
The Benign Clone Causing Aplastic Anaemia

1
Department of Haematology, Trinity College, 2 Dublin, Ireland
2
Northern Ireland Blood Transfusion Service (NIBTS), Belfast BT9 7TS, UK
3
Department of Internal Medicine V, Medical University of Innsbruck, 6020 Innsbruck, Austria
4
Department of Industrial Engineering, University of Trento, 38122 Trento, Italy
*
Author to whom correspondence should be addressed.
Thalass. Rep. 2023, 13(2), 157-164; https://doi.org/10.3390/thalassrep13020015
Submission received: 29 November 2022
/
Revised: 8 June 2023
/
Accepted: 8 June 2023
/
Published: 12 June 2023
(This article belongs to the Special Issue Thalassemia Syndromes as a Benign Cancer of Hematopoietic Stem Cells)
{kind=link}
{kind=link}
Abstract
:Severe Aplastic Anaemia (SAA) is a rare benign disease but carries a high-mortality rate unless treated in a specialised centre. Overwhelming laboratory and clinical evidence points to an autoimmune pathogenesis; although, the aetiology remains obscure in the majority of cases. The differential diagnosis in older patients is problematical and a diagnosis of hypoplastic myelodysplasia remains difficult. This review points out the difficulty in diagnosis without a specific test. Future research needs to define a specific diagnostic test and refine therapeutic interventions.
1. The Benign Clone
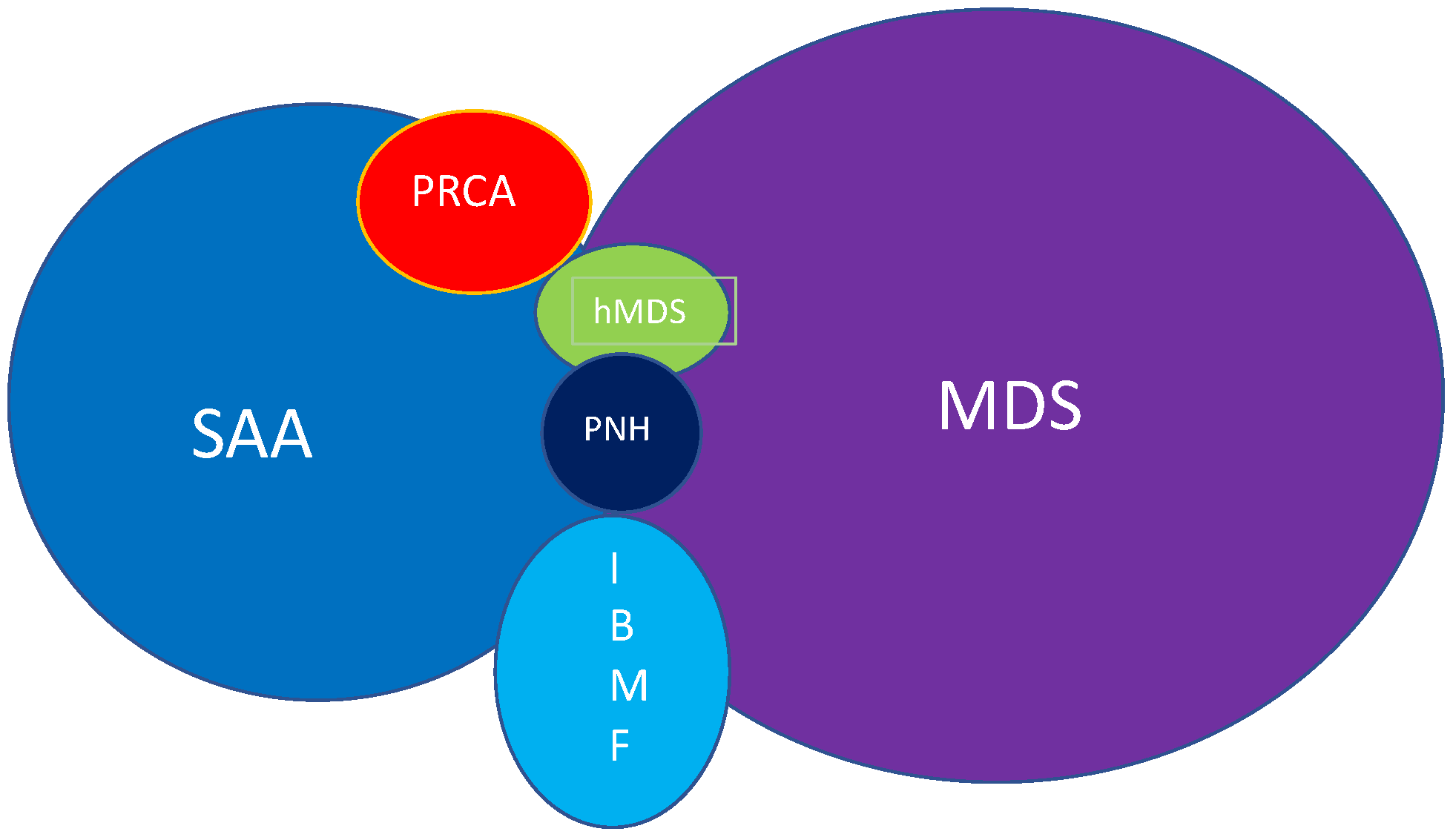
Although acquired SAA is a rare disease, it has been a source of investigation for many haematologists and scientists as it is a curable disease, and intense investigations may provide insights into its pathophysiology and a diagnostic test. Since the ‘genetic revolution’ there has been a race to find a genetic abnormality which is specific to the diagnosis of acquired SAA. Unfortunately, although several cytogenetic and molecular abnormalities have been discovered, we still do not have diagnostic tests [1]. Nevertheless, these progresses in genetic have allowed to discriminate between different types of conditions that may overlap with aplastic anaemia picture, such as myelodisplastic syndrome (MDS) and hypoplastic MDS (hMDS). This is particularly true in paediatric haematology, where a subtype of MDS called refractory cytopenias of childhood (RCC) and inherited bone marrow failure (IBMF), still confuse even expert haematologist. The exact diagnosis of the underlying condition is difficult, but is important to ensure that the correct treatment is delivered [2]. To date, the main difference between SAA, RCC and IBMF is that SAA is driven by an autoimmune clone of lymphocytes directed against the stem cells. Conversely, RCC and IBMF are associated with genetic abnormalities and need to always be treated with haemopoietic stem cell transplant (HSCT) [3]. Other conditions that need to be considered in the differential diagnosis are paroxistic nocturnal haemoglobinuria (PNH), and pure red-cell aplasia (PRA) (Figure 1).
Although clonal evolution to PNH, MDS or acute myeloid leukaemia (AML) has been observed [4], co-existing somatic mutations may predispose this process, but this needs further clarification. Recurrent mutations have been identified in up to 50% of patients with acquired SAA [5] but mutations in ASXL1, DNMT3A are frequently found in myeloid malignancy, limiting their diagnostic usefulness. It is important that molecular and cytogenetic investigations are carried out at diagnosis before the initiation of therapy. It has been demonstrated that in serial samples from patients who have not evolved into MDS, clones with