
Therapeutic Potential of Thymoquinone in Triple-Negative Breast Cancer Prevention and Progression through the Modulation of the Tumor Microenvironment

 ,
,
Abstract
:1. Introduction
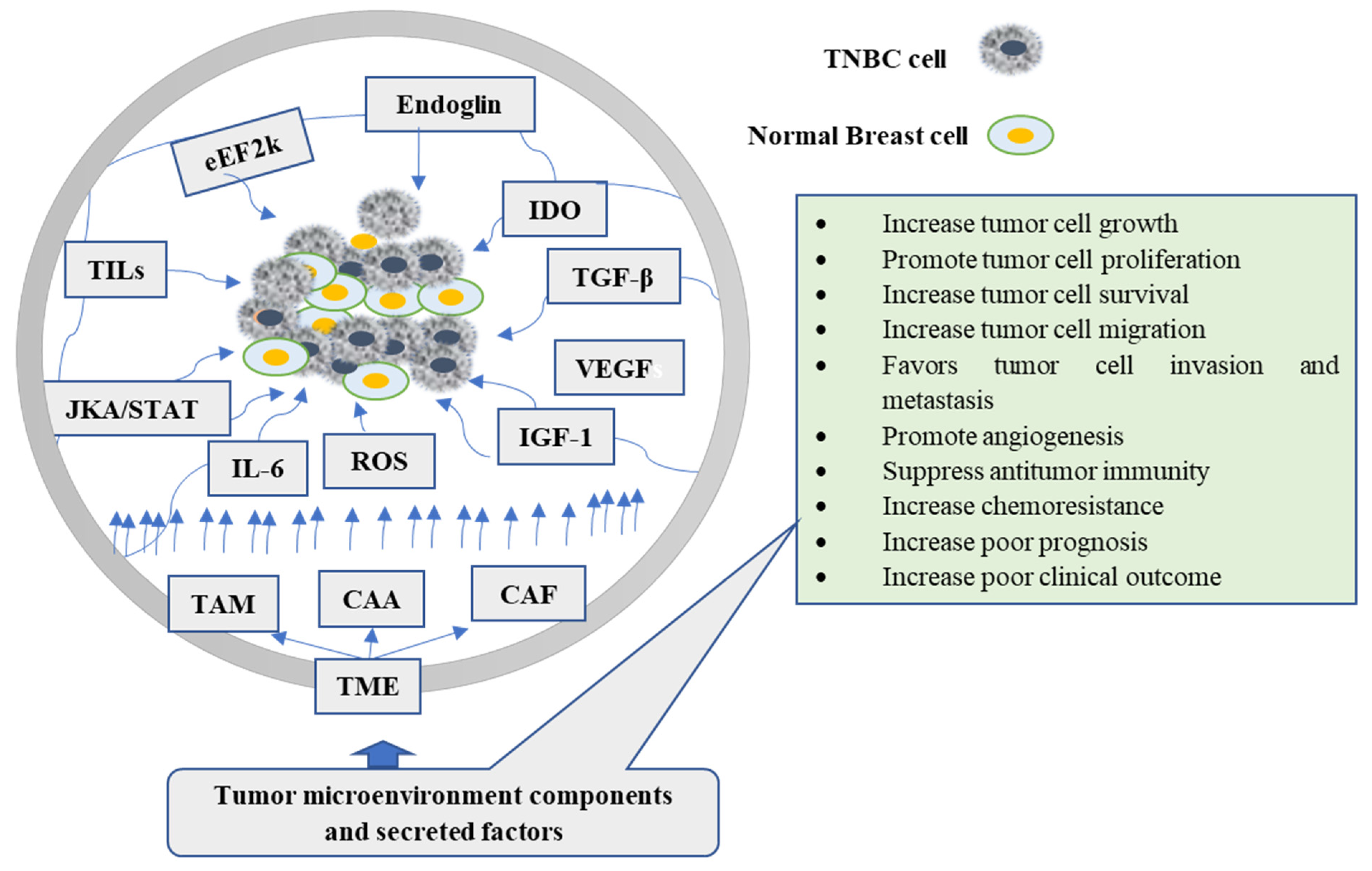
2. Tumor Microenvironment (TME) and TNBC
3. Natural Products Targeting TME
4. Thymoquinone (TQ)
5. Thymoquinone (TQ) Targeting the Cellular Components of TME
5.1. Effect of TQ on Cholesterol Synthesis and Its Metabolites
5.2. Effect of TQ on Reactive Oxygen Species (ROS)
5.3. Effect of TQ on Eukaryotic Elongation Factor-2 Kinase (eEF-2K)
5.4. Effect of TQ on Inflammatory and Immune Cells
5.4.1. Effect of TQ on Tumor-Associated Macrophage (TAM)
5.4.2. Effect of TQ on Cancer-Associated Adipocytes (CAAs)
5.4.3. Effect of TQ on Cancer-Associated Fibroblasts (CAFs)
5.4.4. Effect of TQ on Tumor-Infiltrating Lymphocytes (TILs)
5.4.5. Effect of TQ on Interleukin-6 (IL-6)
5.4.6. Effect of TQ on Janus Kinases-Signal Transducer and Activator of Transcription Factor (JAK-STAT)
5.5. Effect of TQ on Endothelial Cells
5.5.1. Effect of TQ on Vascular Endothelial Growth Factor-A (VEGF-A)
5.5.2. Effect of TQ on Transforming Growth Factor-β (TGFβ)
5.5.3. Effect of TQ on Insulin-Like Growth Factor I (IGF-I)
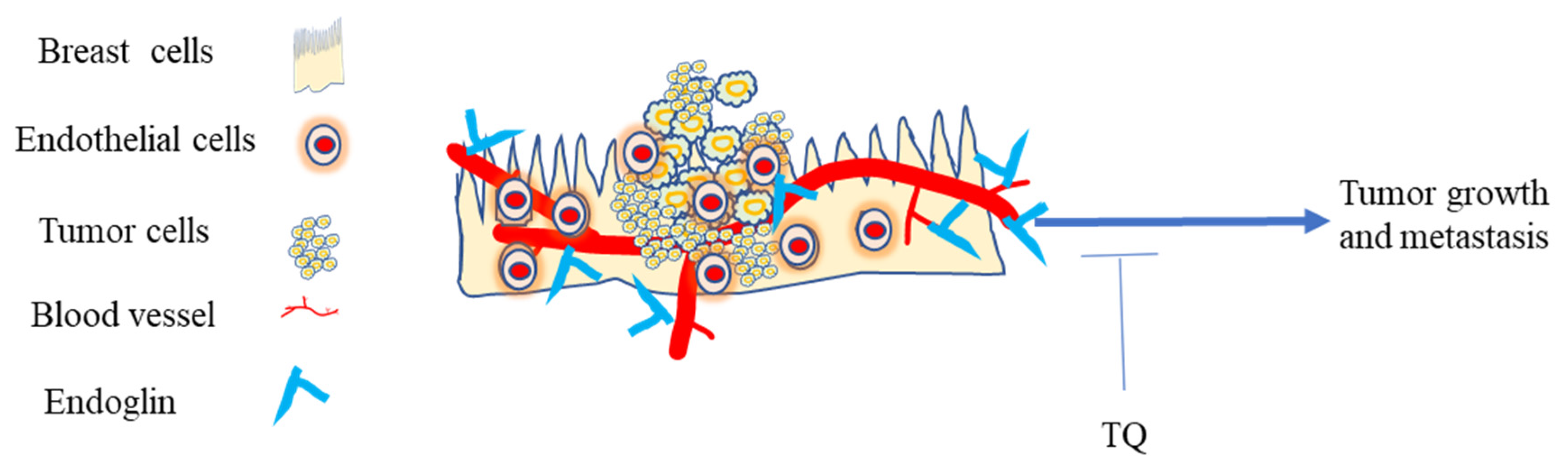
5.5.4. Effect of TQ on Endoglin
5.5.5. Effect of TQ on Indoleamine 2,3-dioxygenase (IDO)
6. Clinical Trials Have Shown the Importance of TQ in the Treatment of a Variety of Diseases
7. Limitations of Thymoquinone (TQ) as a Natural Product
8. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hsu, Y.-L.; Uen, Y.-H.; Chen, Y.; Liang, H.-L.; Kuo, P.-L. Tricetin, a dietary flavonoid, inhibits proliferation of human breast adenocarcinoma mcf-7 cells by blocking cell cycle progression and inducing apoptosis. J. Agric. Food. Chem. 2009, 57, 8688–8695. [Google Scholar] [CrossRef]
- Lei, S.; Zheng, R.; Zhang, S.; Wang, S.; Chen, R.; Sun, K.; Zeng, H.; Zhou, J.; Wei, W. Global patterns of breast cancer incidence and mortality: A population-based cancer registry data analysis from 2000 to 2020. Cancer Commun. 2021, 41, 1183–1194. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Feng, Q.; Yang, H.; Wang, G.; Huang, L.; Ba, Q.; Zhang, C.; Wang, Y.; Chen, Y.; Cheng, Q.; et al. A Light-Triggered Mesenchymal Stem Cell Delivery System for Photoacoustic Imaging and Chemo-Photothermal Therapy of Triple Negative Breast Cancer. Adv. Sci. 2018, 5, 1800382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anders, C.; Carey, L.A. Understanding and Treating Triple-Negative Breast Cancer. Oncology 2008, 22, 1233–1243. [Google Scholar] [PubMed]
- Thakur, K.K.; Bordoloi, D.; Kunnumakkara, A.B. Alarming Burden of Triple-Negative Breast Cancer in India. Clin. Breast Cancer 2018, 18, e393–e399. [Google Scholar] [CrossRef] [PubMed]
- Hwang, K.-T.; Kim, J.; Jung, J.; Chang, J.H.; Chai, Y.J.; Oh, S.W.; Oh, S.; Kim, Y.A.; Park, S.B.; Hwang, K.R. Impact of Breast Cancer Subtypes on Prognosis of Women with Operable Invasive Breast Cancer: A Population-based Study Using SEER Database. Clin. Cancer Res. 2019, 25, 1970–1979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Yu, T.; Di, G. Role of tumor microenvironment in triple-negative breast cancer and its prognostic significance. Chin. J. Cancer Res. 2017, 29, 237–252. [Google Scholar] [CrossRef]
- Lee, A.; Go, S.I.; Lee, W.S.; Lee, U.S.; Kim, M.J.; Kang, M.H.; Lee, G.W.; Kim, H.G.; Kang, J.H.; Jeon, K.N.; et al. Irinotecan and capecitabine combination chemotherapy in a patient with triple-negative breast cancer relapsed after adjuvant chemotherapy with anthracycline and taxane. Tumori 2015, 101, e9–e12. [Google Scholar] [CrossRef]
- Afghahi, A.; Timms, K.M.; Vinayak, S.; Jensen, K.C.; Kurian, A.W.; Carlson, R.W.; Chang, P.-J.; Schackmann, E.; Hartman, A.-R.; Ford, J.M.; et al. Tumor BRCA1 Reversion Mutation Arising During Neoadjuvant Platinum-Based Chemotherapy in Triple-Negative Breast Cancer Is Associated with Therapy Resistance. Clin. Cancer Res. 2017, 23, 3365–3370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, K.D.; Siegel, R.L.; Lin, C.C.; Mariotto, A.B.; Kramer, J.L.; Rowland, J.H.; Stein, K.D.; Alteri, R.; Jemal, A. Cancer treatment and survivorship statistics, 2016. CA Cancer J. Clin. 2016, 66, 271–289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oakman, C.; Moretti, E.; Galardi, F.; Biagioni, C.; Santarpia, L.; Biganzoli, L.; Leo, A.D. Adjuvant systemic treatment for individual patients with triple negative breast cancer. Breast Cancer Res. 2011, 3, S135–S141. [Google Scholar] [CrossRef]
- Hanahan, D.; Coussens, L.M. Accessories to the crime: Functions of cells recruited to the tumor microenvironment. Cancer Cell 2012, 21, 309–322. [Google Scholar] [CrossRef] [Green Version]
- Willumsen, N.; Thomsen, L.B.; Bager, C.L.; Jensen, C.; Karsdal, M.A. Quantification of altered tissue turnover in a liquid biopsy: A proposed precision medicine tool to assess chronic inflammation and desmoplasia associated with a pro-cancerous niche and response to immuno-therapeutic anti-tumor modalities. Cancer Immunol. Immunother. 2018, 67, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Schäfer, M.; Werner, S. Cancer as an overhealing wound: An old hypothesis revisited. Nat. Rev. Mol. Cell Biol. 2008, 9, 628–638. [Google Scholar] [CrossRef] [PubMed]
- Dvorak, H.F. Tumors: Wounds That Do Not Heal-Redux. Cancer Immunol. Res. 2015, 3, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Wong, A.Y.; Whited, J.L. Parallels between wound healing, epimorphic regeneration and solid tumors. Development 2020, 147, dev181636. [Google Scholar] [CrossRef]
- Joyce, J.A.; Pollard, J.W. Microenvironmental regulation of metastasis. Nat. Rev. Cancer 2009, 9, 239–252. [Google Scholar] [CrossRef]
- Liu, S.; Cong, Y.; Wang, D.; Sun, Y.; Deng, L.; Liu, Y.; Martin-Trevino, R.; Shang, L.; McDermott, S.P.; Landis, M.D.; et al. Breast cancer stem cells transition between epithelial and mesenchymal states reflective of their normal counterparts. Stem. Cell Rep. 2014, 2, 78–91. [Google Scholar] [CrossRef]
- Matsumoto, H.; Koo, S.L.; Dent, R.; Tan, P.H.; Iqbal, J. Role of inflammatory infiltrates in triple negative breast cancer. J. Clin. Pathol. 2015, 68, 506–510. [Google Scholar] [CrossRef]
- Esquivel-Velázquez, M.; Ostoa-Saloma, P.; Palacios-Arreola, M.I.; Nava-Castro, K.E.; Castro, J.I.; Morales-Montor, J. The role of cytokines in breast cancer development and progression. J. Interferon Cytokine Res. 2015, 35, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Salehi, B.; Stojanović-Radić, Z.; Matejić, J.; Sharifi-Rad, M.; Anil Kumar, N.V.; Martins, N.; Sharifi-Rad, J. The therapeutic potential of curcumin: A review of clinical trials. Eur. J. Med. Chem. 2019, 163, 527–545. [Google Scholar] [CrossRef] [PubMed]
- Zaynab, M.; Fatima, M.; Abbas, S.; Sharif, Y.; Umair, M.; Zafar, M.H.; Bahadar, K. Role of secondary metabolites in plant defense against pathogens. Microb. Pathog. 2018, 124, 198–202. [Google Scholar] [CrossRef] [PubMed]
- Zubair, H.; Azim, S.; Ahmad, A.; Khan, M.A.; Patel, G.K.; Singh, S.; Singh, A.P. Cancer Chemoprevention by Phytochemicals: Nature’s Healing Touch. Molecules 2017, 22, 395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gali-Muhtasib, H.; Roessner, A.; Schneider-Stock, R. Thymoquinone: A promising anti-cancer drug from natural sources. Int. J. Biochem. Cell Biol. 2006, 38, 1249–1253. [Google Scholar] [CrossRef] [PubMed]
- Vuorela, P.; Leinonen, M.; Saikku, P.; Tammela, P.; Rauha, J.-P.; Wennberge, T.; Vuorela, H. Natural Products in the Process of Finding New Drug Candidates. Curr. Med. Chem. 2004, 11, 1375–1389. [Google Scholar] [CrossRef]
- Newman, D.J.; Cragg, G.M. Natural Products as Sources of New Drugs over the Last 25 Years. J. Nat. Prod. 2007, 70, 461–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Zheng, Z.; Wu, M.; Zhang, L.; Wang, J.; Fu, W.; Xu, N.; Zhao, Z.; Lao, Y.; Xu, H. The natural compound neobractatin inhibits tumor metastasis by upregulating the RNA-binding-protein MBNL2. Cell Death Dis. 2019, 10, 554. [Google Scholar] [CrossRef] [Green Version]
- Casey, S.C.; Amedei, A.; Aquilano, K.; Azmi, A.S.; Benencia, F.; Bhakta, D.; Bilsland, A.E.; Boosani, C.S.; Chen, S.; Ciriolo, M.R.; et al. Cancer prevention and therapy through the modulation of the tumor microenvironment. Semin. Cancer Biol. 2015, 35, S199–S223. [Google Scholar] [CrossRef] [PubMed]
- Deepak, K.G.K.; Vempati, R.; Nagaraju, G.P.; Dasari, V.R.; Nagini, S.; Rao, D.N.; Malla, R.R. Tumor microenvironment: Challenges and opportunities in targeting metastasis of triple negative breast cancer. Pharmacol. Res. 2020, 153, 104683. [Google Scholar] [CrossRef]
- Sharma, S.H.; Thulasingam, S.; Nagarajan, S. Chemopreventive agents targeting tumor microenvironment. Life Sci. 2016, 145, 74–84. [Google Scholar] [CrossRef] [PubMed]
- Trang, N.t.D.; Wanner, M.J.; Phuong, L.V.N.; Koomen, G.J.; Dung, N.X. Thymoquinone from Eupatorium ayapana. Planta Med. 1999, 59, 99. [Google Scholar] [CrossRef] [PubMed]
- Al-Ghamdi, M. A review of pharmaco-therapeutics effects of Nigella Sativa. Pak. J. Med. Res. 2002, 41, 77–83. [Google Scholar]
- Randhawa, M.A.; Alghamdi, M.S. Anticancer Activity of Nigella sativa (Black Seed)—A Review. Am. J. Chin. Med. 2011, 39, 1075–1091. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahmoud, Y.K.; Abdelrazek, H.M. Cancer: Thymoquinone antioxidant/pro-oxidant effect as potential anticancer remedy. Biomed. Pharmacother. 2019, 115, 108783. [Google Scholar] [CrossRef]
