherosclerosis. Cells 2020, 9, 2146. [Google Scholar] [CrossRef] [PubMed]
- Pluquet, O.; Pourtier, A.; Abbadie, C. The unfolded protein response and cellular senescence. A review in the theme: Cellular mechanisms of endoplasmic reticulum stress signaling in health and disease. Am. J. Physiol. Cell Physiol. 2015, 308, C415–C425. [Google Scholar] [CrossRef] [Green Version]
- Abbadie, C.; Pluquet, O. Unfolded Protein Response (UPR) Controls Major Senescence Hallmarks. Trends Biochem. Sci. 2020, 45, 371–374. [Google Scholar] [CrossRef]
- Matsumoto, K.; Yano, Y.; Gabazza, E.C.; Araki, R.; Bruno, N.E.; Suematsu, M.; Akatsuka, H.; Katsuki, A.; Taguchi, O.; Adachi, Y.; et al. Inverse correlation between activated protein C generation and carotid atherosclerosis in Type 2 diabetic patients. Diabet. Med. 2007, 24, 1322–1328. [Google Scholar] [CrossRef]
- Haspula, D.; Vallejos, A.K.; Moore, T.M.; Tomar, N.; Dash, R.K.; Hoffmann, B.R. Influence of a Hyperglycemic Microenvironment on a Diabetic Versus Healthy Rat Vascular Endothelium Reveals Distinguishable Mechanistic and Phenotypic Responses. Front. Physiol. 2019, 10, 558. [Google Scholar] [CrossRef]
- Lv, J.; Yang, L.; Guo, R.; Shi, Y.; Zhang, Z.; Ye, J. Ox-LDL-Induced MicroRNA-155 Promotes Autophagy in Human Endothelial Cells via Repressing the Rheb/ mTOR Pathway. Cell Physiol. Biochem. 2017, 43, 1436–1448. [Google Scholar] [CrossRef] [Green Version]
- Yin, Y.; Li, X.; Sha, X.; Xi, H.; Li, Y.F.; Shao, Y.; Mai, J.; Virtue, A.; Lopez-Pastrana, J.; Meng, S.; et al. Early hyperlipidemia promotes endothelial activation via a caspase-1-sirtuin 1 pathway. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 804–816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dykun, I.; Bayturan, O.; Carlo, J.; Nissen, S.E.; Kapadia, S.R.; Tuzcu, E.M.; Nicholls, S.J.; Puri, R. HbA1c, Coronary atheroma progression and cardiovascular outcomes. Am. J. Prev. Cardiol. 2022, 9, 100317. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.C.; Jandeleit-Dahm, K.; Peter, K. Sodium-Glucose Co-Transporter 2 (SGLT2) Inhibitor Dapagliflozin Stabilizes Diabetes-Induced Atherosclerotic Plaque Instability. J. Am. Heart Assoc. 2022, 11, e022761. [Google Scholar] [CrossRef]
- Zaha, V.G.; Joshi, P.H.; McGuire, D.K. Probing for Vulnerable Plaque in Patients With Diabetes Mellitus. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 124–125. [Google Scholar] [CrossRef] [Green Version]
- Shahzad, K.; Kohli, S.; Al-Dabet, M.M.; Isermann, B. Cell biology of activated protein C. Curr. Opin. Hematol. 2019, 26, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Bock, F.; Shahzad, K.; Vergnolle, N.; Isermann, B. Activated protein C based therapeutic strategies in chronic diseases. Thromb. Haemost. 2014, 111, 610–617. [Google Scholar] [CrossRef]
- Nazir, S.; Gadi, I.; Al-Dabet, M.M.; Elwakiel, A.; Kohli, S.; Ghosh, S.; Manoharan, J.; Ranjan, S.; Bock, F.; Braun-Dullaeus, R.C.; et al. Cytoprotective activated protein C averts Nlrp3 inflammasome-induced ischemia-reperfusion injury via mTORC1 inhibition. Blood 2017, 130, 2664–2677. [Google Scholar] [CrossRef] [Green Version]
- Tufanli, O.; Telkoparan Akillilar, P.; Acosta-Alvear, D.; Kocaturk, B.; Onat, U.I.; Hamid, S.M.; Cimen, I.; Walter, P.; Weber, C.; Erbay, E. Targeting IRE1 with small molecules counteracts progression of atherosclerosis. Proc. Natl. Acad. Sci. USA 2017, 114, E1395–E1404. [Google Scholar] [CrossRef] [Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fatima, S.; Ambreen, S.; Mathew, A.; Elwakiel, A.; Gupta, A.; Singh, K.; Krishnan, S.; Rana, R.; Khawaja, H.; Gupta, D.; et al. ER-Stress and Senescence Coordinately Promote Endothelial Barrier Dysfunction in Diabetes-Induced Atherosclerosis. Nutrients 2022, 14, 2786. https://doi.org/10.3390/nu14142786
Fatima S, Ambreen S, Mathew A, Elwakiel A, Gupta A, Singh K, Krishnan S, Rana R, Khawaja H, Gupta D, et al. ER-Stress and Senescence Coordinately Promote Endothelial Barrier Dysfunction in Diabetes-Induced Atherosclerosis. Nutrients. 2022; 14(14):2786. https://doi.org/10.3390/nu14142786
Chicago/Turabian StyleFatima, Sameen, Saira Ambreen, Akash Mathew, Ahmed Elwakiel, Anubhuti Gupta, Kunal Singh, Shruthi Krishnan, Rajiv Rana, Hamzah Khawaja, Dheerendra Gupta, and et al. 2022. "ER-Stress and Senescence Coordinately Promote Endothelial Barrier Dysfunction in Diabetes-Induced Atherosclerosis" Nutrients 14, no. 14: 2786. https://doi.org/10.3390/nu14142786