GATA2, PHF6, RUNX1, SMC3, TET2 and BCORL1 mutations decrease in size over time, whereas ASXL1, CALR, CUX1, ETV6, EZH2, G3BP1, RIT1, U2AF1 and ZRSR1 expanded [6]. However, several investigators believe that clonal abnormalities do not predict a response to therapy. Telomerase abnormalities have been reported in acquired aplastic anemia, and shortened telomerase length at diagnosis have been shown to correlate with poorer outcomes [7]. Cytogenetic abnormalities especially (−7) are common in acquired aplastic anemia. Recent studies have shown that abnormal cytogenetics is an independent predictor of a poor response to immunosuppressive therapy (HR = 0.255; 95% CI = 0.077–0.839; p = 0.024) [8] This suggests that patients with SAA and abnormal cytogenetics have different clinical characteristics, compared to patients with SAA and normal cytogenetics [8].
2. Insight in the Disease
Acquired aplastic anaemia is a rare, potentially fatal, but curable disease, in which the reported incidence varies from 1.5–2.3 per million in Western countries to 3.0–7.5 per million in Asia [9]. The reasons for the variation in the reported incidence are unknown. In the majority of adults, the aetiology of the disease is unknown and there is no specific test which is diagnostic. Although many patients have abnormal liver blood tests at the time of diagnosis, hepatitis virus A, B and C (HAV, HBV, and HCV) are not believed to be causative in most cases. Benzine toxicity was an etiological agent in the past but is no longer a significant risk factor in Western countries [10]. Numerous factors have been associated with the pathobiology of aplastic anaemia including cytotoxic T cells, cytokines and chemokines, interferon γ and tumour necrosis factor α [11]. However, the diagnosis depends on a number of investigations and the ruling out of IBMF, Fanconi Anaemia (FA), Dyskeratosis Congenita, GATA2 deficiency, Telomere diseases and CTLA4 deficiency, which usually present early in life but may be brought to medical attention for the first time in adult life. A family history may be helpful. An important reason for diagnosing IBMF is that the treatment may be quite different from acquired aplastic anaemia.
The diagnosis of aplastic anaemia is based on the presence of peripheral pancytopenia and a hypoplastic marrow without evidence of malignancy [9,12]. Examination of a blood film is mandatory, although care must be taken as concomitant drug administration (e.g., G-CSF or Epo) may cause misleading dysplastic granulocyte appearances. A marrow biopsy should be carried out to assess cellularity (Figure 2A,B) and cytogenetic analysis and/or FISH should be carried out on a bone marrow aspirate. Pressure should be applied to the biopsy site and a small pressure dressing should also be applied. The site should be examined for bleeding after 30 min. The finding of a deletion of chromosome 7 carries a poor prognosis, but the findings of other abnormalities (+8, +Y) should not alter therapeutic strategies. Whether the uncovering of cytogenetic abnormalities during investigation of a patient with presumed aplastic anaemia alters the diagnosis is debatable [13]. According to Young [10], screening for the approximately 50 genes that cause constitutional marrow failure is particularly useful in moderate and chronic pancytopenia, thrombocytopenia and macrocytic anaemia, in children, adolescents and patients with whom immunosuppressive therapy has been unsuccessful. Next-generation sequencing has identified a number of mutations (DNMT3A, BCOR, BCORL1, and ASXL1) [14].