- El-Mahdy, M.A.; Zhu, Q.; Wang, Q.E.; Wani, G.; Wani, A.A. Thymoquinone induces apoptosis through activation of caspase-8 and mitochondrial events in p53-null myeloblastic leukemia HL-60 cells. Int. J. Cancer 2005, 117, 409–417. [Google Scholar] [CrossRef]
- Rooney, S.; Ryan, M.F. Effects of alpha-hederin and thymoquinone, constituents of Nigella sativa, on human cancer cell lines. Anticancer Res. 2005, 25, 2199–2204. [Google Scholar]
- Adinew, G.M.; Taka, E.; Mendonca, P.; Messeha, S.S.; Soliman, K.F.A. The Anticancer Effects of Flavonoids through miRNAs Modulations in Triple-Negative Breast Cancer. Nutrients 2021, 13, 1212. [Google Scholar] [CrossRef]
- Alhebshi, A.H.; Gotoh, M.; Suzuki, I. Thymoquinone protects cultured rat primary neurons against amyloid#²-induced neurotoxicity. Biochem. Biophys. Res. Commun. 2013, 433, 362–367. [Google Scholar]
- Talib, W.H. Regressions of Breast Carcinoma Syngraft Following Treatment with Piperine in Combination with Thymoquinone. Scientia Pharmaceutica 2017, 85, 27. [Google Scholar] [CrossRef] [Green Version]
- Sutton, K.M.; Greenshields, A.L.; Hoskin, D.W. Thymoquinone, a bioactive component of black caraway seeds, causes G1 phase cell cycle arrest and apoptosis in triple-negative breast cancer cells with mutant p53. Nutr. Cancer 2014, 66, 408–418. [Google Scholar] [CrossRef] [PubMed]
- Rajput, S.; Kumar, B.N.; Sarkar, S.; Das, S.; Azab, B.; Santhekadur, P.K.; Das, S.K.; Emdad, L.; Sarkar, D.; Fisher, P.B.; et al. Targeted apoptotic effects of thymoquinone and tamoxifen on XIAP mediated Akt regulation in breast cancer. PLoS ONE 2013, 8, e61342. [Google Scholar] [CrossRef] [Green Version]
- Woo, C.C.; Hsu, A.; Kumar, A.P.; Sethi, G.; Tan, K.H. Thymoquinone inhibits tumor growth and induces apoptosis in a breast cancer xenograft mouse model: The role of p38 MAPK and ROS. PLoS ONE 2013, 8, e75356. [Google Scholar] [CrossRef] [Green Version]
- Effenberger-Neidnicht, K.; Schobert, R. Combinatorial effects of thymoquinone on the anti-cancer activity of doxorubicin. Cancer Chemother. Pharmacol. 2011, 67, 867–874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arafael, S.A.; Zhu, Q.; Shah, Z.I.; Wani, G.; Barakat, B.M.; Racoma, I.; El-Mahdy, M.A.; Wani, A.A. Thymoquinone up-regulates PTEN expression and induces apoptosis in doxorubicin-resistant human breast cancer cells. Mutat. Res. 2011, 706, 28–35. [Google Scholar] [CrossRef] [Green Version]
- Odeh, F.; Ismail, S.; Abu-Dahab, R.; Mahmoud, I.; Al Bawab, A. Thymoquinone in liposomes: A study of loading efficiency and biological activity towards breast cancer. Drug Deliv. 2012, 19, 371–377. [Google Scholar] [CrossRef]
- Ikonen, E. Cellular cholesterol trafficking and compartmentalization. Nat. Rev. Mol. Cell Biol. 2008, 9, 125–138. [Google Scholar] [CrossRef] [PubMed]
- Murai, T. Cholesterol lowering: Role in cancer prevention and treatment. Biol. Chem. 2015, 396, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Gorin, A.; Gabitova, L.; Astsaturov, I. Regulation of cholesterol biosynthesis and cancer signaling. Curr. Opin. Pharmacol. 2012, 12, 710–716. [Google Scholar] [CrossRef] [Green Version]
- Kuzu, O.F.; Gowda, R.; Noory, M.A.; Robertson, G.P. Modulating cancer cell survival by targeting intracellular cholesterol transport. Br. J. Cancer 2017, 117, 513–524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Z.; Liu, X.; Liu, S.; Cao, Q. Cholesterol promotes the migration and invasion of renal carcinoma cells by regulating the KLF5/miR-27a/FBXW7 pathway. Biochem. Biophys. Res. Commun. 2018, 502, 69–75. [Google Scholar] [CrossRef] [PubMed]
- Kuzu, O.F.; Noory, M.A.; Robertson, G.P. The Role of Cholesterol in Cancer. Cancer Res. 2016, 76, 2063. [Google Scholar] [CrossRef] [Green Version]
- Raccosta, L.; Fontana, R.; Corna, G.; Maggioni, D.; Moresco, M.; Russo, V. Cholesterol metabolites and tumor microenvironment: The road towards clinical translation. Cancer Immunol. Immunother. 2016, 65, 111–117. [Google Scholar] [CrossRef]
- Fararjeh, A.F.S.; Al Khader, A.; Kaddumi, E.; Obeidat, M.; Al-Fawares, O.l. Differential Expression and Prognostic Significance of STARD3 Gene in Breast Carcinoma. Int. J. Mol. Cell Med. 2021, 10, 34–41. [Google Scholar] [CrossRef] [PubMed]
- Montero, J.; Morales, A.; Llacuna, L.; Lluis, J.M.; Terrones, O.; Basañez, G.; Antonsson, B.; Prieto, J.; García-Ruiz, C.; Colell, A.; et al. Mitochondrial Cholesterol Contributes to Chemotherapy Resistance in Hepatocellular Carcinoma. Cancer Res. 2008, 68, 5246–5256. [Google Scholar] [CrossRef] [Green Version]
- Vassilev, B.; Sihto, H.; Li, S.; Hölttä-Vuori, M.; Ilola, J.; Lundin, J.; Isola, J.; Kellokumpu-Lehtinen, P.-L.; Joensuu, e.; Ikonen, E. Elevated levels of StAR-related lipid transfer protein 3 alter cholesterol balance and adhesiveness of breast cancer cells: Potential mechanisms contributing to progression of HER2-positive breast cancers. Am. J. Pathol. 2015, 185, 987–1000. [Google Scholar] [CrossRef]
- Dong, F.; Mo, Z.; Eid, W.; Courtney, K.C.; Zha, X. Akt inhibition promotes ABCA1-mediated cholesterol efflux to ApoA-I through suppressing mTORC1. PLoS ONE 2014, 9, e113789. [Google Scholar] [CrossRef] [Green Version]
- Porstmann, T.; Santos, C.R.; Griffiths, B.; Cully, M.; Wu, M.; Leevers, S.; Griffiths, J.R.; Chung, Y.-L.; Schulze, A. SREBP activity is regulated by mTORC1 and contributes to Akt-dependent cell growth. Cell Metab. 2008, 8, 224–236. [Google Scholar] [CrossRef] [Green Version]
- Freed-Pastor, W.A.; Mizuno, H.; Zhao, X.; Langerød, A.; Moon, S.-H.; Rodriguez-Barrueco, R.; Barsotti, A.; Chicas, A.; Li, W.; Polotskaia, A.; et al. Mutant p53 Disrupts Mammary Tissue Architecture via the Mevalonate Pathway. Cell 2012, 148, 244–258. [Google Scholar] [CrossRef] [Green Version]
- Giró-Perafita, A.; Rabionet, M.; Planas, M.; Feliu, L.; Ciurana, J.; Ruiz-Martínez, S.; Puig, T. EGCG-Derivative G28 Shows High Efficacy Inhibiting the Mammosphere-Forming Capacity of Sensitive and Resistant TNBC Models. Molecules 2019, 24, 1027. [Google Scholar] [CrossRef] [Green Version]
- Giró-Perafita, A.; Palomeras, S.; Lum, D.H.; Blancafort, A.; Viñas, G.; Oliveras, G.; Pérez-Bueno, F.; Sarrats, A.; Welm, A.L.; Puig, T. Preclinical Evaluation of Fatty Acid Synthase and EGFR Inhibition in Triple-Negative Breast Cancer. Clin. Cancer Res. 2016, 22, 4687–4697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antalis, C.J.; Arnold, T.; Rasool, T.; Lee, B.; Buhman, K.K.; Siddiqui, R.A. High ACAT1 expression in estrogen receptor negative basal-like breast cancer cells is associated with LDL-induced proliferation. Breast Cancer Res. Treat. 2010, 122, 661–670. [Google Scholar] [CrossRef]
- Li, J.; Gu, D.; Lee, S.S.Y.; Song, B.; Bandyopadhyay, S.; Chen, S.; Konieczny, S.F.; Ratliff, T.L.; Liu, X.; Xie, J.; et al. Abrogating cholesterol esterification suppresses growth and metastasis of pancreatic cancer. Oncogene 2016, 35, 6378–6388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajput, S.; Kumar, B.N.; Dey, K.K.; Pal, I.; Parekh, A.; Mandal, M. Molecular targeting of Akt by thymoquinone promotes G(1) arrest through translation inhibition of cyclin D1 and induces apoptosis in breast cancer cells. Life Sci. 2013, 93, 783–790. [Google Scholar] [CrossRef] [PubMed]
- Dastjerdi, M.N.; Mehdiabady, E.M.; Iranpour, F.G.; Bahramian, H. Effect of Thymoquinone on P53 Gene Expression and Consequence Apoptosis in Breast Cancer Cell Line. Int. J. Prev. Med. 2016, 7, 66. [Google Scholar] [CrossRef]
- Ray, P.D.; Huang, B.-W.; Tsuji, Y. Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell. Signal. 2012, 24, 981–990. [Google Scholar] [CrossRef] [Green Version]
- Waris, G.; Ahsan, H. Reactive oxygen species: Role in the development of cancer and various chronic conditions. J. Carcinog. 2006, 5, 14. [Google Scholar] [CrossRef]
- Diebold, L.; Chandel, N.S. Mitochondrial ROS regulation of proliferating cells. Free Radic. Biol. Med. 2016, 100, 86–93. [Google Scholar] [CrossRef]
- Semenza, G.L. Hypoxia-inducible factors in physiology and medicine. Cell 2012, 148, 399–408. [Google Scholar] [CrossRef] [Green Version]
- Bao, S.; Wu, Q.; McLendon, R.E.; Hao, Y.; Shi, Q.; Hjelmeland, A.B.; Dewhirst, M.W.; Bigner, D.D.; Rich, J.N. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 2006, 444, 756–760. [Google Scholar] [CrossRef]
- Gorrini, C.; Harris, I.S.; Mak, T.W. Modulation of oxidative stress as an anticancer strategy. Nat. Rev. Drug Discov. 2013, 12, 931–947. [Google Scholar] [CrossRef] [PubMed]
- Liou, G.Y.; Storz, P. Reactive oxygen species in cancer. Free Radic. Res. 2010, 44, 479–496. [Google Scholar] [CrossRef] [Green Version]
- Lee, A.C.; Fenster, B.E.; Ito, H.; Takeda, K.; Bae, N.S.; Hirai, T.; Yu, Z.X.; Ferrans, V.J.; Howard, B.H.; Finkel, T. Ras proteins induce senescence by altering the intracellular levels of reactive oxygen species. J. Biol. Chem. 1999, 274, 7936–7940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, J.-S.; Hong, M.-Z.; Ren, J.-L. Reactive oxygen species: A double-edged sword in oncogenesis. World J. Gastroenterol. 2009, 15, 1702–1707. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, V.; Tuli, H.S.; Varol, A.; Thakral, F.; Yerer, M.B.; Sak, K.; Varol, M.; Jain, A.; Khan, M.A.; Sethi, G. Role of Reactive Oxygen Species in Cancer Progression: Molecular Mechanisms and Recent Advancements. Biomolecules 2019, 9, 735. [Google Scholar] [CrossRef] [Green Version]
- Kodama, R.; Kato, M.; Furuta, S.; Ueno, S.; Zhang, Y.; Matsuno, K.; Yabe-Nishimura, C.; Tanaka, E.; Kamata, T. ROS-generating oxidases Nox1 and Nox4 contribute to oncogenic Ras-induced premature senescence. Genes Cells 2013, 18, 32–41. [Google Scholar] [CrossRef] [PubMed]
