
From Natural Woods to High Density Materials: An Ecofriendly Approach

Abstract
:1. Introduction
2. Materials and Methods
2.1. Preliminary Phase
2.2. Scanning Electron Microscopy (SEM)
2.3. Thermogravimetry (TGA)
2.4. X-ray Fluorescence Spectrometry (XRF)
2.5. Attenuated Total Reflectance Infrared Spectroscopy (ATR-FTIR)
2.6. Micro-Infrared (Micro-IR)
3. Results
3.1. Wood Delignification
3.2. Wood Densification
3.3. Characterization
3.3.1. Scanning Electron Microscopy
3.3.2. X-ray Fluorescence Spectrometry
3.3.3. Thermogravimetric Analysis
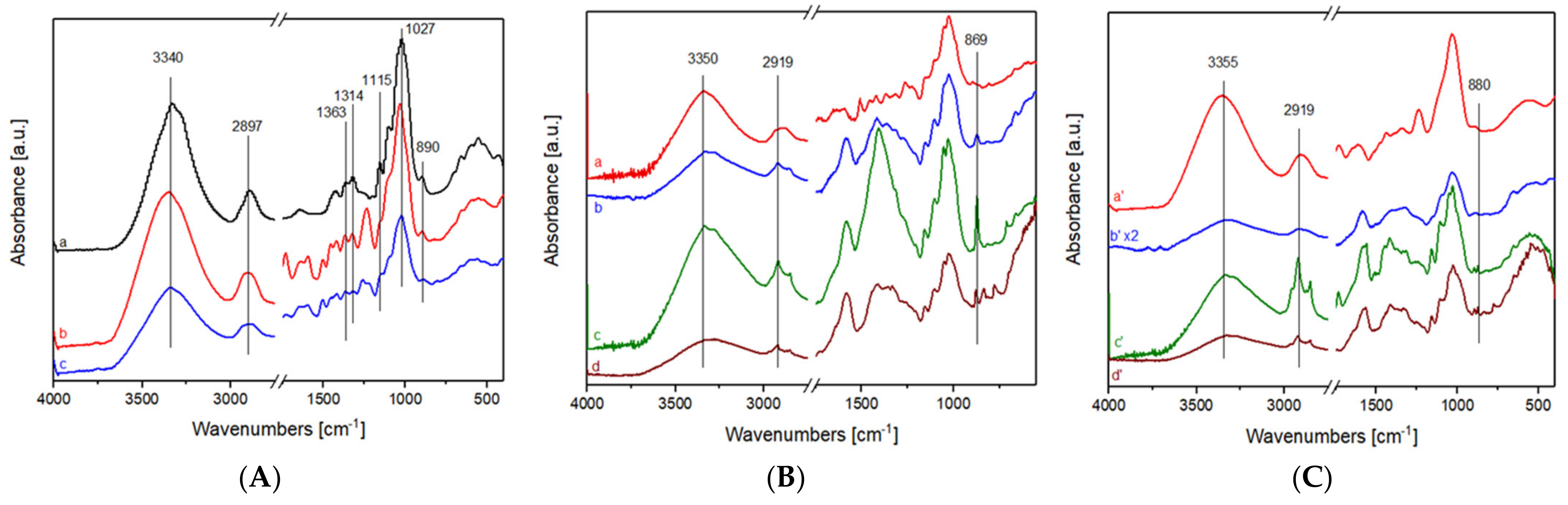
3.3.4. Infrared Spectroscopy in Attenuated Total Reflectance (FTIR-ATR)
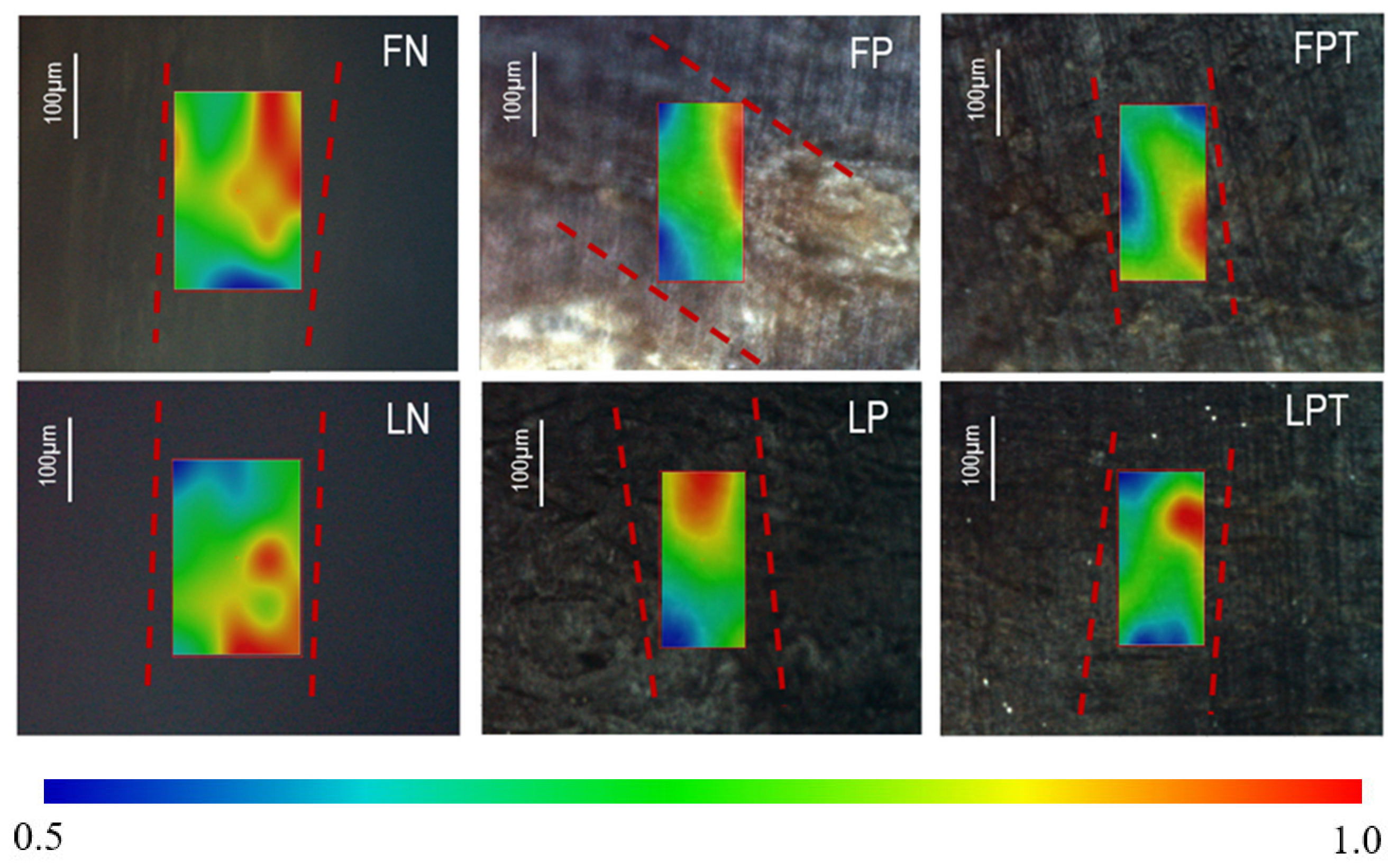
3.3.5. Micro-IR
3.3.6. Mechanical Traction Tests
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Von Arx, G.; Crivellaro, A.; Prendin, A.L.; Čufar, K.; Carrer, M. Quantitative Wood Anatomy—Practical Guidelines. Front. Plant Sci. 2016, 7, 781. [Google Scholar] [CrossRef]
- Zakzeski, J.; Bruijnincx, P.C.A.; Jongerius, A.L.; Weckhuysen, B.M. The Catalytic Valorization of Lignin for the Production of Renewable Chemicals. Chem. Rev. 2010, 110, 3552–3599. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Luo, J.; Qin, W.; Tong, Z. Elemental Analysis, Chemical Composition, Cellulose Crystallinity, and FT-IR Spectra of Toona Sinensis Wood. Mon. Chem. Chem. Mon. 2014, 145, 175–185. [Google Scholar] [CrossRef]
- Jones, P.D.; Schimleck, L.R.; Peter, G.F.; Daniels, R.F.; Clark, A. Nondestructive Estimation of Wood Chemical Composition of Sections of Radial Wood Strips by Diffuse Reflectance near Infrared Spectroscopy. Wood Sci. Technol. 2006, 40, 709–720. [Google Scholar] [CrossRef]
- Chandrasekaran, S.R.; Hopke, P.K.; Rector, L.; Allen, G.; Lin, L. Chemical Composition of Wood Chips and Wood Pellets. Energy Fuels 2012, 26, 4932–4937. [Google Scholar] [CrossRef]
- Curling, S.F.; Clausen, C.A.; Winandy, J.E. Relationships between Mechanical Properties, Weight Loss, and Chemical Composition of Wood during Incipient Brown-Rot Decay. For. Prod. J. 2002, 52, 34–39. [Google Scholar]
- Fodil Cherif, M.; Trache, D.; Brosse, N.; Benaliouche, F.; Tarchoun, A.F. Comparison of the Physicochemical Properties and Thermal Stability of Organosolv and Kraft Lignins from Hardwood and Softwood Biomass for Their Potential Valorization. Waste Biomass Valorization 2020, 11, 6541–6553. [Google Scholar] [CrossRef]