MDS and SAA are two distinct conditions that share same similar feature. Up to 20% of patients with MDS have hypocellular bone marrow and this is known as hypoplastic MDS [15]. This entity is still classified within myeloid neoplasm; however, it represents an overlapping condition between SAA and MDS. Therefore, is important that clinicians are aware of it. hMDS is more commonly reported in patients with low risk MDS. The implementation of NGS has allowed to better discriminate between SAA and hMDS. The genes mutation more typically seen in SAA are: (i) phosphatidylinositol glycan anchor biosynthesis class A (PIGA), (ii) BCOR/BCOR-like 1 (BCORL1), (iii) DNMT3A and (iv) ASXL1 [16], while the following mutations are rarely seen in SAA JAK2/JAK3, RUNX1, TP53, TET2, CUB, and sushi multiple domains 1 genes, SRSF2, U2AF1, MPL and Erb-B2 receptor tyrosine kinase 2 [17].
3. Paroxysmal Nocturnal Hemoglobinuria and Aplastic Anaemia
Heretofore, clonality was associated with malignant disease but PNH is an example of a clonal disorder which is benign and is the result of a clonal expansion of GPI-deficient hemopoietic cells, leading to severe symptoms and premature mortality. Many clonal disorders affect the elderly disproportionally, such as cancer and dementia [18]. Moreover, the presence of a clonal hemopoietic cell per se does not necessarily imply an evolution towards malignancy (e.g., leukaemia). Similarly, somatic mutations tend to be found over time in haematopoietic stem cell precursors (HSCP). This is particularly true in SAA, where the evolution of the disease is believed to be caused by an emerging clone of lymphocytes capable of attacking the stem cells. Previous studies have suggested that the increase in a PNH clone in SAA may be the consequence of an advantage mechanism against auto-immunity (theory of GPI-advantage). However, recent studies have shown that PNH clones are also present in normal healthy people [19].
Identification of a PNH clone by flow cytometry is important, but it must be remembered that a small clone of GPI-deficient cells does not make a diagnosis of PNH, as this rare disease is characterised by complement-mediated intravascular haemolysis [20] and an increased thrombogenic risk. The morbidity and outcome for PNH has been radically altered by the introduction of the humanised-monoclonal antibody, eculizumab, aimed at component 5 of the complement cascade [21]. Other contenders, Crovalimab, Ravulizumab and Pegcetacoplan, all of which prevent complement-mediated haemolysis, have since entered the race. These monoclonal antibodies also reduce or eliminate the thrombotic risk and have reduced the requirement for HCT in PNH, other than in rare patients in whom aplasia is the predominant feature. All these anti-complementary agents necessitate the administration of vaccines against Gram-positive encapsulated organisms and prophylactic penicillin.
A negative diepoxybutane test (DEB) will exclude FA. The major difficulty is to distinguish aplastic anaemia from hypoplastic myelodysplastic syndrome, which may be very difficult. The increase in frequency of aplastic anaemia in elderly patients may make the differentiation between hypoplastic MDS and aplastic anaemia more difficult, as MDS incidence increases with age. Clonal evolution, which occurs in 30% of patients [22] treated with IS, may represent undiagnosed MDS at the time of initiation of treatment or may be, in some way, related to the treatment.
The absolute neutrophil count in the peripheral blood at diagnosis enables a classification of very severe (neutrophils 0–0.2 × 109/L), or severe (neutrophils 0.21–0.5 × 109/L) aplastic anaemia. Measurement of lymphocyte telomere length, in acquired aplastic anaemia, has been championed by Neal Young and colleagues, but may occur in IBMF; however, this investigation may not be diagnostic [10], and the expertise may not be available in many haematology laboratories.
4. Clonal Haematopoiesis and Malignancy
Clonal haematopoiesis increases in frequency with advancing age. Although clonality has been associated with malignancy, the relationship is not completely clear. PNH is a good example of a non-malignant clonal disorder. Clonal haematopoiesis of indeterminate potential (CHIP) is the presence of a clonally expanded hematopoietic stem cell caused by a leukaemogenic mutation in individuals without evidence of hematologic malignancy, dysplasia, or cytopenia. CHIP is associated with a 0.5–1.0% risk per year of leukaemia [23]. The majority of individuals have mutations in epigenetic regulators: DNMT3A, TET2, ASXL1; DNA damage repair genes: PPM1D, TP53; the regulatory tyrosine kinase: JAK2; or mRNA spliceosome components: SF3B1, and SRSF2. CHIP may be involved in the development of solid cancers or immune disorders, such as aplastic anaemia and MDS.
The correct and early diagnosis is important, as the length of time to diagnose and provide treatment influences the outcome [24].