- Scherz-Shouval, R.; Elazar, Z. Regulation of autophagy by ROS: Physiology and pathology. Trends Biochem. Sci. 2011, 36, 30–38. [Google Scholar] [CrossRef] [PubMed]
- Chandra, J.; Samali, A.; Orrenius, S. Triggering and modulation of apoptosis by oxidative stress. Free Radic. Biol. Med. 2000, 29, 323–333. [Google Scholar] [CrossRef]
- López-Lázaro, M. Dual role of hydrogen peroxide in cancer: Possible relevance to cancer chemoprevention and therapy. Cancer Lett. 2007, 252, 1–8. [Google Scholar] [CrossRef]
- Perillo, B.; Di Donato, M.; Pezone, A.; Di Zazzo, E.; Giovannelli, P.; Galasso, G.; Castoria, G.; Migliaccio, A. ROS in cancer therapy: The bright side of the moon. Exp. Mol. Med. 2020, 52, 192–203. [Google Scholar] [CrossRef]
- Son, Y.; Cheong, Y.-K.; Kim, N.-H.; Chung, H.-T.; Kang, D.G.; Pae, H.-O. Mitogen-Activated Protein Kinases and Reactive Oxygen Species: How Can ROS Activate MAPK Pathways? J. Signal. Transduct. 2011, 2011, 792639. [Google Scholar] [CrossRef]
- Leslie, N.R.; Bennett, D.; Lindsay, Y.E.; Stewart, H.; Gray, A.; Downes, C.P. Redox regulation of PI 3-kinase signalling via inactivation of PTEN. EMBO J. 2003, 22, 5501–5510. [Google Scholar] [CrossRef]
- Bernatoniene, J.; Kopustinskiene, D.M. The Role of Catechins in Cellular Responses to Oxidative Stress. Molecules 2018, 23, 965. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.H.; Huang, C.W.; Chang, P.C.; Shiau, J.P.; Lin, I.P.; Lin, M.Y.; Chen, C.Y. Reactive oxygen species mediate the chemopreventive effects of syringin in breast cancer cells. Phytomedicine 2019, 61, 152844. [Google Scholar] [CrossRef]
- Zhou, F.; Feng, B.; Wang, T.; Wang, D.; Cui, Z.; Wang, S.; Ding, C.; Zhang, Z.; Liu, J.; Yu, H.; et al. Theranostic Prodrug Vesicles for Reactive Oxygen Species-Triggered Ultrafast Drug Release and Local-Regional Therapy of Metastatic Triple-Negative Breast Cancer. Adv. Funct. Mater. 2017, 27, 1703674. [Google Scholar] [CrossRef]
- Khader, M.; Eckl, P.M. Thymoquinone: An emerging natural drug with a wide range of medical applications. Iran. J. Basic Med. Sci. 2014, 17, 950–957. [Google Scholar]
- Khan, M.A.; Tania, M.; Fu, S.; Fu, J. Thymoquinone, as an anticancer molecule: From basic research to clinical investigation. Oncotarget 2017, 8, 1907–51919. [Google Scholar]
- Kassab, R.B.; El-Hennamy, R.E. The role of thymoquinone as a potent antioxidant in ameliorating the neurotoxic effect of sodium arsenate in female rat. Egypt. J. Basic Appl. Sci. 2017, 4, 160–167. [Google Scholar] [CrossRef] [Green Version]
- Sayed-Ahmed, M.M.; Aleisa, A.M.; Al-Rejaie, S.S.; Al-Yahya, A.A.; Al-Shabanah, O.A.; Hafez, M.M.; Nagi, M.N. Thymoquinone attenuates diethylnitrosamine induction of hepatic carcinogenesis through antioxidant signaling. Oxid. Med. Cell Longev. 2010, 3, 254–261. [Google Scholar] [CrossRef]
- Badary, O.A.; Taha, R.A.; Gamal el-Din, A.M.; Abdel-Wahab, M.H. Thymoquinone is a potent superoxide anion scavenger. Drug Chem. Toxicol. 2003, 26, 87–98. [Google Scholar] [CrossRef]
- Kundu, J.; Kim, D.-H.; Kundu, J.K.; Chun, K.-S. Thymoquinone induces heme oxygenase-1 expression in HaCaT cells via Nrf2/ARE activation: Akt and AMPKα as upstream targets. Food Chem. Toxicol. 2014, 65, 18–26. [Google Scholar] [CrossRef] [PubMed]
- Ushio-Fukai, M.; Ash, D.; Nagarkoti, S.; Belin de Chantemèle, E.J.; Fulton, D.J.R.; Fukai, T. Interplay Between Reactive Oxygen/Reactive Nitrogen Species and Metabolism in Vascular Biology and Disease. Antioxid. Redox Signal. 2021, 34, 1319–1354. [Google Scholar] [CrossRef] [PubMed]
- Ryazanov, A.G. Ca2+/calmodulin-dependent phosphorylation of elongation factor 2. FEBS Lett. 1987, 214, 331–334. [Google Scholar] [CrossRef] [Green Version]
- Leprivier, G.; Remke, M.; Rotblat, B.; Dubuc, A.; Mateo, A.-R.F.; Kool, M.; Agnihotri, S.; El-Naggar, A.; Yu, B.; Somasekharan, S.P.; et al. The eEF2 Kinase Confers Resistance to Nutrient Deprivation by Blocking Translation Elongation. Cell 2013, 153, 1064–1079. [Google Scholar] [CrossRef] [Green Version]
- Kenney, J.W.; Moore, C.E.; Wang, X.; Proud, C.G. Eukaryotic elongation factor 2 kinase, an unusual enzyme with multiple roles. Adv. Biol. Regul. 2014, 55, 15–27. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.H.; Riazy, M.; Smith, E.M.; Proud, C.G.; Steinbrecher, U.P.; Duronio, V. Oxidized LDL-mediated macrophage survival involves elongation factor-2 kinase. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 92–98. [Google Scholar] [CrossRef] [Green Version]
- Xu, Z.; Zhao, L.; Yang, X.; Ma, S.; Ge, Y.; Liu, Y.; Liu, S.; Shi, J.; Zheng, D. Mmu-miR-125b overexpression suppresses NO production in activated macrophages by targeting eEF2K and CCNA2. BMC Cancer 2016, 16, 252. [Google Scholar] [CrossRef] [Green Version]
- Moore, C.E.; Mikolajek, H.; Regufe da Mota, S.; Wang, X.; Kenney, J.W.; Werner, J.M.; Proud, C.G. Elongation Factor 2 Kinase Is Regulated by Proline Hydroxylation and Protects Cells during Hypoxia. Mol. Cell Biol. 2015, 35, 1788–1804. [Google Scholar] [CrossRef] [Green Version]
- Kenney, J.W.; Genheden, M.; Moon, K.M.; Wang, X.; Foster, L.J.; Proud, C.G. Eukaryotic elongation factor 2 kinase regulates the synthesis of microtubule-related proteins in neurons. J. Neurochem. 2016, 136, 276–284. [Google Scholar] [CrossRef] [Green Version]
- Neri, D.; Supuran, C.T. Interfering with pH regulation in tumours as a therapeutic strategy. Nat. Rev. Drug Discov. 2011, 10, 767–777. [Google Scholar] [CrossRef] [Green Version]
- Xie, J.; Mikolajek, H.; Pigott, C.R.; Hooper, K.J.; Mellows, T.; Moore, C.E.; Mohammed, H.; Werner, J.M.; Thomas, G.J.; Proud, C.G. Molecular Mechanism for the Control of Eukaryotic Elongation Factor 2 Kinase by pH: Role in Cancer Cell Survival. Mol. Cell. Biol. 2015, 35, 1805–1824. [Google Scholar] [CrossRef] [Green Version]
- Gschwendt, M.; Muller, H.J.; Kielbassa, K.; Zang, R.; Kittstein, W.; Rincke, G.; Marks, F. Rottlerin, a Novel Protein Kinase Inhibitor. Biochem. Biophys. Res. Commun. 1994, 199, 93–98. [Google Scholar] [CrossRef]
- Gschwendt, M.; Kittstein, W.; Marks, F. Elongation factor-2 kinase: Effective inhibition by the novel protein kinase inhibitor rottlerin and relative insensitivity towards staurosporine. FEBS Lett. 1994, 338, 85–88. [Google Scholar] [CrossRef] [Green Version]
- Tekedereli, I.; Alpay, S.N.; Tavares, C.D.J.; Cobanoglu, Z.E.; Kaoud, T.S.; Sahin, I.; Sood, A.K.; Lopez-Berestein, G.; Dalby, K.N.; Ozpolat, B. Targeted silencing of elongation factor 2 kinase suppresses growth and sensitizes tumors to doxorubicin in an orthotopic model of breast cancer. PLoS ONE 2012, 7, e41171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bayraktar, R.; Pichler, M.; Kanlikilicer, P.; Ivan, C.; Bayraktar, E.; Kahraman, N.; Aslan, B.; Oguztuzun, S.; Ulasli, M.; Arslan, A.; et al. MicroRNA 603 acts as a tumor suppressor and inhibits triple-negative breast cancer tumorigenesis by targeting elongation factor 2 kinase. Oncotarget 2017, 8, 11641–11658. [Google Scholar] [CrossRef] [Green Version]
- Kabil, N.; Bayraktar, R.; Kahraman, N.; Mokhlis, H.A.; Calin, G.A.; Lopez-Berestein, G.; Ozpolat, B. Thymoquinone inhibits cell proliferation, migration, and invasion by regulating the elongation factor 2 kinase (eEF-2K) signaling axis in triple-negative breast cancer. Breast Cancer Res. Treat. 2018, 171, 593–605. [Google Scholar] [CrossRef] [PubMed]
- Balkwill, F.; Mantovani, A. Inflammation and cancer: Back to Virchow? Lancet 2001, 357, 539–545. [Google Scholar] [CrossRef]
- Mantovani, A.; Bussolino, F.; Dejana, E. Cytokine regulation of endothelial cell function. FASEB J. 1992, 6, 2591–2599. [Google Scholar] [CrossRef]
- Pathria, P.; Louis, T.L.; Varner, J.A. Targeting Tumor-Associated Macrophages in Cancer. Trends Immunol. 2019, 40, 177. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Z.-Y.; Luo, R.-Z.; Peng, R.-J.; Wang, S.-S.; Xue, C. High infiltration of tumor-associated macrophages in triple-negative breast cancer is associated with a higher risk of distant metastasis. Onco Targets Ther. 2014, 7, 1475–1480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leek, R.D.; Lewis, C.E.; Whitehouse, R.; Greenall, M.; Clarke, J.; Harris, A.L. Association of Macrophage Infiltration with Angiogenesis and Prognosis in Invasive Breast Carcinoma. Cancer Res. 1996, 56, 4625–4629. [Google Scholar]
- Priceman, S.J.; Sung, J.L.; Shaposhnik, Z.; Burton, J.B.; Torres-Collado, A.X.; Moughon, D.L.; Johnson, M.; Lusis, A.J.; Cohen, D.A.; Iruela-Arispe, M.L.; et al. Targeting distinct tumor-infiltrating myeloid cells by inhibiting CSF-1 receptor: Combating tumor evasion of antiangiogenic therapy. Blood 2010, 115, 1461–1471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harney, A.S.; Arwert, E.N.; Entenberg, D.; Wang, Y. Real-Time Imaging Reveals Local, Transient Vascular Permeability, and Tumor Cell Intravasation Stimulated by TIE2hi Macrophage-Derived VEGFA. Cancer Discov. 2015, 5, 932–943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hughes, R.; Qian, B.-Z.; Rowan, C.; Muthana, M.; Keklikoglou, I.; Olson, O.C.; Tazzyman, S.; Danson, S.; Addison, C.; Clemons, M.; et al. Perivascular M2 Macrophages Stimulate Tumor Relapse after Chemotherapy. Cancer Res. 2015, 75, 3479–3491. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Yang, B.; Huang, J.; Lin, Y.; Xiang, T.; Wan, J.; Li, H.; Chouaib, S.; Ren, G. Cyclooxygenase-2 in tumor-associated macrophages promotes breast cancer cell survival by triggering a positive-feedback loop between macrophages and cancer cells. Oncotarget 2015, 6, 29637–29650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qian, B.-Z.; Zhang, H.; Li, J.; He, T.; Yeo, E.-J.; Soong, D.Y.H.; Carragher, N.O.; Munro, A.; Chang, A.; Bresnick, A.R.; et al. FLT1 signaling in metastasis-associated macrophages activates an inflammatory signature that promotes breast cancer metastasis. J. Exp. Med. 2015, 212, 1433–1448. [Google Scholar] [CrossRef] [Green Version]