- Wang, J.; Minami, E.; Asmadi, M.; Kawamoto, H. Effect of Delignification on Thermal Degradation Reactivities of Hemicellulose and Cellulose in Wood Cell Walls. J. Wood Sci. 2021, 67, 19. [Google Scholar] [CrossRef]
- Chen, Y.; Dang, B.; Jin, C.; Sun, Q. Processing Lignocellulose-Based Composites into an Ultrastrong Structural Material. ACS Nano 2019, 13, 371–376. [Google Scholar] [CrossRef]
- Liu, Y.; Li, B.; Mao, W.; Hu, W.; Chen, G.; Liu, Y.; Fang, Z. Strong Cellulose-Based Materials by Coupling Sodium Hydroxide–Anthraquinone (NaOH–AQ) Pulping with Hot Pressing from Wood. ACS Omega 2019, 4, 7861–7865. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.; Dang, B.; Luo, X.; Li, W.; Li, J.; Yu, H.; Liu, S.; Li, S. Deep Eutectic Solvent-Assisted In Situ Wood Delignification: A Promising Strategy To Enhance the Efficiency of Wood-Based Solar Steam Generation Devices. ACS Appl. Mater. Interfaces 2019, 11, 26032–26037. [Google Scholar] [CrossRef] [PubMed]
- Akkuş, M.; Budakçı, M. Determination of Color-Changing Effects of Bleaching Chemicals on Some Heat-Treated Woods. J. Wood Sci. 2020, 66, 68. [Google Scholar] [CrossRef]
- Cencin, A.; Zanetti, M.; Urso, T.; Crivellaro, A. Effects of an Innovative Densification Process on Mechanical and Physical Properties of Beech and Norway Spruce Veneers. J. Wood Sci. 2021, 67, 15. [Google Scholar] [CrossRef]
- Sonderegger, W.; Kránitz, K.; Bues, C.-T.; Niemz, P. Aging Effects on Physical and Mechanical Properties of Spruce, Fir and Oak Wood. J. Cult. Herit. 2015, 16, 883–889. [Google Scholar] [CrossRef]
- Krisdianto, K.; Balfas, J. Anatomical Changes of Kekabu Wood (Bombax ceiba L.) Due to Mechanical Densification. Indones. J. For. Res. 2005, 2, 27–36. [Google Scholar] [CrossRef]
- Darwis, A.; Wahyudi, I.; Dwianto, W.; Cahyono, T.D. Densified Wood Anatomical Structure and the Effect of Heat Treatment on the Recovery of Set. J. Indian Acad. Wood Sci. 2017, 14, 24–31. [Google Scholar] [CrossRef]
- Mayer, I.; Koch, G. Element Content and PH Value in American Black Cherry (Prunus serotina) with Regard to Colour Changes during Heartwood Formation and Hot Water Treatment. Wood Sci. Technol. 2007, 41, 537–547. [Google Scholar] [CrossRef]