Some clinicians contend that observational databases combined with an expert opinion provide reliable and effective ways of determining effective new therapies in the transplant setting [25]. However, despite the aetiology of acquired aplastic anaemia, it seems that the pathogenesis, in the majority of patients, is auto-immune in nature. Laboratory data have shown increased interferon-producing T cells, in the marrow of AA patients. More recently, the “escape mechanisms of stem cells from an autoimmune T cells attack” has been strongly suggested by the following results in AA patients: loss of heterozygosity of chromosome arm 6p [26], loss of HLA DR 15 [27], and structural homology of HLA evolutionary divergence, defining patterns of autoreactivity [26]. This in vitro evidence further supports the autoimmune pathogenesis already suggested to explain the expansion of GPI negative cells. Additional in vivo observation is the response of patients to immunosuppressive treatment, although the effect of anti-thymocyte globulin (ATG) is more complex than immunosuppression alone [28].
5. SAA Treatment
The standard therapy for first-line IST is ATG (antithymocyte globulin) and CsA (cyclosporine A). Horse ATG is preferred to rabbit ATG, as it produces better results, but horse ATG is not available in many centres/countries. Failure to respond to this (30–50%) should initiate a search for a matched-unrelated donor or proceed with a second course of IST with ATG + CsA + G-CSF. In the context of IST (immunosuppressive therapy), the addition of Eltrombopag, an oral thrombopoietin-receptor agonist, has been shown to increase the rate of haematological responses [29].
CsA should be continued for one year post-HCT with gradual dose reduction. Chimeric studies should be carried out as a progressive reduction in donor cell proportions may herald graft rejection/relapse. Results of a second HCT provide conflicting results [30,31]. In a retrospective analysis, published in 1994 [32], second HCT for graft rejection had a poor outcome of 33%, but appeared to improve in another retrospective report in 2015 [33]. In general, SAA response to treatment is dependent on the age of the patient and the severity of the disease [26]. Haematopietic stem cell transplants (HSCT) from a matched sibling donor is currently the gold standard for SAA patients < 50 years of age. The use of a matched-unrelated donor (MUD) should be considered for patients who relapse after immunosuppression (IS). The availability of new conditioning regimes has recently allowed the acquisition of excellent results, even with haploidentical donors [34].
Cyclophosphamide and ATG remain the conditioning therapy. In older patients (>30 years), a combination of fludarabine, cyclophosphamide with ATG or alemtuzumab (CAMPATH) is recommended, with a reduction in the cyclophosphamide dose to alleviate toxicity. There is no doubt that bone marrow, rather than mobilised-peripheral blood, is the stem cell source of choice [26]. In the absence of an HLA matched sibling donor, the use of a fully matched-unrelated donor transplant has been shown to be successful [26]. Haploidentical transplantation is now associated with excellent results; however, the use of irradiation in some conditioning regimens is somewhat disconcerting in view of the earlier finding of secondary malignancies in patients with aplastic anaemia, who received irradiation as part of their conditioning [35].
6. Chimerism Issues in SAA
Laboratory animals which had the haematopoietic system of the donor following haemopoietic cell transplantation, HCT, were called ‘radiation’ chimaeras. Following successful HCT, the recipient is known as a chimaera. The degree of chimerism (i.e., the % of donor and recipient haematopoiesis) following HCT for aplastic anaemia may be very important [36] in determining Graft versus Host Disease (GvHD), graft rejection, disease recurrence or autologous recovery. There are many methods of ascertaining the degree of hemopoietic chimerism following HCT, but the measurement of short tandem repeats, which are highly polymorphic, have proved useful and are now measured by fluorometric methods for clinical use.
Autologous reconstitution (AR) represents a unique event after HSC for aplastic anaemia. AR consists of the recovery of host haematopoiesis and peripheral blood counts after HSCT. AR differs from graft rejection, as in the former there is a complete recovery of peripheral blood counts of recipient origin, and in the latter, pancytopenia occurs as the most probable reflecting-disease relapse. Graft rejection has a poor outcome despite treatment with a second HSCT, immunosuppression or donor lymphocyte infusion. AR has an incidence of 4.2%. and may be higher, as chimerism follow up may not be regularly performed following HSCT. Patients who developed AR showed a better OS after HSCT 84% (versus 74% of the control group) at 10 years of follow up [37].
7. SAA Guidelines
A number of guidelines have been published aimed at facilitating the diagnosis and management of aplastic anaemia [38,39]. However, we must admit that we have a problem with ‘guidelines’ for any clinical situation. Firstly, guidelines cannot possibly deal with all clinical problems. Secondly, guidelines are not directives but, unfortunately, are believed to be by some doctors and members of the legal profession. Guidelines should be brief (the guidelines by Marsh and colleagues and Killick and colleagues are approximately 20 pages each). Although guidelines are very useful in determining strategy, they may inhibit innovation and critical thinking [40] when applied to individual clinical situations.