- Qian, B.Z.; Pollard, J.W. Macrophage diversity enhances tumor progression and metastasis. Cell 2010, 141, 39–51. [Google Scholar] [CrossRef] [Green Version]
- Hollmén, M.; Roudnicky, F.; Karaman, S.; Detmar, M. Characterization of macrophage--cancer cell crosstalk in estrogen receptor positive and triple-negative breast cancer. Sci. Rep. 2015, 5, 9188. [Google Scholar] [CrossRef]
- Robinson, S.C.; Scott, K.A.; Wilson, J.L.; Thompson, R.G.; Proudfoot, A.E.I.; Balkwill, F.R. A Chemokine Receptor Antagonist Inhibits Experimental Breast Tumor Growth. Cancer Res. 2003, 63, 8360. [Google Scholar]
- Vukanovic, J.; Isaacs, J.T. Linomide Inhibits Angiogenesis, Growth, Metastasis, and Macrophage Infiltration within Rat Prostatic Cancers. Cancer Res. 1995, 55, 1499. [Google Scholar]
- Whitehurst, B.; Flister, M.J.; Bagaitkar, J.; Volk, L.; Bivens, C.M.; Pickett, B.; Castro-Rivera, E.; Brekken, R.A.; Gerard, R.D.; Ran, S. Anti-VEGF-A therapy reduces lymphatic vessel density and expression of VEGFR-3 in an orthotopic breast tumor model. Int. J. Cancer 2007, 121, 2181–2191. [Google Scholar] [CrossRef]
- Dineen, S.P.; Lynn, K.D.; Holloway, S.E.; Miller, A.F.; Sullivan, J.P.; Shames, D.S.; Beck, A.W.; Barnett, C.C.; Fleming, J.B.; Brekken, R.A. Vascular Endothelial Growth Factor Receptor 2 Mediates Macrophage Infiltration into Orthotopic Pancreatic Tumors in Mice. Cancer Res. 2008, 68, 4340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Mahmoudy, A.; Matsuyama, H.; Borgan, M.A.; Shimizu, Y.; El-Sayed, M.G.; Minamoto, N.; Takewaki, T. Thymoquinone suppresses expression of inducible nitric oxide synthase in rat macrophages. Int. Immunopharmacol. 2002, 2, 1603–1611. [Google Scholar] [CrossRef]
- Wilson, A.J.; Saskowski, J.; Barham, W.; Khabele, D.; Yull, F. Microenvironmental effects limit efficacy of thymoquinone treatment in a mouse model of ovarian cancer. Mol. Cancer 2015, 14, 192. [Google Scholar] [CrossRef] [Green Version]
- Paramasivam, A.; Kalaimangai, M.; Sambantham, S.; Anandan, B.; Jayaraman, G. Anti-angiogenic activity of thymoquinone by the down-regulation of VEGF using zebrafish (Danio rerio) model. Biomed. Prev. Nutr. 2012, 2, 169–173. [Google Scholar] [CrossRef]
- Shanmugam, M.K.; Ahn, K.S.; Hsu, A.; Woo, C.C.; Yuan, Y.; Tan, K.H.B.; Chinnathambi, A.; Alahmadi, T.A.; Alharbi, S.A.; Koh, A.P.F.; et al. Thymoquinone Inhibits Bone Metastasis of Breast Cancer Cells Through Abrogation of the CXCR4 Signaling Axis. Front. Pharmacol. 2018, 9, 1294. [Google Scholar] [CrossRef] [PubMed]
- Nieman, K.M.; Romero, I.L.; Houten, B.V.; Lengyel, E. Adipose tissue and adipocytes support tumorigenesis and metastasis. Biochim. Biophys. Acta 2013, 1831, 1533–1541. [Google Scholar] [CrossRef] [Green Version]
- Poulos, S.P.; Hausman, D.B.; Hausman, G.J. The development and endocrine functions of adipose tissue. Mol. Cell Endocrinol. 2010, 323, 20–34. [Google Scholar] [CrossRef]
- Vona-Davis, L.; Rose, D.P. Adipokines as endocrine, paracrine, and autocrine factors in breast cancer risk and progression. Endocr. Relat. Cancer 2007, 14, 189–206. [Google Scholar] [CrossRef]
- Bifulco, M.; Pisanti, S. “Adiponcosis”: A new term to name the obesity and cancer link. J. Clin. Endocrinol. Metab. 2013, 98, 4664–4665. [Google Scholar] [CrossRef] [Green Version]
- Dirat, B.; Bochet, L.; Dabek, M.; Daviaud, D.; Dauvillier, S.; Majed, B.; Wang, Y.Y.; Meulle, A.; Salles, B.; Gonidec, S.L.; et al. Cancer-Associated Adipocytes Exhibit an Activated Phenotype and Contribute to Breast Cancer Invasion. Cancer Res. 2011, 71, 2455–2465. [Google Scholar] [CrossRef] [Green Version]
- Wagner, M.; Bjerkvig, R.; Wiig, H.; Melero-Martin, J.M.; Lin, R.-Z.; Klagsbrun, M.; Dudley, A.C. Inflamed tumor-associated adipose tissue is a depot for macrophages that stimulate tumor growth and angiogenesis. Angiogenesis 2012, 15, 481–495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Q.; Li, B.; Li, Z.; Li, J.; Sun, S.; Sun, S. Cancer-associated adipocytes: Key players in breast cancer progression. J. Hematol. Oncol. 2019, 12, 95. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Tiruthani, K.; Wang, M.; Zhou, X.; Qiu, N.; Xiong, Y.; Pecot, C.V.; Liu, R.; Huang, L. Tumor-targeted gene therapy with lipid nanoparticles inhibits tumor-associated adipocytes and remodels the immunosuppressive tumor microenvironment in triple-negative breast cancer. Nanoscale Horiz. 2021, 6, 319–329. [Google Scholar] [CrossRef] [PubMed]
- Palma, M.D.; Biziato, D.; Petrova, T.V. Microenvironmental regulation of tumour angiogenesis. Nat. Rev. Cancer 2017, 17, 457–474. [Google Scholar] [CrossRef]
- D’Esposito, V.; Liguoro, D.; Ambrosio, M.R.; Collina, F.; Cantile, M.; Spinelli, R.; Raciti, G.A.; Miele, C.; Valentino, R.; Campiglia, P.; et al. Adipose microenvironment promotes triple negative breast cancer cell invasiveness and dissemination by producing CCL5. Oncotarget 2016, 7, 24495–24509. [Google Scholar] [CrossRef] [Green Version]
- Choia, J.; Chab, Y.J.; Koo, J.S. Adipocyte biology in breast cancer: From silent bystander to active facilitator. Prog. Lipid Res. 2018, 69, 11–20. [Google Scholar] [CrossRef]
- Pandolfi, J.B.; Ferraro, A.A.; Sananez, I.; Gancedo, M.C.; Baz, P.; Billordo, L.A.; Fainboim, L.; Arruvito, L. ATP-induced inflammation drives tissue-resident Th17 cells in metabolically unhealthy obesity. J. Immunol. 2016, 196, 3287–3296. [Google Scholar] [CrossRef] [Green Version]
- Shaterzadeh-Yazdi, H.; Noorbakhsh, M.-F.; Hayati, F.; Samarghandian, S.; Farkhondeh, T. Immunomodulatory and Anti-inflammatory Effects of Thymoquinone. Cardiovasc. Hematol. Disord Drug Targets 2018, 18, 52–60. [Google Scholar] [CrossRef]
- Pannu, A.; Goyal, R.K.; Ojha, S.; Nandave, M. Chapter 33—Therapeutic Potential of Thymoquinone in Treatment of Rheumatoid Arthritis and Related Autoimmune Diseases. In Bioactive Food as Dietary Interventions for Arthritis and Related Inflammatory Diseases, 2nd ed.; Watson, R.R., Preedy, V.R., Eds.; Academic Press: Cambridge, MA, USA, 2019; pp. 575–587. [Google Scholar] [CrossRef]
- Shen, H.H.; Peterson, S.J.; Bellner, L.; Choudhary, A.; Levy, L.; Gancz, L.; Sasson, A.; Trainer, J.; Rezzani, R.; Resnick, A.; et al. Cold-Pressed Nigella Sativa Oil Standardized to 3% Thymoquinone Potentiates Omega-3 Protection against Obesity-Induced Oxidative Stress, Inflammation, and Markers of Insulin Resistance Accompanied with Conversion of White to Beige Fat in Mice. Antioxidants 2020, 9, 489. [Google Scholar] [CrossRef] [PubMed]
- Virchow, R. As Based upon Physiological and Pathological Histology. Nutr. Rev. 1989, 47, 23–25. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R.; Zeisberg, M. Fibroblasts in cancer. Nat. Rev. Cancer 2006, 6, 392–401. [Google Scholar] [CrossRef] [PubMed]
- Tuxhorn, J.A.; McAlhany, S.J.; Dang, T.D.; Ayala, G.E.; Rowley, D.R. Stromal Cells Promote Angiogenesis and Growth of Human Prostate Tumors in a Differential Reactive Stroma (DRS) Xenograft Model. Cancer Res. 2002, 62, 3298–3307. [Google Scholar] [PubMed]
- Gerber, H.P.; Kowalski, J.; Sherman, D.; Eberhard, D.A.; Ferrara, N. Complete inhibition of rhabdomyosarcoma xenograft growth and neovascularization requires blockade of both tumor and host vascular endothelial growth factor. Cancer Res. 2000, 60, 6253–6258. [Google Scholar] [PubMed]
- Crawford, Y.; Kasman, I.; Yu, L.; Zhong, C.; Wu, X.; Modrusan, Z.; Kaminker, J.; Ferrara, N. PDGF-C mediates the angiogenic and tumorigenic properties of fibroblasts associated with tumors refractory to anti-VEGF treatment. Cancer Cell 2009, 15, 21–34. [Google Scholar] [CrossRef] [Green Version]
- Pietras, K.; Pahler, J.; Bergers, G.; Hanahan, D. Functions of paracrine PDGF signaling in the proangiogenic tumor stroma revealed by pharmacological targeting. PLoS Med. 2008, 5, e19. [Google Scholar] [CrossRef]
- Egeblad, M.; Rasch, M.G.; Weaver, V.M. Dynamic interplay between the collagen scaffold and tumor evolution. Curr. Opin. Cell Biol. 2010, 22, 697–706. [Google Scholar] [CrossRef] [Green Version]
- Orimo, A.; Gupta, P.B.; Sgroi, D.C.; Arenzana-Seisdedos, F.; Delaunay, T.; Naeem, R.; Carey, V.J.; Richardson, A.L.; Weinberg, R.A. Stromal Fibroblasts Present in Invasive Human Breast Carcinomas Promote Tumor Growth and Angiogenesis through Elevated SDF-1/CXCL12 Secretion. Cell 2005, 121, 335–348. [Google Scholar] [CrossRef]
- Wang, M.; Zhang, J.; Huang, Y.; Ji, S.; Shao, G.; Feng, S.; Chen, D.; Zhao, K.; Wang, Z.; Wu, A. Cancer-Associated Fibroblasts Autophagy Enhances Progression of Triple-Negative Breast Cancer Cells. Med. Sci. Monit. 2017, 23, 3904–3912. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Wu, H.; Fan, S.; Bi, Y.; Hao, M.; Shang, J. Cancer-associated fibroblast-derived LRRC15 promotes the migration and invasion of triple-negative breast cancer cells via Wnt/β-catenin signalling pathway regulation. Mol. Med. Rep. 2022, 25, 2. [Google Scholar] [CrossRef]
- Bhowmick, N.A.; Chytil, A.; Plieth, D.; Gorska, A.E.; Dumont, N.; Shappell, S.; Washington, M.K.; Neilson, E.G.; Moses, H.L. TGF-beta signaling in fibroblasts modulates the oncogenic potential of adjacent epithelia. Science 2004, 3030, 848–851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, X.; Zhang, L.; He, X.; Zhang, P.; Sun, C.; Xu, X.; Lu, Y.; Li, F. TGF-β plays a vital role in triple-negative breast cancer (TNBC) drug-resistance through regulating stemness, EMT and apoptosis. Biochem. Biophys. Res. Commun. 2018, 502, 160–165. [Google Scholar] [CrossRef] [PubMed]