- Morgan, T.J.; George, A.; Boulamanti, A.K.; Álvarez, P.; Adanouj, I.; Dean, C.; Vassilev, S.V.; Baxter, D.; Andersen, L.K. Quantitative X-Ray Fluorescence Analysis of Biomass (Switchgrass, Corn Stover, Eucalyptus, Beech, and Pine Wood) with a Typical Commercial Multi-Element Method on a WD-XRF Spectrometer. Energy Fuels 2015, 29, 1669–1685. [Google Scholar] [CrossRef]
- Sebio-Puñal, T.; Naya, S.; López-Beceiro, J.; Tarrío-Saavedra, J.; Artiaga, R. Thermogravimetric Analysis of Wood, Holocellulose, and Lignin from Five Wood Species. J. Therm. Anal. Calorim. 2012, 109, 1163–1167. [Google Scholar] [CrossRef]
- Wang, J.; Minami, E.; Asmadi, M.; Kawamoto, H. Thermal Degradation of Hemicellulose and Cellulose in Ball-Milled Cedar and Beech Wood. J. Wood Sci. 2021, 67, 32. [Google Scholar] [CrossRef]
- Leclerc, D.F.; Trung, T.P. Vibrational Spectroscopy in the Pulp and Paper Industry. In Handbook of Vibrational Spectroscopy; Chalmers, J.M., Ed.; John Wiley and Sons, Ltd.: Chichester, UK, 2006. [Google Scholar]
- Colom, X.; Carrillo, F.; Nogués, F.; Garriga, P. Structural Analysis of Photodegraded Wood by Means of FTIR Spectroscopy. Polym. Degrad. Stab. 2003, 80, 543–549. [Google Scholar] [CrossRef]
- Adel, A.M.; El–Wahab, Z.H.A.; Ibrahim, A.A.; Al–Shemy, M.T. Characterization of Microcrystalline Cellulose Prepared from Lignocellulosic Materials. Part I. Acid Catalyzed Hydrolysis. Bioresour. Technol. 2010, 101, 4446–4455. [Google Scholar] [CrossRef] [PubMed]
- Schwanninger, M.; Rodrigues, J.C.; Pereira, H.; Hinterstoisser, B. Effects of Short-Time Vibratory Ball Milling on the Shape of FT-IR Spectra of Wood and Cellulose. Vib. Spectrosc. 2004, 36, 23–40. [Google Scholar] [CrossRef]
- Hu, G.; Cateto, C.; Pu, Y.; Samuel, R.; Ragauskas, A.J. Structural Characterization of Switchgrass Lignin after Ethanol Organosolv Pretreatment. Energy Fuels 2012, 26, 740–745. [Google Scholar] [CrossRef]
- Pandey, K.; Pitman, A. FTIR Studies of the Changes in Wood Chemistry Following Decay by Brown-Rot and White-Rot Fungi. Int. Biodeterior. Biodegrad. 2003, 52, 151–160. [Google Scholar] [CrossRef]
- Pandey, K.K. A Study of Chemical Structure of Soft and Hardwood and Wood Polymers by FTIR Spectroscopy. J. Appl. Polym. Sci. 1999, 71, 1969–1975. [Google Scholar] [CrossRef]