Anecdote: a colleague in a large university hospital was conducting a ward round in the stem cell transplant unit. She came to a very ill post-transplant patient and asked her accompanying staff (who were all considered to be “high flyers”): what should we do? The answer was: consult the guidelines; hold a multi-disciplinary conference. The patient clearly needed immediate and urgent action and a decision needed to be made quickly.
8. Conclusions
The survival for patients who are treated with HCT (haemopoietic cell transplantation) or immunosuppressive therapy has significantly improved. The interrelationship between SAA and PNH are not fully elucidated. In spite of molecular abnormalities, there is still no diagnostic test for SAA. Since SAA is a very rare condition, patients with a suspected diagnosis of SAA should be referred to a malignant haematology centre capable of performing specific genetic testing and with large experience in performing HSCT.
Overall, the prognosis for patients with severe acquired aplastic anaemia has improved dramatically, and newer techniques in HCT may facilitate even better outcomes. The difference between hypoplastic MDS and aplastic anaemia in the elderly is extremely difficult. Most patients with aplastic anaemia are curable or manageable. Finally, we agree with Andrea Bacigalupo: patients should be treated, preferentially, in experienced centres, and at best, in the context of clinical trials [30].
Author Contributions
Conceptualization, S.R.M. and AP.; writing—review and editing, S.R.M. and A.P. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
The authors are extremely grateful to Andrea Bacigalupo (Policlinico Gemelli, La Sapienza University, Rome, Italy) for his help in the preparation of this manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Brzezniakiewicz-Janus, K.; Rupa-Matysek, J.H.; Hoppe, A.; Gil, L. Impact of clonal hematopoiesis on outcomes in patients with aplastic anemia. Acta Haematol. Pol. 2021, 52, 543–551. [Google Scholar] [CrossRef]
- Niemeyer, C.M.; Baumann, I. Classification of childhood aplastic anemia and myelodysplastic syndrome. Hematol. Am. Soc. Hematol. Educ. Program. 2011, 2011, 84–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strahm, B.; Locatelli, F.; Bader, P.; Ehlert, K.; Kremens, B.; Zintl, F.; Führer, M.; Stachel, D.; Sykora, K.W.; Sedlacek, P.; et al. Reduced intensity conditioning in unrelated donor transplantation for refractory cytopenia in childhood. Bone Marrow Transpl. 2007, 40, 329–333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Socié, G.; Rosenfeld, S.; Frickhofen, N.; Gluckman, E.; Tichelli, A. Late clonal diseases of treated aplastic anemia. Seminal. Hematol. 2000, 37, 91–101. [Google Scholar] [CrossRef]
- Babushok, D.V.; Perdigones, N.; Perin, J.C.; Olson, T.S.; Ye, W.; Roth, J.J.; Lind, C.; Cattier, C.; Li, Y.; Hartung, H.; et al. Emergence of clonal hematopoiesis in the majority of patients with acquired aplastic anemia. Cancer Genet. 2015, 208, 115–128. [Google Scholar] [CrossRef] [Green Version]
- Negoro, E.; Negata, Y.; Clemente, M.J.; Hosono, N.; Shen, W.; Nazha, S.; Yoshizato, T.; Hirsch, C.; Przychodzen, B.; Mahfouz, R.Z.; et al. Origins of myelodysplastic syndrome after aplastic anemia. Blood 2017, 130, 1953–1957. [Google Scholar] [CrossRef] [Green Version]
- Gadalla, S.M.; Wang, T.; Dagnall, C.; Spellman, S.R.; Lee, S.J.; Williams, K.M.; Wong, J.Y.; De Vivo, I.; Savage, S.A. Association between donor leukocyte telomere length and survival after unrelated allogeneic hematopoietic cell transplantation for severe aplastic anemia. JAMA 2015, 313, 594–602. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.Y.; Lee, J.W.; Lee, S.E.; Cho, B.S.; Kim, M.; Eom, K.S.; Kim, Y.J.; Kim, H.J.; Lee, S.; Min, C.K.; et al. The characteristics and clinical outcome of adult patients with aplastic anemia and abnormal cytogenetics at diagnosis. Genes. Chromosomes Cancer 2010, 49, 844–850. [Google Scholar] [CrossRef]
- Norasetthada, L.; Wongkhantee, S.; Chaipokam, J.; Charoenpeasset, K.; Chuncharunee, S.; Rojnuckarin, P.; Sirijerechai, C.; Wanachiwanawin, W.; Issaragisil, S. Thai Aplastic Anemia Study Group. Adult aplastic anemia in Thailand: Incidence and treatment outcome from a prospective nationwide population-based study. Ann. Hematol. 2021, 1000, 2443–2452. [Google Scholar] [CrossRef]