- Takai, K.; Le, A.; Weaver, V.M.; Werb, Z. Targeting the cancer-associated fibroblasts as a treatment in triple-negative breast cancer. Oncotarget 2016, 7, 82889–82901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, N.; Xiang, Y.; Zhao, X.; Cai, C.; Chen, H.; Jiang, W.; Wang, Y.; Zeng, C. Thymoquinone suppresses platelet-derived growth factor-BB–induced vascular smooth muscle cell proliferation, migration and neointimal formation. J. Cell. Mol. Med. 2019, 23, 8482–8492. [Google Scholar] [CrossRef]
- Imrana, M.; Raufb, A.; Khanc, I.A.; Shahbazd, M.; Qaisranie, T.B.; Fatmawatif, S.; Abu-Izneidg, T.; Imranh, A.; Rahmanb, K.U.; Gondal, T.A. Thymoquinone: A novel strategy to combat cancer: A review. Biomed. Pharmacother. 2018, 106, 390–402. [Google Scholar] [CrossRef]
- Haiaty, S.; Rashidi, M.-R.; Akbarzadeh, M.; Bazmani, A.; Mostafazadeh, M.; Nikanfar, S.; Zibaei, Z.; Rahbarghazi, R.; Nouri, M. Thymoquinone inhibited vasculogenic capacity and promoted mesenchymal-epithelial transition of human breast cancer stem cells. BMC Complement. Med. Ther. 2021, 21, 83. [Google Scholar] [CrossRef]
- Cruz-Merino, L.d.l.; Barco-Sánchez, A.; Carrasco, F.H.; Fernández, E.N.; Benítez, A.V.; Molina, J.B.; Peinado, A.M.; López, A.G.; Borrego, M.R.; Villena, M.C.M.d.; et al. New insights into the role of the immune microenvironment in breast carcinoma. Clin. Dev. Immunol. 2013, 2013, 785317. [Google Scholar] [CrossRef]
- Stanton, S.E.; Disis, M.L. Clinical significance of tumor-infiltrating lymphocytes in breast cancer. J. Immunother. Cancer 2016, 4, 59. [Google Scholar] [CrossRef] [Green Version]
- Dieci, M.V.; Mathieu, M.C.; Guarneri, V.; Conte, P.; Delaloge, S.; Andre, F.; Goubar, A. Prognostic and predictive value of tumor-infiltrating lymphocytes in two phase III randomized adjuvant breast cancer trials. Ann. Oncol. 2015, 26, 1698–1704. [Google Scholar] [CrossRef]
- Loi, S.; Sirtaine, N.; Piette, F.; Salgado, R.; Viale, G.; Eenoo, F.V.; Rouas, G.; Francis, P.; Crown, J.P.A.; Hitre, E.; et al. Prognostic and predictive value of tumor-infiltrating lymphocytes in a phase III randomized adjuvant breast cancer trial in node-positive breast cancer comparing the addition of docetaxel to doxorubicin with doxorubicin-based chemotherapy: BIG 02-98. J. Clin. Oncol. 2013, 31, 860–867. [Google Scholar] [CrossRef] [PubMed]
- Adams, S.; Gray, R.J.; Demaria, S.; Goldstein, L.; Perez, E.A.; Shulman, L.N.; Martino, S.; Wang, M.; Jones, V.E.; Saphner, T.J.; et al. Prognostic Value of Tumor-Infiltrating Lymphocytes in Triple-Negative Breast Cancers From Two Phase III Randomized Adjuvant Breast Cancer Trials: ECOG 2197 and ECOG 1199. J. Clin. Oncol. 2014, 32, 2959–2966. [Google Scholar] [CrossRef] [PubMed]
- Loi, S.; Dushyanthen, S.; Beavis, P.A.; Salgado, R.; Denkert, C.; Savas, P.; Combs, S.; Rimm, D.L.; Giltnane, J.M.; Estrada, M.V.; et al. RAS/MAPK activation is associated with reduced tumor-infiltrating lymphocytes in triple-negative breast cancer: Therapeutic cooperation between MEK and PD-1/PD-L1 immune checkpoint inhibitors. Clin. Cancer Res. 2016, 22, 1499–1509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guida, M.S.; El-Aal, A.A.; Kafafy, Y.; Salama, S.F.; Badr, B.M.; Badr, G. Thymoquinone Rescues T Lymphocytes from Gamma Irradiation-Induced Apoptosis and Exhaustion by Modulating Pro-Inflammatory Cytokine Levels and PD-1, Bax, and Bcl-2 Signaling. Cell. Physiol. Biochem. 2016, 38, 786–800. [Google Scholar] [CrossRef] [Green Version]
- Kheirouri, S.; Hadi, V.; Alizadeh, M. Immunomodulatory Effect of Nigella sativa Oil on T Lymphocytes in Patients with Rheumatoid Arthritis. Immunol. Investig. 2016, 45, 271–283. [Google Scholar] [CrossRef]
- Badr, G.; Alwasel, S.; Ebaid, H.; Mohany, M.; Alhazza, I. Perinatal supplementation with thymoquinone improves diabetic complications and T cell immune responses in rat offspring. Cell. Immunol. 2011, 267, 133–140. [Google Scholar] [CrossRef]
- Zheng, X.; Jin, W.; Wang, S.; Ding, H. Progression on the Roles and Mechanisms of Tumor-Infiltrating T Lymphocytes in Patients With Hepatocellular Carcinoma. Front. Immunol. 2021, 12, 729705. [Google Scholar] [CrossRef]
- Loi, S.; Michiels, S.; Salgado, R.; Sirtaine, N.; Jose, V.; Fumagalli, D.; KellokumpuLehtinen, P.-L.; Bono, V.K.; Desmedt, C.; Piccart, M.J.; et al. Tumor infiltrating lymphocytes are prognostic in triple negative breast cancer and predictive for trastuzumab benefit in early breast cancer: Results from the FinHER trial. Ann. Oncol. 2014, 25, 1544–1550. [Google Scholar] [CrossRef]
- Nicolini, A.; Carpi, A.; Rossi, G. Cytokine & Growth Factor Reviews. Cytokine Growth Factor Rev. 2006, 17, 325–337. [Google Scholar]
- Karin, M.; Greten, F.R. NF-kappaB: Linking inflammation and immunity to cancer development and progression. Nat. Rev. Immunol. 2005, 5, 749–759. [Google Scholar] [CrossRef]
- Kuilman, T.; Michaloglou, C.; Vredeveld, L.C.W.; Douma, S.; Doorn, R.v.; Desmet, C.J.; Aarden, L.A.; Mooi, W.J.; Peeper, D.S. Oncogene-induced senescence relayed by an interleukin-dependent inflammatory network. Cell 2008, 133, 1019–1031. [Google Scholar] [CrossRef] [Green Version]
- Guo, Y.; Xu, F.; Lu, T.; Duan, Z.; Zhang, Z. Interleukin-6 signaling pathway in targeted therapy for cancer. Cancer Treat. Rev. 2012, 38, 904–910. [Google Scholar] [CrossRef]
- Bharti, R.; Dey, G.; Mandal, M. Cancer development, chemoresistance, epithelial to mesenchymal transition and stem cells: A snapshot of IL-6 mediated involvement. Cancer Lett. 2016, 375, 51–61. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Tunon, I.; Ricote, M.; Ruiz, A.; Fraile, B.; Paniagua, R.; Royuela, M. IL-6, its receptors and its relationship with bcl-2 and bax proteins in infiltrating and in situ human breast carcinoma. Histopathology 2005, 47, 82–89. [Google Scholar] [CrossRef] [PubMed]
- Hartman, Z.C.; Poage, G.M.; Hollander, P.d.; Tsimelzon, A.; Hill, J.; Panupinthu, N.; Zhang, Y.; Mazumdar, A.; Hilsenbeck, S.G.; Mills, G.B.; et al. Growth of triple-negative breast cancer cells relies upon coordinate autocrine expression of the pro-inflammatory cytokines IL-6 and IL-8. Cancer Res. 2013, 73, 3470–3480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, E.; Fertig, E.J.; Jin, K.; Sukumar, S.; Pandey, N.B.; Popela, A.S. Breast cancer cells condition lymphatic endothelial cells within pre-metastatic niches to promote metastasis. Nat. Commun. 2014, 5, 4715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Won, H.S.; Kim, Y.A.; Lee, J.S.; Jeon, E.K.; An, H.J.; Sun, D.S.; Ko, Y.H.; Kim, J.S. Soluble Interleukin-6 Receptor is a Prognostic Marker for Relapse-Free Survival in Estrogen Receptor-Positive Breast Cancer. J. Cancer Investig. 2013, 31, 516–521. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.A.; Scheller, J.; Rose-John, S. Therapeutic strategies for the clinical blockade of IL-6/gp130 signaling. J. Clin. Investig. 2011, 121, 3375–3383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saini, U.; Naidu, S.; ElNaggar, A.C.; Bid, H.K.; Wallbillich, J.J.; Bixel, K.; Bolyard, C.; Suarez, A.A.; Kaur, B.; Kuppusamy, P.; et al. Elevated STAT3 expression in ovarian cancer ascites promotes invasion and metastasis: A potential therapeutic target. Oncogene 2017, 36, 168–181. [Google Scholar] [CrossRef] [Green Version]
- Jin, K.; Pandey, N.B.; Popel, A.S. Simultaneous blockade of IL-6 and CCL5 signaling for synergistic inhibition of triple-negative breast cancer growth and metastasis. Breast Cancer Res. Treat. 2018, 20, 54. [Google Scholar] [CrossRef] [Green Version]
- Oshima, T.; Cao, X.; Grande, F.; Yamada, R.; Garofalo, A.; Louie, S.; Neamati, N. Combination effects of SC144 and cytotoxic anticancer agents. Anticancer Drugs 2009, 20, 312–320. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Grande, F.; Garofalo, A.; Neamati, N. Discovery of a Novel Orally Active Small-Molecule gp130 Inhibitor for the Treatment of Ovarian Cancer. Mol. Cancer Ther. 2013, 12, 937–949. [Google Scholar] [CrossRef] [Green Version]
- Heo, T.-H.; Wahler, J.; Suh, N. Potential therapeutic implications of IL-6/IL-6R/gp130-targeting agents in breast cancer. Oncotarget 2016, 7, 15460–15473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, F.; Rajendran, P.; Sethi, G. Thymoquinone inhibits proliferation, induces apoptosis and chemosensitizes human multiple myeloma cells through suppression of signal transducer and activator of transcription 3 activation pathway. Br. J. Pharmacol. 2010, 161, 541–554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tu, Y.; Gardner, A.; Lichtenstein, A. The phosphatidylinositol 3-kinase/AKT kinase pathway in multiple myeloma plasma cells: Roles in cytokine-dependent survival and proliferative responses. Cancer Res. 2000, 60, 6763–6770. [Google Scholar] [PubMed]
- Shuai, K.; Liu, B. Regulation of JAK-STAT signalling in the immune system. Nat. Rev. Immunol. 2003, 3, 900–911. [Google Scholar] [CrossRef]
- Sonnenblick, A.; Uziely, B.; Nechushtan, H.; Kadouri, L.; Galun, E.; Axelrod, J.H.; Katz, D.; Daum, H.; Hamburger, T.; Maly, B.; et al. Tumor STAT3 tyrosine phosphorylation status, as a predictor of benefit from adjuvant chemotherapy for breast cancer. Breast Cancer Res. Treat. 2013, 138, 407–413. [Google Scholar] [CrossRef]
- Shields, B.J.; Wiede, F.; Gurzov, E.N.; Wee, K.; Hauser, C.; Zhu, H.-J.; Molloy, T.J.; O’Toole, S.A.; Daly, R.J.; Sutherland, R.L.; et al. TCPTP Regulates SFK and STAT3 Signaling and Is Lost in Triple-Negative Breast Cancers. Mol. Cell. Biol. 2013, 3, 557–570. [Google Scholar] [CrossRef] [Green Version]
