
Thalass. Rep. 2024, 14(2), 26-32; https://doi.org/10.3390/thalassrep14020004 - 10 Apr 2024
Abstract
►
Show Figures
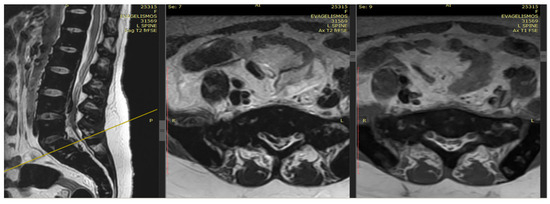
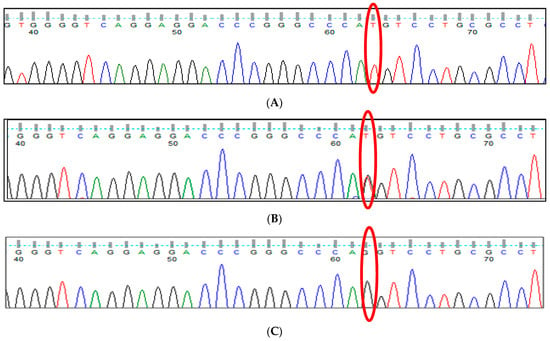
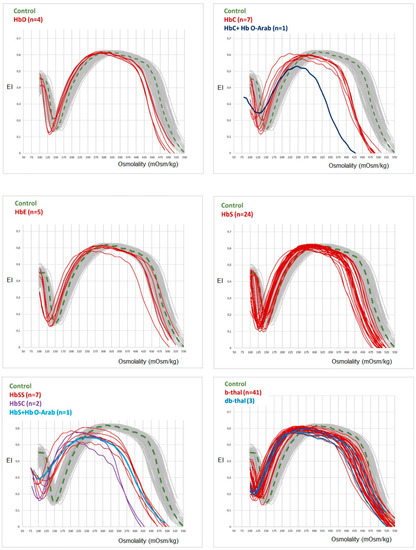
Extramedullary hematopoiesis (EMH) serves as a compensatory mechanism in chronic hemolytic anemias, such as thalassemia, and can result in spinal cord compression. This case report highlights a 36-year-old woman with transfusion-dependent β-thalassemia (TDT) who presented with lower extremity motor deficiency, pelvic paresthesia, and
[...] Read more.
Extramedullary hematopoiesis (EMH) serves as a compensatory mechanism in chronic hemolytic anemias, such as thalassemia, and can result in spinal cord compression. This case report highlights a 36-year-old woman with transfusion-dependent β-thalassemia (TDT) who presented with lower extremity motor deficiency, pelvic paresthesia, and bladder dysfunction. The patient had a history of lower back pain, bilateral lower limb weakness, and demonstrated poor compliance with iron chelation therapy. MRI findings indicated spinal cord compression attributable to extramedullary hematopoiesis. Due to the infeasibility of surgical intervention, the patient underwent hypertransfusion and iron chelation therapy. While neurological symptoms improved, urinary retention persisted. The patient continues to receive iron chelation treatment and undergo transfusions. Managing extramedullary hematopoiesis in thalassemia necessitates an individualized treatment approach.
Full article
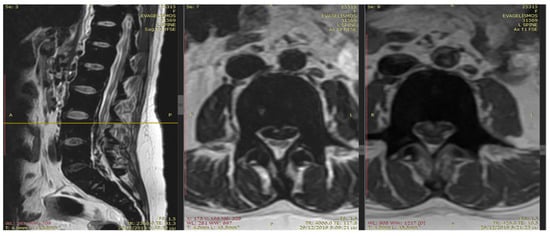
Figure 1





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}