- Young, N.S. Aplastic Anemia. N. Engl. J. Med. 2018, 379, 1843–1856. [Google Scholar] [CrossRef]
- Guidice, V.; Selleri, C. Aplastic anemia: Pathophysiology. Semin. Hematol. 2022, 59, 13–20. [Google Scholar] [CrossRef] [PubMed]
- Camitta, B.M.; Rappeport, J.M.; Parkman, R.; Nathan, D.G. Selection of patients for bone marrow transplantation in severe aplastic anemia. Blood 1975, 45, 355–363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maciejewski, J.P.; Risitano, A.; Sloand, E.M.; Nunez, O.; Young, N.S. Distinct clinical outcomes for cytogenetic abnormalities evolving from aplastic anemia. Blood 2002, 99, 3129–3135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeZem, A.E.; Churpek, J.E. Approach to the diagnosis of aplastic anemia. Blood Adv. 2021, 5, 2660–2671. [Google Scholar] [CrossRef]
- Durrani, J.; Maciejewski, J.P. Idiopathic aplastic anemia vs hypocellular myelodysplastic syndrome. Hematol. Am. Soc. Hematol. Educ. Program. 2019, 2019, 97–104. [Google Scholar] [CrossRef]
- Votavova, H.; Belickova, M. Hypoplastic myelodysplastic syndrome and acquired aplastic anemia: Immune-mediated bone marrow failure syndromes. Int. J. Oncol. 2022, 60, 7. [Google Scholar] [CrossRef]
- Yoshizato, T.; Dumitriu, B.; Hosokawa, K.; Makishima, H.; Yoshida, K.; Townsley, D.; Sato-Otsubo, A.; Sato, Y.; Liu, D.; Suzuki, H.; et al. Somatic mutations and clonal hematopoiesis in aplastic anemia. N. Engl. J. Med. 2015, 373, 35–47. [Google Scholar] [CrossRef] [Green Version]
- Kinoshita, T.; Normitsu, I. Relationship between Aplastic Anemia and Paroxysmal Nocturnal Hemoglobinuria. Int. J. Hematol. 2002, 75, 117–122. [Google Scholar] [CrossRef]
- Babushok, D.V. When does a PNH clone have clinical significance? Hematol. Am. Soc. Educ. Program. 2021, 2021, 143–152. [Google Scholar] [CrossRef]
- McCann, S.R. Blood Matters; Interview with Lucio Luzzatto: Stockholm, Sweden, 2013; ISBN 978-90-823759-1-6. [Google Scholar]
- Rother, R.P.; Rollins, S.A.; Mojcik, C.F.; Brodsky, R.A.; Bell, L. Discovery and development of the complement inhibitor eculizumab for the treatment of paroxysmal nocturnal hemoglobinuria. Nat. Biotechnol. 2007, 25, 1256–1264. [Google Scholar] [CrossRef]
- Afable, M.G.; Tiu, R.V.; Maciejewski, J.P. Clonal evolution in aplastic anemia. Am. Soc. Hematol. Educ. Program. 2011, 128, 90–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marnell, C.S.; Bick, A.; Nataragjan, P. Clonal hematopoiesis of indeterminate potential (CHIP): Linking somatic mutations, hematopoiesis, chronic inflammation and cardiovascular disease. J. Mol. Cell. Cardiol. 2021, 161, 98–105. [Google Scholar] [CrossRef] [PubMed]
- Locasciulli, A.; Oneto, R.; Bacigalupo, A.; Socié, G.; Korthof, E.; Bekassy, A.; Schrezenmeier, H.; Passweg, J.; Fuhrer, M. Severe Aplastic Anemia Working Party of the European Blood and Marrow Transplant Group. Outcome of patients with acquired aplastic anemia given first line bone marrow transplantation or immunosuppressive treatment in the last decade: A report from the European Group for Blood and Marrow Transplantation (EBMT). Haematologica 2007, 92, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Gale, R.P.; Eapen, M.; Logan, B.; Zhang, M.; Lazarus, H.M. Are there roles for observational database studies and structured quantification of expert opinion to answer therapy controversies in Transplants? Bone Marrow Transplant. 2009, 43, 435–446. [Google Scholar] [CrossRef] [Green Version]
- Tsugi, N.; Hosokawa, K.; Urushihara, R.; Tanabe, M.; Katagiri, T.; Ozawa, T.; Takamatsu, H.; Ishiyama, K.; Yamazaki, H.; Kishi, H.; et al. Frequent HLA-DR loss on hemopoietic stem progenitor cells in patients with cyclosporine-dependent aplastic anemia carrying HLA-DR15. Leukemia 2022, 36, 1666–1675. [Google Scholar] [CrossRef]
- Zaimoku Patel, B.A.; Adams, S.; Shaloub, R.; Groarke, E.; Lee, A.I.C.; Kajigaya, S.; Feng, X.; Rios, O.J.; Eager, H.; Alemu, L.; et al. HLA associations, somatic loss of HLA expression and clinical outcomes in immune aplastic anemia. Blood 2021, 138, 2781–2898. [Google Scholar] [CrossRef]
- Young, N.L. Current concepts in the pathophysiology and treatment of aplastic anemia. Am. Soc. Hematol. Educ. Program. 2013, 76, 76–78. [Google Scholar] [CrossRef] [Green Version]