- Yokogami, K.; Wakisaka, S.; Avruch, J.; Reeves, S.A. Serine phosphorylation and maximal activation of STAT3 during CNTF signaling is mediated by the rapamycin target mTOR. Curr. Biol. 2000, 10, 47–50. [Google Scholar] [CrossRef] [Green Version]
- Sutherland, K.D.; Lindeman, G.J.; Choong, D.Y.H.; Wittlin, S.; Brentzell, L.; Phillips, W.; Campbell, I.G.; Visvader, J.E. Differential hypermethylation of SOCS genes in ovarian and breast carcinomas. Oncogene 2004, 23, 7726–7733. [Google Scholar] [CrossRef] [Green Version]
- Bachelot, T.; Ray-Coquard, I.; Menetrier-Caux, C.; Rastkha, M.; Duc, A.; Blay, J.-Y. Prognostic value of serum levels of interleukin 6 and of serum and plasma levels of vascular endothelial growth factor in hormone-refractory metastatic breast cancer patients. Br. J. Cancer 2003, 88, 1721–1726. [Google Scholar] [CrossRef] [Green Version]
- Bowman, T.; Garcia, R.; Turkson, J.; Jove, R. STATs in oncogenesis. Oncogene 2000, 19, 2474–2488. [Google Scholar] [CrossRef] [Green Version]
- Yu, H.; Jove, R. The STATs of cancer--new molecular targets come of age. Nat. Rev. Cancer 2004, 4, 97–105. [Google Scholar] [CrossRef]
- Balko, J.M.; Schwarz, L.J.; Luo, N.; Estrada, M.V.; Giltnane, J.M.; Dávila-González, D.; Wang, K.; Sánchez, V.; Dean, P.T.; Combs, S.E.; et al. Triple-negative breast cancers with amplification of JAK2 at the 9p24 locus demonstrate JAK2-specific dependence. Sci. Transl. Med. 2016, 8, 334ra353. [Google Scholar] [CrossRef] [Green Version]
- Cikman, O.; Taysi, S.; Gulsen, M.; Demir, E.; Akan, M.; Diril, H.; Kiraz, H.; Karaayvaz, M.; Tarakcioglu, M. The Radioprotective Effects of Caffeic Acid Phenethyl Ester and Thymoquinone on Oxidative and Nitrosative Stress in Liver Tissue of Rats Exposed to Total Head Irradiation. West. Indian Med. J. 2016, 65, 1–7. [Google Scholar]
- Zhu, W.-Q.; Wang, J.; Guo, X.-F.; Liu, Z.; Dong, W.-G. Thymoquinone inhibits proliferation in gastric cancer via the STAT3 pathway in vivo and in vitro. World J. Gastroenterol. 2016, 22, 4149–4159. [Google Scholar] [CrossRef] [PubMed]
- Thomas, J.-L.; Eichmann, A. The power of VEGF (vascular endothelial growth factor) family molecules. Cell Mol. Life Sci. 2013, 70, 1673–1674. [Google Scholar] [CrossRef]
- Chung, A.S.; Lee, J.; Ferrara, N. Targeting the tumour vasculature: Insights from physiological angiogenesis. Nat. Rev. Cancer 2010, 10, 505–514. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Sullivan, C.A.W.; Zerkowski, M.P.; Molinaro, A.M.; Rimm, D.L.; Camp, R.L.; Chung, G.G. High levels of vascular endothelial growth factor and its receptors (VEGFR-1, VEGFR-2, neuropilin-1) are associated with worse outcome in breast cancer. Hum. Pathol. 2008, 39, 1835–1843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.C.; Voisin, V.; Wang, S.; Wang, D.-Y.; Jones, R.A.; Datti, A.; Uehling, D.; Al-awar, R.; Egan, S.E.; Bader, G.D.; et al. Combined deletion of Pten and p53 in mammary epithelium accelerates triple-negative breast cancer with dependency on eEF2K. EMBO Mol. Med. 2014, 6, 1542–1560. [Google Scholar] [CrossRef]
- Hamurcu, Z.; Ashour, A.; Kahraman, N.; Ozpolat, B. FOXM1 regulates expression of eukaryotic elongation factor 2 kinase and promotes proliferation, invasion and tumorgenesis of human triple negative breast cancer cells. Oncotarget 2016, 7, 16619–16635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, D.; Pan, C.; Sun, J.; Gilbert, C.; Drews-Elger, K.; Azzam, D.J.; Picon-Ruiz, M.; Kim, M.; Ullmer, W.; El-Ashry, D.; et al. VEGF drives cancer-initiating stem cells through VEGFR-2/Stat3 signaling to upregulate Myc and Sox2. Oncogene 2015, 34, 3107–3119. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.-X.; Chen, S.; Huang, L.; Zhou, Y.; Shao, Z.-M. Monitoring Serum VEGF in Neoadjuvant Chemotherapy for Patients with Triple-Negative Breast Cancer: A New Strategy for Early Prediction of Treatment Response and Patient Survival. Oncologist 2019, 24, 753–761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrara, N.; Gerber, H.-P.; LeCouter, J. The biology of VEGF and its receptors. Nat. Med. 2003, 9, 669–676. [Google Scholar] [CrossRef]
- Marty, M.; Pivot, X. The potential of anti-vascular endothelial growth factor therapy in metastatic breast cancer: Clinical experience with anti-angiogenic agents, focusing on bevacizumab. Eur. J. Cancer 2008, 44, 912–920. [Google Scholar] [CrossRef] [PubMed]
- Almatroodi, S.A.; Almatroudi, A.; Alsahli, M.A.; Khan, A.A.; Rahmani, A.H. Thymoquinone, an Active Compound of Nigella sativa: Role in Prevention and Treatment of Cancer. Curr. Pharm. Biotechnol. 2020, 21, 1028–1041. [Google Scholar] [CrossRef]
- Rashid, M.; Sanjarin, F.; Sabouni, F. Thymoquinone Effects on Cell Viability, Apoptosis and VEGF-A Gene Expression Level in AGS(CRL-1739) Cell Line. Anti-Cancer Agents Med. Chem. 2019, 19, 820–826. [Google Scholar] [CrossRef]
- Peng, L.; Liu, A.; Shen, Y.; Xu, H.-Z.; Yang, S.-Z.; Ying, X.-Z.; Liao, W.; Liu, H.-X.; Lin, Z.-Q.; Chen, Q.-Y.; et al. Antitumor and anti-angiogenesis effects of thymoquinone on osteosarcoma through the NF-κB pathway. Oncol. Rep. 2013, 29, 571–578. [Google Scholar] [CrossRef] [Green Version]
- Yegin, Z.; Duran, T.; Yildirim, I.H. Thymoquinone Down-regulates VEGFA and Up-regulates FLT1 Transcriptional Levels in Human Breast Cancer Cells. Int. J. Hum. Genet. 2020, 20, 19–24. [Google Scholar] [CrossRef] [Green Version]
- Alobaedi, O.H.; Alib, W.H.T.; Basheti, I.A. Antitumor effect of thymoquinone combined with resveratrol on mice transplanted with breast cancer. Asian Pac. J. Trop. Med. 2017, 10, 400–408. [Google Scholar] [CrossRef]
- Bierie, B.; Moses, H.L. Gain or loss of TGFbeta signaling in mammary carcinoma cells can promote metastasis. Cell Cycle 2009, 8, 3319–3327. [Google Scholar] [CrossRef] [Green Version]
- Papageorgis, P.; Stylianopoulos, T. Role of epithelial-mesenchymal transition markers in triple-negative breast cancer (Review). Int. J. Oncol. 2015, 46, 933–943. [Google Scholar] [CrossRef] [Green Version]
- Dallas, N.A.; Samuel, S.; Xia, L.; Fan, F.; Gray, M.J.; Lim, S.J.; Ellis, L.M. Endoglin (CD105): A Marker of Tumor Vasculature and Potential Target for Therapy. Clin. Cancer Res. 2008, 14, 1931–1937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thiery, J.P.; Acloque, H.; Huang, R.Y.J.; Nieto, M.A. Epithelial-mesenchymal transitions in development and disease. Cell 2009, 139, 871–890. [Google Scholar] [CrossRef] [PubMed]
- Joshi, A.; Cao, D. TGF-beta signaling, tumor microenvironment and tumor progression: The butterfly effect. Front. Biosci. 2010, 15, 180–194. [Google Scholar] [CrossRef] [Green Version]
- Band, A.M.; Laiho, M. Crosstalk of TGF-β and Estrogen Receptor Signaling in Breast Cancer. J. Mammary Gland. Biol. Neoplasia 2011, 16, 109–115. [Google Scholar] [CrossRef]
- Lin, S.; Yang, J.; Elkahloun, A.G.; Bandyopadhyay, A.; Wang, L.; Cornell, J.E.; Yeh, I.-T.; Agyin, J.; Tomlinson, G.; Suna, L.-Z. Attenuation of TGF-β signaling suppresses premature senescence in a p21-dependent manner and promotes oncogenic Ras-mediated metastatic transformation in human mammary epithelial cells. Biol. Cell 2012, 23, 1569–1581. [Google Scholar] [CrossRef] [PubMed]
- Datto, M.B.; Li, Y.; Panus, J.F.; Howe, D.J.; Xiong, Y.; Wang, X.F. Transforming growth factor beta induces the cyclin-dependent kinase inhibitor p21 through a p53-independent mechanism. Proc. Natl. Acad. Sci. USA 1995, 92, 5545–5549. [Google Scholar] [CrossRef] [Green Version]
- Hannon, G.J.; Beach, D. pl5INK4B is a potentia| effector of TGF-β-induced cell cycle arrest. Nature 1994, 371, 257–261. [Google Scholar] [CrossRef]
- Pietenpol, J.A.; Stein, R.W.; Moran, E.; Yaciuk, P.; Schlegel, R.; Lyons, R.M.; Pittelkow, M.R.; Münger, K.; Howley, P.M.; Moses, H.L. TGF-β1 inhibition of c-myc transcription and growth in keratinocytes is abrogated by viral transforming proteins with pRB binding domains. Cell 1990, 61, 777–785. [Google Scholar] [CrossRef]
- Tang, B.; Vu, M.; Booke, T.; Santner, S.J.; Miller, F.R.; Anver, M.R.; Wakefield, L.M. TGF-beta switches from tumor suppressor to prometastatic factor in a model of breast cancer progression. J. Clin. Investig. 2003, 112, 1116–1124. [Google Scholar] [CrossRef] [Green Version]
- Yi, T.; Cho, S.-G.; Yi, Z.; Pang, X.; Rodriguez, M.; Wang, Y.; Sethi, G.; Aggarwal, B.B.; Liu, M. Thymoquinone inhibits tumor angiogenesis and tumor growth through suppressing AKT and extracellular signal-regulated kinase signaling pathways. Mol. Cancer Ther. 2008, 7, 1789–1796. [Google Scholar] [CrossRef] [Green Version]
- Kou, B.; Liu, W.; Zhao, W.; Duan, P.; Yang, Y.; Yi, Q.; Guo, F.; Li, J.; Zhou, J.; Kou, Q. Thymoquinone inhibits epithelial-mesenchymal transition in prostate cancer cells by negatively regulating the TGF-β/Smad2/3 signaling pathway. Oncol. Rep. 2017, 38, 3592–3598. [Google Scholar] [CrossRef] [Green Version]
- Pollak, M.N.; Schernhammer, E.S.; Hankinson, S.E. Insulin-like growth factors and neoplasia. Nat. Rev. Cancer 2004, 4, 505–518. [Google Scholar] [CrossRef] [PubMed]
- Pollak, M. Insulin and insulin-like growth factor signalling in neoplasia. Nat. Rev. Cancer 2008, 8, 915–928. [Google Scholar] [CrossRef]
- Yang, Y.; Yee, D. Targeting Insulin and Insulin-Like Growth Factor Signaling in Breast Cancer. J. Mammary Gland. Biol. Neoplasia 2012, 17, 251–261. [Google Scholar] [CrossRef] [PubMed]
- Crudden, C.; Song, D.; Cismas, S.; Trocmé, E.; Pasca, S.; Calin, G.A.; Girnita, A.; Girnita, L. Below the Surface: IGF-1R Therapeutic Targeting and Its Endocytic Journey. Cells 2019, 8, 1223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MyersJr, M.G.; Sun, X.J.; White, M.F. The IRS-1 signaling system. Trends Biochem. Sci. 1994, 19, 289–293. [Google Scholar] [CrossRef]