- Popescu, C.-M.; Singurel, G.; Popescu, M.-C.; Vasile, C.; Argyropoulos, D.S.; Willför, S. Vibrational Spectroscopy and X-Ray Diffraction Methods to Establish the Differences between Hardwood and Softwood. Carbohydr. Polym. 2009, 77, 851–857. [Google Scholar] [CrossRef]
- Rautkari, L.; Hill, C.A.S.; Curling, S.; Jalaludin, Z.; Ormondroyd, G. What Is the Role of the Accessibility of Wood Hydroxyl Groups in Controlling Moisture Content? J. Mater. Sci. 2013, 48, 6352–6356. [Google Scholar] [CrossRef]
- Sun, J.; Zhao, R.; Zhong, Y.; Chen, Y. Compressive Mechanical Properties of Larch Wood in Different Grain Orientations. Polymers 2022, 14, 3771. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Weight (g) | Volume (cm3) | Density (g/cm3) |
|---|---|---|---|
| Fir | 7.00 ± 0.26 | 15.20 ± 0.45 | 0.488 ± 0.014 |
| Larch | 8.77 ± 0.38 | 11.90 ± 0.47 | 0.735 ± 0.011 |
| Sample | Starting Density (g/cm3) | Final Density (g/cm3) | Increase (%) |
|---|---|---|---|
| Densified fir | 0.48 ± 0.01 | 1.60 ± 0.09 | 340.32 ± 24.45 |
| Densified fir with TEOS | 1.46 ± 0.19 | 303.60 ± 38.60 | |
| Densified larch | 0.73 ± 0.02 | 1.64 ± 0.03 | 224.86 ± 5.05 |
| Densified larch with TEOS | 1.48 ± 0.12 | 197.78 ± 16.68 |
| Sample | Si (Atomic %) |
|---|---|
| FP | - |
| FPT | 0.166 ± 0.061 |
| LP | - |
| LPT | 0.043 ± 0.005 |
| Sample | Force at Yield (N) |
|---|---|
| Natural fir | 225.3 |
| Densified fir densified | 911.1 ± 42.1 |
| Densified fir with TEOS | 841.9 ± 269.6 |
| Natural larch | 1060.9 |
| Densified larch | 1166.9 ± 72.1 |
| Densified larch with TEOS | 1005.2 ± 119.3 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gullo, F.; Marangon, A.; Croce, A.; Gatti, G.; Aceto, M. From Natural Woods to High Density Materials: An Ecofriendly Approach. Sustainability 2023, 15, 2055. https://doi.org/10.3390/su15032055
Gullo F, Marangon A, Croce A, Gatti G, Aceto M. From Natural Woods to High Density Materials: An Ecofriendly Approach. Sustainability. 2023; 15(3):2055. https://doi.org/10.3390/su15032055
Chicago/Turabian StyleGullo, Francesca, Andrea Marangon, Alessandro Croce, Giorgio Gatti, and Maurizio Aceto. 2023. "From Natural Woods to High Density Materials: An Ecofriendly Approach" Sustainability 15, no. 3: 2055. https://doi.org/10.3390/su15032055