- Peffault de Latour, P.; Kulasekarara, A.; Iacobelli, S.; Trewel, S.R.; Cook, R.; Griffin, M.; Halkes, C.J.M.; Recher, C.; Barraco, F.; Forcade, E.; et al. for the Severe Aplastic Anemia Working Party of the European Society for Blood and marrow Transplantation. Eltrombopag Added to Immunosuppression in Severe Aplastic Anemia. N. Engl. J. Med. 2022, 286, 11–23. [Google Scholar] [CrossRef]
- Bacigalupo, A. How I treat acquired aplastic anemia. Blood 2017, 129, 1428–1436. [Google Scholar] [CrossRef]
- Cesaro, S.; Peffault de Latour, R.; Tridello, G.; Pillon, M.; Carlson, K.; Fagioli, F.; Jouet, J.-P.; Koh, M.B.C.; Panizzolo, I.R.; Kyrcz-Krzemien, S.; et al. Second allogeneic stem cell transplant for aplastic anaemia; a retrospective study by the severe aplastic anaemia Working Party of the EBMT. Br. J. Haematol. 2015, 171, 606–614. [Google Scholar] [CrossRef] [PubMed]
- McCann, S.R.; Bacigalupo, A.; Gluckman, E.; Hinterberger, W.; Hows, J.; Ljungman, P.; Marin, P.; Nissen, C.; van’t Veer Kerthof, E.; Raghavachar, A.; et al. Graft rejection and second bone marrow transplantation for acquired aplastic anaemia: A report from the aplastic anaemia Working Party of the EBMT. Bone Marrow Transplant. 1994, 13, 233–237. [Google Scholar]
- Dufour, C.; Veys, P.; Carraro, E.; Bhatnagar, N.; Pillon, M.; Wynn, R.; Gibson, B.; Vora, A.J.; Steward, C.G.; Ewins, A.M.; et al. Similar outcomes of upfront-unrelated and matched sibling stem cell transplantation in idiopathic paediatric aplastic anaemia. A study on behalf of the UK Paediatric BMT Working Party, Paediatric Diseases and Severe Aplastic Working Party of the EBMT. Br. J. Haematol. 2015, 171, 585–594. [Google Scholar] [CrossRef]
- Young, M.E.; Potter, V.; Kulasekararaj, A.G.; Mufti, G.J.; Marsh, J.C. Haematopoietic stem cell transplantation for acquired aplastic anaemia. Curr. Opin. Hematol. 2013, 20, 515–520. [Google Scholar] [CrossRef] [PubMed]
- Sociè, G.; Henry-Amar, M.; Cossett, J.M.; Devergie, A.; Grinsky, T.; Gluckman, E. Increased Incidence of Solid Malignant tumors after Bone Marrow Transplantation for Severe Aplastic Anemia. Blood 1991, 78, 277–279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lawler, M.; McCann, S.R.; March, J.C.; Ljungman, P.; Hows, J.; Vandenberghe, E.; O’Riordan, J.; Locasciulli, A.; Socié, G.; Kelly, A.; et al. Severe Aplastic Anaemia Working Party of the European Blood and Marrow Transplant Group. Serial chimerism analyses indicate that mixed hematopoietic chimerism influences the probability of graft rejection and disease recurrence following allogeneic stem cell transplantation (SCT) for severe aplastic anaemia (SAA): Indication for routine assessment of chimerism post SCT for SAA. Br. J. Haematol. 2009, 144, 933–934. [Google Scholar] [PubMed]
- Piccin, A.; McCann, S.; Socié, G.; Oneto, R.; Bacigalupo, A.; Locasciulli, A.; Marsh, J.; Schrezenmeier, H.; Tichelli, A.; Hand, E.; et al. Survival of patients with documented autologous recovery after SCT for severe aplastic anaemia: A study by the WPSAA of the EBMT. Bone Marrow Transpl. 2010, 45, 1008–1013. [Google Scholar] [CrossRef] [Green Version]
- Marsh, J.C.; Ball, S.E.; Cavenagh, J.; Darbyshire, P.; Dokal, I.; Gordon-Smith, E.C.; Keidan, J.; Laurie, A.; Martin, A.; Merciera, J.; et al. Guidelines for the diagnosis and management of aplastic anaemia. Br. J. Haematol. 2009, 147, 43–70. [Google Scholar] [CrossRef] [PubMed]
- Killick, S.B.; Brown, N.; Cavenagh, J.; Dokal, I.; Foukaneli, T.; Hill, A.; Hillmen, P.; Ireland, R.; Kulasekaraj, A.; Mufti, G.; et al. Wood Guidelines for the diagnosis and management of adult aplastic anaemia. British Society for Standards in Haematology. Br. J. Haematol. 2016, 172, 187–207. [Google Scholar] [CrossRef]
- McCann, S.R. Can guidelines inhibit innovation and critical thinking? Bone Marrow Transplant. 2020, 55, 1217–1219. [Google Scholar] [CrossRef]
Figure 1.
Overlapping between SAA and other conditions with similar features. These conditions need to be out ruled during differential diagnosis. SAA: severe aplastic anaemia; PRCA: pure red-cell anaemia; MDS: myelodisplastic syndrome; hMDS: hypoplastic myelodisplastic syndrome; PNH: paroxisitc nocturnal haemoglobinuria; IBMF: inherited bone marrow failure.