- Rosenzweig, S.A.; Atreya2, H.S. Defining the pathway to insulin-like growth factor system targeting in cancer. Biochem. Pharmacol. 2010, 10, 1115–1124. [Google Scholar] [CrossRef] [Green Version]
- LeRoith, D. Insulin-like growth factor I receptor signaling--overlapping or redundant pathways? Endocrinology 2000, 141, 1287–1288. [Google Scholar] [CrossRef]
- Wu, Y.; Brodt, P.; Sun, H.; Mejia, W.; Novosyadlyy, R.; Nunez, N.; Chen, X.; Mendoza, A.; Hong, S.H.; Khanna, C.; et al. Insulin-like growth factor-I regulates the liver microenvironment in obese mice and promotes liver metastasis. Cancer Res. 2010, 70, 57–67. [Google Scholar] [CrossRef] [Green Version]
- Law, J.H.; Habibi, G.; Kaiji Hu, H.M.; Wang, M.Y.C.; Stratford, A.L.; Park, E.; Gee, J.M.W.; Finlay, P.; Jones, H.E.; Nicholson, R.I.; et al. Phosphorylated Insulin-Like Growth Factor-I/Insulin Receptor Is Present in All Breast Cancer Subtypes and Is Related to Poor Survival. Cancer Res. 2008, 68, 10238–10246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartog, H.; Horlings, H.M.; Vegt, B.v.d.; Kreike, B.; Ajouaou, A.; Vijver, M.J.v.d.; Boezen, H.M.; Bock, G.H.d.; Graaf, W.T.A.v.d.; Wesseling, J. Divergent effects of insulin-like growth factor-1 receptor expression on prognosis of estrogen receptor positive versus triple negative invasive ductal breast carcinoma. Breast Cancer Res. Treat. 2011, 129, 725–736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reiss, K.; Wang, J.-Y.; Romano, G.; Furnari, F.B.; Cavenee, W.K.; Morrione, A.; Tu, X.; Baserga, R. IGF-I receptor signaling in a prostatic cancer cell line with a PTEN mutation. Oncogene 2000, 19, 2687–2694. [Google Scholar] [CrossRef] [Green Version]
- Hankinson, S.E. Circulating levels of sex steroids and prolactin in premenopausal women and risk of breast cancer. Adv. Exp. Med. Biol. 2008, 617, 161–169. [Google Scholar]
- Bahhnassy, A.; Mohanad, M.; Shaarawy, S.; Ismail, M.F.; El-Bastawisy, A.; Ashmawy, A.M.; Zekri, A.-R. Transforming growth factor-β, insulin-like growth factor I/insulin-like growth factor I receptor and vascular endothelial growth factor-A: Prognostic and predictive markers in triple-negative and non-triple-negative breast cancer. Mol. Med. Rep. 2015, 12, 851–864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Resnik, J.L.; Reichart, D.B.; Huey, K.; Webster, N.J.; Seely, B.L. Elevated insulin-like growth factor I receptor autophosphorylation and kinase activity in human breast cancer. Cancer Res. 1998, 58, 1159–1164. [Google Scholar]
- Ma, J.; Hu, X.; Li, J.; Wu, D.; Lan, Q.; Wang, Q.; Tian, S.; Dong, W. Enhancing conventional chemotherapy drug cisplatin-induced anti-tumor effects on human gastric cancer cells both in vitro and in vivo by Thymoquinone targeting PTEN gene. Oncotarget 2017, 8, 85926–85939. [Google Scholar] [CrossRef] [PubMed]
- Gougos, A.; Letarte, M. Primary Structure of Endoglin, an RGD-containing Glycoprotein of Human Endothelial Cells. J. Biol. Chem. 1990, 265, 8361–8364. [Google Scholar] [CrossRef]
- Miller, D.W.; Graulich, W.; Karges, B.; Stahl, S.; Ernst, M.; Ramaswamy, A.; Sedlacek, H.H.; Müller, R.; Adamkiewicz, J. Elevated expression of endoglin, a component of the TGF-β-receptor complex, correlates with proliferation of tumor endothelial cells. IJC 1999, 81, 568–572. [Google Scholar] [CrossRef]
- Li, D.Y.; Sorensen, L.K.; Brooke, B.S.; Urness, L.D.; Davis, E.C.; Taylor, D.G.; Boak, B.B.; Wendel, D.P. Defective Angiogenesis in Mice Lacking Endoglin. Science 1999, 284, 1534–1537. [Google Scholar] [CrossRef] [PubMed]
- Lebrin, F.; Goumans, M.-J.; Jonker, L.; Carvalho, R.L.C.; Valdimarsdottir, G.; Thorikay, M.; Mummery, C.; Arthur, H.M.; Dijke, P.t. Endoglin promotes endothelial cell proliferation and TGF-beta/ALK1 signal transduction. EMBO J. 2004, 23, 4018–4028. [Google Scholar] [CrossRef] [Green Version]
- Bernabeu, C.; Conley, B.A.; Vary, C.P.H. Novel Biochemical Pathways of Endoglin in Vascular Cell Physiology. J. Cell Biochem. 2007, 102, 1375–1388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beresford, M.J.; Harris, A.L.; Ah-See, M.; Daley, F.; Padhani, A.R.; Makris, A. The relationship of the neo-angiogenic marker, endoglin, with response to neoadjuvant chemotherapy in breast cancer. Br. J. Cancer 2006, 95, 1683–1688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dales, J.-P.; Garcia, S.; Bonnier, P.; Duffaud, F.; Andrac-Meyer, L.; Ramuz, O.; Lavaut, M.-N.; Allasia, C.; Charpin, C. CD105 Expression Is a Marker of High Metastatic Risk and Poor Outcome in Breast Carcinomas: Correlations Between Immunohistochemical Analysis and Long-Term Follow-up in a Series of 929 Patients. Am. J. Clin. Pathol. 2003, 119, 374–380. [Google Scholar] [CrossRef] [Green Version]
- Schoonderwoerd, M.J.A.; Koops, M.F.M.; Angela, R.A.; Koolmoes, B.; Toitou, M.; Paauwe, M.; Barnhoorn, M.C.; Liu, Y.; Sier, C.F.M.; Hardwick, J.C.H.; et al. Targeting Endoglin-Expressing Regulatory T Cells in the Tumor Microenvironment Enhances the Effect of PD1 Checkpoint Inhibitor Immunotherapy. Clin. Cancer Res. 2020, 26, 3831–3842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosen, L.S.; Gordon, M.S.; Robert, F.; Matei, D.E. Endoglin for Targeted Cancer Treatment. Curr. Oncol. Rep. 2014, 16, 365. [Google Scholar] [CrossRef] [PubMed]
- Bergers, G.; Hanahan, D. Modes of resistance to anti-angiogenic therapy. Nat. Rev. Cancer 2008, 8, 592–603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paauwe, M.; Heijkants, R.; Oudt, C.; Pelt, G.v.; Cui, C.; Theuer, C.; Hardwick, J.; Sier, C.; Hawinkels, L. Endoglin targeting inhibits tumor angiogenesis and metastatic spread in breast cancer. Oncogene 2016, 35, 4069–4079. [Google Scholar] [CrossRef] [PubMed]
- Folkman, J. New perspectives in clinical oncology from angiogenesis research. Eur. J. Cancer 1996, 32A, 2534–2539. [Google Scholar] [CrossRef]
- Lim, S.O.; Li, C.W.; Xia, W.; Lee, H.H.; Chang, S.S.; Shen, J.; Hsu, J.L.; Raftery, D.; Djukovic, D.; Gu, H.; et al. EGFR Signaling Enhances Aerobic Glycolysis in Triple-Negative Breast Cancer Cells to Promote Tumor Growth and Immune Escape. Cancer Res. 2016, 76, 1284–1296. [Google Scholar] [CrossRef] [Green Version]
- Munn, D.H.; Shafizadeh, E.; Attwood, J.T.; Bondarev, I.; Pashine, A.; Mellor, A.L. Inhibition of T Cell Proliferation by Macrophage Tryptophan Catabolism. J. Exp. Med. 1999, 189, 1363–1372. [Google Scholar] [CrossRef] [PubMed]
- Prendergast, G.C. Immune escape as a fundamental trait of cancer: Focus on IDO. Oncogene 2008, 27, 3889–3900. [Google Scholar] [CrossRef] [Green Version]
- Fallarino, F.; Grohmann, U.; Vacca, C.; Bianchi, R.; Orabona, C.; Spreca, A.; Fioretti, M.C.; Puccetti, P. T cell apoptosis by tryptophan catabolism. Cell Death Differ. 2002, 9, 1069–1077. [Google Scholar] [CrossRef]
- Dill, E.A.; Dillon, P.M.; Bullock, T.N.; Mills, A.M. IDO expression in breast cancer: An assessment of 281 primary and metastatic cases with comparison to PD-L1. Mod. Pathol. 2018, 31, 1513–1522. [Google Scholar] [CrossRef] [PubMed]
- Grohmann, U.; Fallarino, F.; Puccetti, P. Tolerance, DCs and tryptophan: Much ado about IDO. Trends Immunol. 2003, 24, 242–248. [Google Scholar] [CrossRef]
- Munn, D.H.; Sharma, M.D.; Hou, D.; Baban, B.; Lee, J.R.; Antonia, S.J.; Messina, J.L.; Chandler, P.; Koni, P.A.; Mellor, A.L. Expression of indoleamine 2,3-dioxygenase by plasmacytoid dendritic cells in tumor-draining lymph nodes. J. Clin. Investig. 2004, 114, 280–290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, J.; Du, W.; Yan, F.; Wang, Y.; Li, H.; Cao, S.; Yu, W.; Shen, C.; Liu, J.; Ren, X. Myeloid-derived suppressor cells suppress antitumor immune responses through IDO expression and correlate with lymph node metastasis in patients with breast cancer. J. Immunol. 2013, 190, 3783–3797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, T.; Rowswell-Turner, R.B.; Kilinc, M.O.; Egilmez, N.K. Central role of IFNgamma-indoleamine 2,3-dioxygenase axis in regulation of interleukin-12-mediated antitumor immunity. Cancer Res. 2010, 70, 129–138. [Google Scholar] [CrossRef] [Green Version]
- Sakurai, K.; Amano, S.; Enomoto, K.; Kashio, M.; Saito, Y.; Sakamoto, A.; Matsuo, S.; Suzuki, M.; Kitajima, A.; Hirano, T.; et al. Study of indoleamine 2,3-dioxygenase expression in patients with breast cancer. Gan Kagaku Ryoho. Cancer Chemother. 2005, 32, 1546–1549. [Google Scholar]
- Théate, I.; Baren, N.v.; Pilotte, L.; Moulin, P.; Larrieu, P.; Renauld, J.-C.; Hervé, C.; Gutierrez-Roelens, I.; Marbaix, E.; Sempoux, C.; et al. Extensive profiling of the expression of the indoleamine 2,3-dioxygenase 1 protein in normal and tumoral human tissues. Cancer Immunol. Res. 2015, 3, 161–172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Godin-Ethier, J.; Pelletier, S.; Hanafi, L.A.; Gannon, P.O.; Forget, M.A.; Routy, J.P.; Boulassel, M.R.; Krzemien, U.; Tanguay, S.; Lattouf, J.B.; et al. Human activated T lymphocytes modulate IDO expression in tumors through Th1/Th2 balance. J. Immunol. 2009, 183, 7752–7760. [Google Scholar] [CrossRef]
- Asghar, K.; Loya, A.; Rana, I.A.; Tahseen, M.; Ishaq, M.; Farooq, A.; Bakar, M.A.; Masood, I. Indoleamine 2,3-dioxygenase expression and overall survival in patients diagnosed with breast cancer in Pakistan. Cancer Manag. Res. 2019, 11, 475–481. [Google Scholar] [CrossRef] [Green Version]