Figure 1.
Overlapping between SAA and other conditions with similar features. These conditions need to be out ruled during differential diagnosis. SAA: severe aplastic anaemia; PRCA: pure red-cell anaemia; MDS: myelodisplastic syndrome; hMDS: hypoplastic myelodisplastic syndrome; PNH: paroxisitc nocturnal haemoglobinuria; IBMF: inherited bone marrow failure.

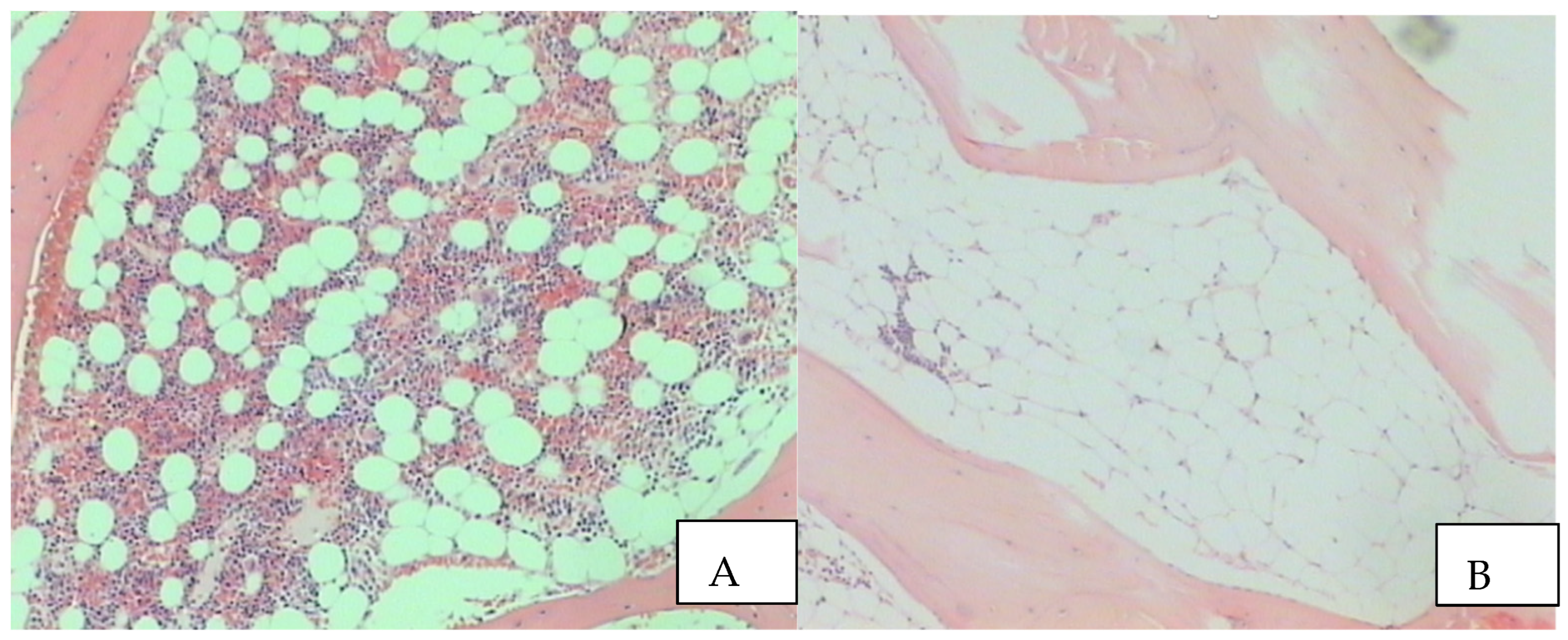
Figure 2.
Low power magnification of a bone marrow biopsy showing a normal bone marrow (A). Note, the cellularity is clearly present and is in a ratio 1-2/1 with the adipose tissue. Low power magnification of a bone marrow biopsy in a case of SAA. Please note the minimal residual cellularity which is fully replaced by fatty cells (B).
Figure 2.
Low power magnification of a bone marrow biopsy showing a normal bone marrow (A). Note, the cellularity is clearly present and is in a ratio 1-2/1 with the adipose tissue. Low power magnification of a bone marrow biopsy in a case of SAA. Please note the minimal residual cellularity which is fully replaced by fatty cells (B).

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
McCann, S.R.; Piccin, A. The Benign Clone Causing Aplastic Anaemia. Thalass. Rep. 2023, 13, 157-164. https://doi.org/10.3390/thalassrep13020015
AMA Style
McCann SR, Piccin A. The Benign Clone Causing Aplastic Anaemia. Thalassemia Reports. 2023; 13(2):157-164. https://doi.org/10.3390/thalassrep13020015
Chicago/Turabian StyleMcCann, Shaun R., and Andrea Piccin. 2023. "The Benign Clone Causing Aplastic Anaemia" Thalassemia Reports 13, no. 2: 157-164. https://doi.org/10.3390/thalassrep13020015