- Brandacher, G.; Perathoner, A.; Ladurner, R.; Schneeberger, S.; Obrist, P.; Winkler, C.; Werner, E.R.; Werner-Felmayer, G.; Weiss, H.G.; Göbel, G.; et al. Prognostic value of indoleamine 2,3-dioxygenase expression in colorectal cancer: Effect on tumor-infiltrating T cells. Clin. Cancer Res. 2006, 12, 1144–1151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ino, K.; Yoshida, N.; Kajiyama, H.; Shibata, K.; Yamamoto, E.; Kidokoro, K.; Takahashi, N.; Terauchi, M.; Nawa, A.; Nomura, S.; et al. Indoleamine 2,3-dioxygenase is a novel prognostic indicator for endometrial cancer. Br. J. Cancer 2006, 95, 1555–1561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okamoto, A.; Nikaido, T.; Ochiai, K.; Takakura, S.; Saito, M.; Aoki, Y.; Ishii, N.; Yanaihara, N.; Yamada, K.; Takikawa, O.; et al. Indoleamine 2,3-dioxygenase serves as a marker of poor prognosis in gene expression profiles of serous ovarian cancer cells. Clin. Cancer Res. 2005, 11, 6030–6039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.; Liu, L.; Liu, K.; Bizargity, P.; Hancock, W.W.; Visner, G.A. Reduced cytotoxic function of effector CD8+ T cells is responsible for indoleamine 2,3-dioxygenase-dependent immune suppression. J. Immunol. 2009, 183, 1022–1031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whiteside, T.L. Immune suppression in cancer: Effects on immune cells, mechanisms and future therapeutic intervention. Semin. Cancer Biol. 2006, 16, 3–15. [Google Scholar] [CrossRef]
- Ling, W.; Zhang, J.; Yuan, Z.; Ren, G.; Zhang, L.; Chen, X.; Rabson, A.B.; Roberts, A.I.; Wang, Y.; Shi, Y. Mesenchymal stem cells use IDO to regulate immunity in tumor microenvironment. Cancer Res. 2014, 74, 1576–1587. [Google Scholar] [CrossRef] [Green Version]
- Muller, A.J.; DuHadaway, J.B.; Donover, P.S.; Sutanto-Ward, E.; Prendergast, G.C. Inhibition of indoleamine 2,3-dioxygenase, an immunoregulatory target of the cancer suppression gene Bin1, potentiates cancer chemotherapy. Nat. Med. 2005, 11, 312–319. [Google Scholar] [CrossRef]
- Fujigaki, H.; Saito, K.; Fujigaki, S.; Takemura, M.; Sudo, K.; Ishiguro, H.; Seishima, M. The signal transducer and activator of transcription 1alpha and interferon regulatory factor 1 are not essential for the induction of indoleamine 2,3-dioxygenase by lipopolysaccharide: Involvement of p38 mitogen-activated protein kinase and nuclear factor-kappaB pathways, and synergistic effect of several proinflammatory cytokines. J. Biochem. 2006, 139, 655–662. [Google Scholar]
- Alam, M.; Zameer, S.; Najmi, A.K.; Ahmad, F.J.; Imam, S.S.; Akhtar, M. Thymoquinone Loaded Solid Lipid Nanoparticles Demonstrated Antidepressant-Like Activity in Rats via Indoleamine 2, 3- Dioxygenase Pathway. Drug Res. 2020, 70, 206–213. [Google Scholar] [CrossRef] [PubMed]
- Sethi, G.; Ahn, K.S.; Aggarwal, B.B. Targeting nuclear factor-kappa B activation pathway by thymoquinone: Role in suppression of antiapoptotic gene products and enhancement of apoptosis. Mol. Cancer Res. 2008, 6, 1059–1070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- NIH.US. National Library of Medicinev ClnicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/results?cond=thymoquinone&term=&cntry=&state=&city=&dist= (accessed on 20 December 2021).
- Akhondian, J.; Parsa, A.; Rakhshande, H. The effect of Nigella sativa L. (black cumin seed) on intractable pediatric seizures. Med. Sci. Monit. 2007, 13, Cr555–Cr559. [Google Scholar] [PubMed]
- Al Amri, A.M.; Bamosa, A.O. Phase i safety and clinical activity study of thymoquinone in patients with advanced refractory malignant disease. Shiraz E Med. J. 2009, 10, 107–111. [Google Scholar]
- Ammar, I.M.M.; Salem, M.A.A. Amelioration of polycystic ovary syndrome-related disorders by supplementation of thymoquinone and metformin. Middle East. Fertil. Soc. J. 2021, 26, 29. [Google Scholar] [CrossRef]
- Heshmati, J.; Namazi, N.; Memarzadeh, M.-R.; Taghizadeh, M.; Kolahdooz, F. Nigella sativa oil affects glucose metabolism and lipid concentrations in patients with type 2 diabetes: A randomized, double-blind, placebo-controlled trial. Food Res. Int. 2015, 70, 87–93. [Google Scholar] [CrossRef]
- Nikkhah-Bodaghi, M.; Darabi, Z.; Agah, S.; Hekmatdoost, A. The effects of Nigella sativa on quality of life, disease activity index, and some of inflammatory and oxidative stress factors in patients with ulcerative colitis. Phytother. Res. 2019, 33, 1027–1032. [Google Scholar] [CrossRef]
- Boskabady, M.H.; Javan, H.; Sajady, M.; Rakhshandeh, H. The possible prophylactic effect of Nigella sativa seed extract in asthmatic patients. Fundam. Clin. Pharmacol. 2007, 21, 559–566. [Google Scholar] [CrossRef]
- Alkharfy, K.M.; Ahmad, A.; Khan, R.M.; Al-Shagha, W.M. Pharmacokinetic plasma behaviors of intravenous and oral bioavailability of thymoquinone in a rabbit model. Eur. J. Drug Metab. Pharmacokinet. 2015, 40, 319–323. [Google Scholar] [CrossRef]
- Ng, W.K.; Saiful Yazan, L.; Yap, L.H.; Wan Nor Hafiza, W.A.; How, C.W.; Abdullah, R. Thymoquinone-loaded nanostructured lipid carrier exhibited cytotoxicity towards breast cancer cell lines (MDA-MB-231 and MCF-7) and cervical cancer cell lines (HeLa and SiHa). BioMed Res. Int. 2015, 2015, 263131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdelwahab, S.I.; Sheikh, B.Y.; Taha, M.M.; How, C.W.; Abdullah, R.; Yagoub, U.; El-Sunousi, R.; Eid, E.E. Thymoquinone-loaded nanostructured lipid carriers: Preparation, gastroprotection, in vitro toxicity, and pharmacokinetic properties after extravascular administration. Int. J. Nanomed. 2013, 8, 2163–2172. [Google Scholar] [CrossRef] [Green Version]
- Dehghani, H.; Hashemi, M.; Entezari, M.; Mohsenifar, A. The comparison of anticancer activity of thymoquinone and nanothymoquinone on human breast adenocarcinoma. Iran. J. Pharm. Res. 2015, 14, 539–546. [Google Scholar]
- Mu, G.G.; Zhang, L.L.; Li, H.Y.; Liao, Y.; Yu, H.G. Thymoquinone Pretreatment Overcomes the Insensitivity and Potentiates the Antitumor Effect of Gemcitabine Through Abrogation of Notch1, PI3K/Akt/mTOR Regulated Signaling Pathways in Pancreatic Cancer. Dig. Dis. Sci. 2015, 60, 1067–1080. [Google Scholar] [CrossRef]
- Vance, S.H.; Benghuzzi, H.; Wilson-Simpson, F.; Tucci, M. Thymoquinone supplementation and its effect on kidney tubule epithelial cells in vitro. Biomed. Sci. Instrum. 2008, 44, 477–482. [Google Scholar]
- Abukhader, M.M. The effect of route of administration in thymoquinone toxicity in male and female rats. Indian J. Pharm. Sci. 2012, 74, 195–200. [Google Scholar] [CrossRef] [Green Version]
- Al-Ali, A.; Alkhawajah, A.A.; Randhawa, M.A.; Shaikh, N.A. Oral and intraperitoneal LD50 of thymoquinone, an active principle of Nigella sativa, in mice and rats. J. Ayub. Med. Coll. Abbottabad. 2008, 20, 25–27. [Google Scholar]
- Gali-Muhtasib, H.; Kuester, D.; Mawrin, C.; Bajbouj, K.; Diestel, A.; Ocker, M.; Habold, C.; Foltzer-Jourdainne, C.; Schoenfeld, P.; Peters, B.; et al. Thymoquinone triggers inactivation of the stress response pathway sensor CHEK1 and contributes to apoptosis in colorectal cancer cells. Cancer Res. 2008, 68, 5609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khader, M.; Bresgen, N.; Eckl, P. In Vitro toxicological properties of thymoquinone. Food Chem. Toxicol. Int. J. Publ. Br. Ind. Biol. Res. Assoc. 2008, 47, 129–133. [Google Scholar] [CrossRef]
- Khan, M.A.; Tania, M.; Wei, C.; Mei, Z.; Fu, S.; Cheng, J.; Xu, J.; Fu, J. Thymoquinone inhibits cancer metastasis by downregulating TWIST1 expression to reduce epithelial to mesenchymal transition. Oncotarget 2015, 6, 19580–19591. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Disease | Study Type | Major Clinical Findings | Ref. |
|---|---|---|---|
| Intractable pediatric seizures | Double-blinded crossover clinical trial | The frequency of seizures decreased significantly with extract of Nigella sativa | [276] |
| Advanced Refractory Malignant Disease | Open-label, non-randomized cohort study | TQ had no adverse effects and no anti-cancer effects | [277] |
| Polycystic Ovary Syndrome (PCOS) | Randomized clinical trial | Supplementing black cumin oil with metformin improves PCOS-related symptoms | [278] |
| Type II Diabetes millets | Randomized double-blind, placebo-controlled trial | Fasting blood sugar, glycated hemoglobin, triglyceride, and low-density lipoprotein–cholesterol levels all changed considerably in the intervention group (Nigella sativa) compared to the placebo group. | [279] |
| Ulcerative colitis | Prospective, randomized, double-blind, placebo-controlled trial | No significant difference between the two groups (Placebo vs. Nigella sativa) | [280] |
| Asthma disease | Double-blind, placebo-controlled trial | All asthma symptoms, frequency of asthma symptoms/week, chest wheezing, and pulmonary function tests values in the study group (Nigella sativa) had significantly improved | [281] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Adinew, G.M.; Taka, E.; Mochona, B.; Badisa, R.B.; Mazzio, E.A.; Elhag, R.; Soliman, K.F.A. Therapeutic Potential of Thymoquinone in Triple-Negative Breast Cancer Prevention and Progression through the Modulation of the Tumor Microenvironment. Nutrients 2022, 14, 79. https://doi.org/10.3390/nu14010079
Adinew GM, Taka E, Mochona B, Badisa RB, Mazzio EA, Elhag R, Soliman KFA. Therapeutic Potential of Thymoquinone in Triple-Negative Breast Cancer Prevention and Progression through the Modulation of the Tumor Microenvironment. Nutrients. 2022; 14(1):79. https://doi.org/10.3390/nu14010079
Chicago/Turabian StyleAdinew, Getinet M., Equar Taka, Bereket Mochona, Ramesh B. Badisa, Elizabeth A. Mazzio, Rashid Elhag, and Karam F. A. Soliman. 2022. "Therapeutic Potential of Thymoquinone in Triple-Negative Breast Cancer Prevention and Progression through the Modulation of the Tumor Microenvironment" Nutrients 14, no. 1: 79. https://doi.org/10.3390/nu14010079